 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of European pharmacopoeial impurities A, B, C, and D of cabergoline†
Jernej Wagger*a, Aljaž Požesb and Franc Požganbc
aLek d.d., a Sandoz Company, Kolodvorska cesta 27, 1234 Mengeš, Slovenia. E-mail: jernej.wagger@sandoz.com; Tel: +386 1 721 7206
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, 1000 Ljubljana, Slovenia
cEN→FIST Centre of Excellence, Dunajska c. 156, 1000 Ljubljana, Slovenia
First published on 25th September 2013
Abstract
For the use of analytics, European pharmacopoeial impurities A, B, C, and D of cabergoline were synthesized. Ergocryptine was chosen as a starting material and synthesis was accomplished via two approaches, different in length and stereochemical outcome. A longer, indirect approach was realized through otherwise problematic oxidations of the 9,10-dihidrolysergol derivative, to the corresponding aldehyde and carboxylic acid. This was achieved by the use of activated DMSO and a Pinnick oxidation sequence. All four synthesized impurities are used as analytical standards in cabergoline manufacturing processes.
Introduction
Cabergoline (1) is a natural product-based drug, representing a complex, branched amide of 6-N-allylated 9,10-dihydrolysergic acid (cabergolinic acid) A (Fig. 1). Primarily acting as a dopaminergic D2 receptor agonist, cabergoline is most widely used for treatment of hyperprolactinaemic disorders and both early and advanced Parkinson's disease.1 The structure of cabergoline also encodes for different biogenic amines than dopamine, therefore displaying modest pharmacodynamic properties towards adrenergic and serotonergic receptors.1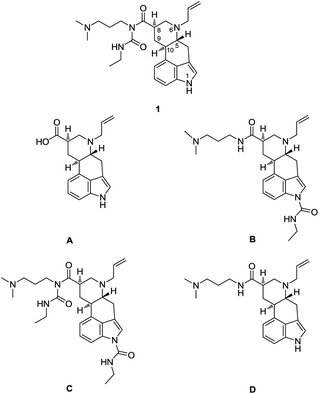 | ||
| Fig. 1 Cabergoline (1) and Ph. Eur. impurities of cabergoline A, B, C and D. | ||
One of the fundamental roles of the pharmaceutical industry is to develop new drugs that are safe, effective and of high quality when they reach the patients. With respect to this, delivering an impurity profile of an active pharmaceutical ingredient (API) is a must for fulfilling the aforementioned criteria.2 For the purpose of qualifying and/or quantifying the impurity profile of cabergoline (1) as the API, we have synthesized four known impurities, qualified by European Pharmacopoeia (Ph. Eur.)3 as impurities A, B, C and D (Fig. 1).
The genesis of these impurities can be found in the syntheses and other manufacturing process activities of cabergoline (Scheme 1).4
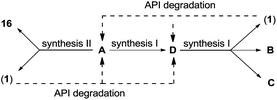 | ||
| Scheme 1 Possible routes of cabergoline impurities formation. Some impurities (A, D) can represent syntheses intermediates or are formed via hydrolysis of amide bonds from cabergoline (1) or other impurities (B, C). | ||
Since cabergoline (1), with the maximum weekly dosage of 2.0 mg, falls into low dosing drugs, the threshold for identification and qualification of impurities in API is usually 0.10% and 0.15% of mass, respectively.2 These limits are low and efficient production processes should not exceed them. Therefore production syntheses are usually neither suitable nor cost effective means to produce impurities for the use of analytics. Often, new synthesis routes have to be designed to obtain desired impurities and this was also our task. Synthesis routes for Ph. Eur. impurities A, B, C and D of cabergoline (1) were designed and results are presented in this paper.
Results and discussion
9,10-Dihydrolysergic acid (2) was selected as one of the key synthetic intermediates. Although there are several total syntheses of lysergic acid available, through which 9,10-dihydrolysergic acid (2) could be prepared, α-ergocryptine (3) was chosen as starting material. The reason is that α-ergocryptine (3) is biosynthetically produced at our production site and way of preparing 9,10-dihydrolysergic acid (2) from α-ergocryptine (3) is advantageous over total synthesis because it is shorter, easier and more economical. Also, there is a longstanding tradition of ergot alkaloids chemistry within Sandoz company, pioneered by Albert Hofmann.Starting with α-ergocryptine (3), the double bond between C9–C10 was diastereoselectively hydrogenated and the reduced ergopeptine 4 was cleaved by alkaline hydrolysis to give 9,10-dihydrolysergic acid (2).5 To increase solubility of 9,10-dihydrolysergic acid (2) in organic solvents, the corresponding methyl ester 5 was prepared (Scheme 2).6
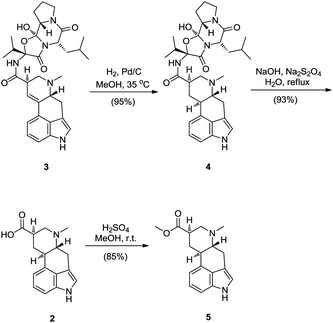 | ||
| Scheme 2 Synthesis of 9,10-dihydrolysergic acid methyl ester 5. | ||
In the next steps allylic substituent on 6-N of 5 had to be introduced. To introduce the allylic substituent on 6-N of 5, 9,10-dihydrolysergic acid (2) had to be N-demethylated first. 2,2,2-Trichloroethyl chloroformate (TrocCl) was used, in a transformation mechanistically analogues to the von Braun reaction,7 to N-demethylate 9,10-dihydrolysergic acid methyl ester (5). Subsequent zinc mediated reductive cleavage of 6-N-Troc substituted compound 6 gave the desired 6-N-demethylated intermediate 7. This N-demethylation sequence was already reported earlier and slight procedure modifications were used in our case.8 We have noticed that intermediate 7 decomposes slowly in acetic acid and is photolabile and therefore, the reaction yield depends on the swiftness of work-up and isolation. 6-N-Allylation of 7 was then realized by reaction with allylbromide and potassium carbonate in dimethylformamide, giving allylated intermediate 8 in a high yield (Scheme 3).9
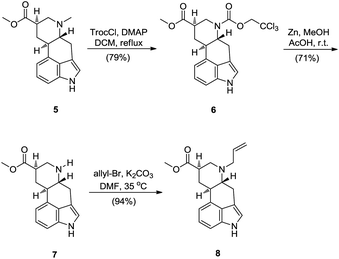 | ||
| Scheme 3 Synthesis of 6-N-allylated 9,10-dihydrolysergic acid methyl ester 8. | ||
At this point, intermediate 8 served us for the synthesis of impurities A and D. Alkaline hydrolysis of methyl ester 8 gave cabergolinic acid, termed as impurity A by Ph. Eur., in 82% yield. The impurity D was prepared directly from methyl ester 8 as well. By reacting 8 with 3-(dimethylamino)-1-propylamine in acetic acid, direct amidation of ester gave impurity D in 74% yield (Scheme 4).4
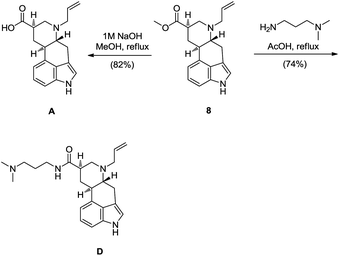 | ||
| Scheme 4 Synthesis of Ph. Eur. impurities A and D from intermediate 8. | ||
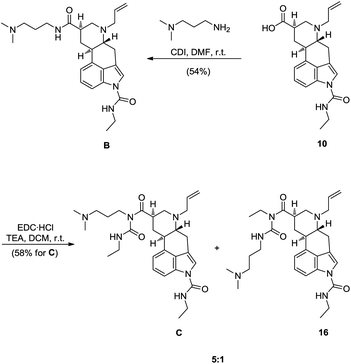
Compared to A and D, both impurities B and C have an ethylcarbamoyl functionality on the indole nitrogen. Since such functionalization of indole nitrogen requires the use of base, epimerization at the C8 position of ester 8 was possible. Although the stereocenter at C8 in 9,10-dihydrolysergic acid (2) is less prone to epimerization under basic conditions than in lysergic acid, due to the absence of C9–C10 double bond, epimerization does occur.10,11 For example, it is known from the literature, that exposing compound 5 to 4 equivalents of NaH in THF at 20 °C, app. 30% of the opposite, thermodynamically less favourable, axial epimer is observed, and by exposing compound 8 to 4 equivalents of LDA in THF at −20 °C, almost 71% of axial epimer is generated.11 Therefore, for installing the ethylcarbamoyl fragment on the indole nitrogen N-1 we have envisioned two different approaches. In the first one, a direct functionalization of N-1 of 8, under basic conditions with EtNCO was planned. Being aware of the potential epimerisation problems under these conditions, a second indirect approach was envisioned. In this, indirect approach it was planned to reduce C8 carbonyl group of 8 to corresponding alcohol, do the functionalization of N-1 with EtNCO under basic conditions and then reoxidise the alcohol back to the corresponding carboxylic acid. Although longer, this reduction/oxidation approach, in comparison to the first direct one, would ensure us with the epimerisation free preparation of the desired carboxylic acid. Since both approaches for functionalization of N-1, direct and indirect one, have advantages and disadvantages, we have decided to test both of them. Subjecting methyl ester 8 to the 1.1 equivalents of NaH and EtNCO afforded the desired epimer 9a in 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio and with 47% isolated yield (Scheme 5).12 With the methyl ester 9a in hand we were only one step short of the key intermediate 9,10-dihydrolysergic acid 10, which would enable us to prepare impurities B and C. Luckily saponification of the methyl ester 9a proceeded selectively with respect to the N-1 ethylcarbamoyl fragment. This has provided us with the desired 9,10-dihydrolysergic acid derivative 10 in 77% yield. We found 9,10-dihydrolysergic acid 10 rather difficult to isolate, since it displays modest solubility in both organic solvents and water, but with acid 10 in hand, the door towards completion of the synthesis plan was open (Scheme 5).
:1 diastereomeric ratio and with 47% isolated yield (Scheme 5).12 With the methyl ester 9a in hand we were only one step short of the key intermediate 9,10-dihydrolysergic acid 10, which would enable us to prepare impurities B and C. Luckily saponification of the methyl ester 9a proceeded selectively with respect to the N-1 ethylcarbamoyl fragment. This has provided us with the desired 9,10-dihydrolysergic acid derivative 10 in 77% yield. We found 9,10-dihydrolysergic acid 10 rather difficult to isolate, since it displays modest solubility in both organic solvents and water, but with acid 10 in hand, the door towards completion of the synthesis plan was open (Scheme 5).
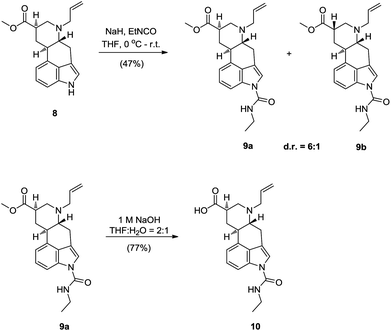 | ||
| Scheme 5 Direct synthesis of the key 9,10-dihydrolysergic acid 10—functionalization of the 9,10-dihydrolysergic acid methyl ester 8. | ||
For the indirect approach, methyl ester 8 had to be reduced first. Reacting ester 8 with the LiAlH4 in THF proceeded smoothly and in high yield to give 9,10-dihydrolysergol derivative 11.13 Hydroxy group in 9,10-dihydrolysergol derivative 11 was protected as TBDMS ether 12 and a carbamoyl moiety was introduced by reacting 12 with sodium hydride and ethylisocyanate to give TBDMS-protected 9,10-dihydrolysergol derivative 13 (Scheme 6).
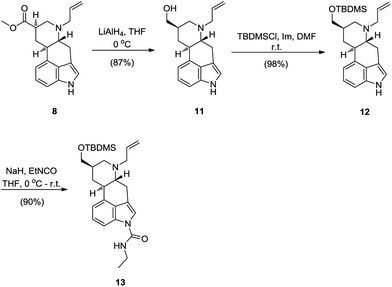 | ||
| Scheme 6 Synthesis of 13—introduction of an ethylcarbamoyl fragment onto the indole nitrogen of lysergol derivative 12. | ||
For the next steps, TBDMS-protected alcohol group in 13 had to be removed and free alcohol reoxidized back to carboxylic acid. Deprotection of silyl ether in 13 with TBAF gave 9,10-dihydrolysergol derivative 14 in very good yield, although some temperature dependent chemoselectivity was observed. When silyl deprotection was performed at 0 °C, slow but clean conversion of 13 to 14 was achieved. On the other hand, when the reaction was done at room temperature, removal of the ethylcarbamoyl group was observed in addition to silyl deprotection. 9,10-Dihydrolysergol derivative 14 was then used for subsequent oxidation reaction. Ideally a one-step oxidation procedure from primary alcohol 14 to the corresponding carboxylic acid would be a method of choice. But surprisingly, while searching the literature, we learned that examples covering oxidations of 9,10-dihydrolysergols and their derivatives to the corresponding 9,10-dihydrolysergals and/or 9,10-dihydrolysergic acids are very scarce. In the most recent literature, Křen et al. reported on their efforts to oxidize N-1-protected lysergol derivatives to aldehydes and acids.14 Beside that, patent literature covers two examples of 9,10-dihydrolysergol oxidations. In the first one BAIB/TEMPO was used for direct oxidation of 9,10-dihydrolysergol derivative to the corresponding 9,10-dihydrolysergic acid.15 Unfortunately, BAIB/TEMPO mediated oxidation did not work in our case, giving only traces of the desired acid, and failure to oxidize the alcohol 14 is in agreement with the results reported in the literature.14 In the second literature example, slightly modified Parikh–Doering oxidation (SO3·TEA, DMSO, TEA) was successfully used to oxidize 9,10-dihydrolysergol to the 9,10-dihydrolysergal,16 but as reported by Křen, they were unsuccessful in repeating this type of oxidation under standard Parikh–Doering conditions (SO3·Py, DMSO, TEA).14 Since 9,10-dihidrolysergol derivatives are poorly soluble in various solvents, we anticipated that DMSO mediated oxidations could be a good choice amongst the plethora of alcohol oxidation methods. DMSO should address the solubility issue of 9,10-dihydrolysergol derivative and concurrently serve as an oxidant. Since contrary reports about effectiveness of the Parikh–Doering oxidation of 9,10-dihidrolysergols were available in the literature, we have decided to try other analogues reaction. Luckily one can chose among different DMSO mediated oxidations. Indeed, when alcohol 14 was subjected to Pfitzner–Moffatt oxidation conditions, a clean and high yielding conversion to the corresponding 9,10-dihydrolysergal 15 was observed.17 Here again temperature dependence on reaction selectivity was observed. Below 5 °C the reaction was clean, giving only aldehyde as a product, while at room temperature or higher several side products could be observed by TLC. Since we were successful with Pfitzner–Moffatt oxidation of 14, the question was raised, whether the outcome of Parikh–Doering reaction, on the same alcohol substrate would be analogues? Oxidation of the alcohol 14, under the Parikh–Doering conditions, proceeded smoothly, affording aldehyde 15 in slightly lower yield than via Pfitzner–Moffatt reaction (Scheme 7).18
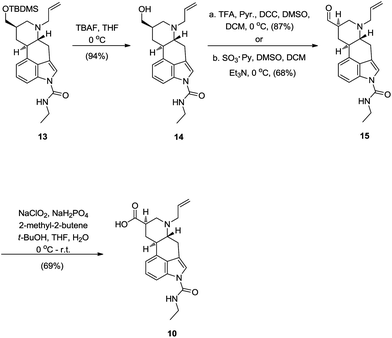 | ||
| Scheme 7 Indirect, oxidative route to the 9,10-dihydrolysergic acid 10. | ||
An added value of this oxidation transformations also lies in the 9,10-dihydrolysergal itself. Knowing that aldehydes of this type are difficult to prepare and that pharmacological activity of synthetically modified ergot alkaloids varies mainly because of the nature and configuration of substituents on C8, such aldehydes also represent valuable medicinal chemistry intermediates.4a,16a,19
Oxidations of 9,10-dihydrolysergals to the corresponding 9,10-dihydrolysergic acids are hardly reported in the literature. This is mainly due to the fact that 9,10-dihydrolysergic acid derivatives are easily accessible from naturally occurring lysergic acid. The literature reports on oxidation of 9,10-dihydrolysergal with Tollens's reagent or MCPBA, but both methods have their limitations.14 The first one unfortunately gives poor yields while the second one suffers from chemoselectivity. Peroxy acid oxidizes not only the aldehyde, but also the nitrogen on position 6 to the corresponding N-oxide. Although reduction of N-oxide to the desired 9,10-dihydrolysergic acid was demonstrated,14 this method suffers from functional group compatibility, since allylated lysergal derivatives are not tolerated by peroxy acids nor catalytic hydrogenation. On the other hand there are some examples of lysergal oxidations to lysergic acid derivatives. In the final steps of total synthesis of lysergic acid by Fujii and Ohno, Pinnick oxidation was used to oxidize 1-N-6-N-ditosylated isolysergal into the corresponding lysergic acid.20 Indeed, upon subjecting aldehyde 15 to Pinnick oxidation, the desired 9,10-dihydrolysergic acid 10 was obtained, in 69% yield (Scheme 7).21
9,10-Dihydrolysergic acid 10 was then used to synthesize both impurities B and C. The coupling of 10 with 3-(dimethylamino)-1-propylamine by the use of CDI furnished the impurity B in 54% yield. For the synthesis of impurity C a more complex N-acylurea appendage on C8 had to be installed. In the original synthesis of cabergoline, this side chain was installed with the use of EDC·HCl, by exploiting the usually undesired ability of carbodiimide coupling reagents to form N-acylureas as intramolecular rearrangement side products.22 Reacting cabergolinic acid A with EDC·HCl, in the absence of an external nucleophile, Mantegani and co-workers synthesized cabergoline (1).4 Although the use of EDC·HCl gives the desired N-acylurea appendage in one step, a disadvantage of using this asymmetric carbodiimide is concomitant formation of regioisomeric N-acylurea and tedious separation of both isomers.
To compensate for disadvantage of applying EDC·HCl, different synthetic strategy was utilized in which excess ethylisocyanate was reacted with compound D to install N-acylurea appendage. Unfortunately the use of ethylisocyanate is also the root cause for formation of impurities B and C as side products in cabergoline (1) synthesis (Scheme 1).4 Other methods for N-acylurea installation in cabergoline (1) synthesis were also developed, although none of them introduces N-acylurea appendage in one step from the corresponding 9,10-dihydrolysergic acid.23 Therefore, compromising lower yield, we have used EDC·HCl in DCM24 for the synthesis of impurity C, thus preparing the desired impurity and its regioisomer 16 in ratio 5:1 respectively.25 After separation of regioisomers, the impurity C was obtained in 58% yield (Scheme 8).
 | ||
| Scheme 8 Synthesis of Ph. Eur. impurities B and C. | ||
For the synthesis of C, another synthetic strategy would be viable, by following linear route 10 → B → C. By reacting B with an excess of EtNCO, impurity C could be prepared. In the original synthesis of cabergoline (1) up to 40 equivalents of EtNCO were reacted with compound D to get the desired API. The amount of EtNCO was later reduced in consequent synthesis optimizations, by the use of CuI and Ph3P catalysis, to as low as 3 equivalents. Under these, optimised conditions cabergoline (1) was prepared along with the impurities B and C in the ratio of 82:6:12 respectively.26 As mentioned earlier, other syntheses of cabergoline (1) were developed to avoid the use of toxic EtNCO and they are all based on the functionalization of the carboxamide side chain of D.23 In our case the impurity B was prepared from precursor 10 in a modest 54% yield. Therefore, if the reaction of B with EtNCO, or any other suitable reagent, would work quantitatively, the impurity C, starting linearly from 10, would be prepared in not greater than 54% yield. Following this, we have never tried to prepare impurity C from 10via impurity B. Instead, the impurity C was prepared in one step, directly from carboxylic acid 10 with 58% yield (Scheme 8).
The structure of proper N-acylurea appendage in the impurity C was determined using 2D COSY NMR technique, where expected interactions between ethylamido protons were observed (Fig. 2). The proton NMR of the impurity C is also in accordance with the previous report of Mantegani and co-workers.4
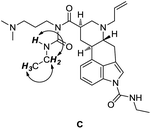 | ||
| Fig. 2 Detected COSY interactions in ethylamido side chain (CH3, 1.18 ppm; CH2, 3.30 ppm; NH, 9.45 ppm) in impurity C are indicated by arrows. | ||
Conclusions
A synthesis of four European pharmacopoeial impurities A, B, C and D of cabergoline (1) was demonstrated and the synthesized impurities are used as HPLC analytical standards. To the best of our knowledge, this is the first report on selective synthesis of these impurities. From α-ergocryptine (3), 9,10-dihydrolysergic acid methyl ester (5) was prepared, which served as the key starting material. Using 5, N-1 ethylcarbamoyl 9,10-dihydrolysergic acid 10 was prepared via two different routes. First route, a direct one, proceeds through direct N-1 ethylcarbamoylation of 10, causing partial epimerisation at C8 position. In spite of epimerisation, this route is shorter and therefore advantageous over the indirect one. Second route, an indirect one, follows reduction–oxidation of 9,10-dihydrolysergic acid derivative 8, for epimerisation free installation of N-1 ethylcarbamoyl moiety. Notwithstanding the fact that oxidations of 9,10-dihydrolysergol derivatives to the corresponding 9,10-dihydrolysergals and further on to 9,10-dihydrolysergic acids are very rare, we have shown that by combination of activated DMSO and Pinnick reactions this oxidation sequence can be achieved successfully. Furthermore, such oxidation method of preparing otherwise scarcely presented 9,10-dihydrolysergals, can be useful in medicinal chemistry, for preparing novel pharmacologically active C8-substituted derivatives of ergoline.Experimental
General experimental details
Where stated, anhydrous reactions were carried out in vacuum oven dried glassware, under stream of nitrogen, passed through Drierite™ drying column. Anhydrous solvents were purchased form Sigma-Aldrich and all reagents were commercial grade, used without any purification. Exception was α-ergocryptine which was acquired through in-house biofermentation process and was purified by crystallization from toluene. Reactions were monitored by thin layer chromatography (TLC) performed on pre-coated aluminum-baked silica gel (60 F254, Merck) or aluminum oxide (60 F254, Merck) plates. Developed TLC plates were visualized under UV light and/or by aqueous cerium ammonium molybdate and aqueous potassium permanganate. Flash chromatography was performed on ZEOPrep 60 Eco 40–63 μm silica gel and aluminum oxide activated, basic, with indicated mobile phase. Lower-temperature reactions were performed on ice/water bath or on Mettler-Toledo EasyMax™ synthesis workstation. Infrared spectra were recorded on a Bruker Alpha (Platinum ATR) FT-IR spectrometer and are reported in reciprocal centimeters (cm−1). Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 500 and Bruker Avance DPX 300 spectrometers. Chemical shifts for 1H NMR spectra are reported in parts per million from tetramethylsilane, with the solvent resonance as the internal standard (CDCl3, δ 7.26 ppm, DMSO-d6, δ 2.50 ppm). Data are reported as follows: chemical shift, multiplicity, (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, m = multiplet, br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra are reported in parts per million from tetramethylsilane, using solvent central peak as the internal standard (CDCl3, δ 77.16 ppm, DMSO-d6, δ 39.52 ppm). Where stated 2D NMR spectra were recorded. For pH measurements of water solutions, Seven Easy Mettler-Toledo pH meter was used. Optical rotations were determined at 589 nm at ambient temperature on Perkin-Elmer 2400 241MC polarimeter. Data reported as follows: [α]D, concentration (c in g per 100 mL) and solvent. Elemental microanalyses were performed on Perkin-Elmer Analyser 2400. Mass spectra were obtained using an AutoSpecQ, Q-TOF Premier and Agilent 6224 Accurate TOF LC/MS spectrometers. Melting points were determined on a Kofler micro hot stage and are uncorrected.:10%); mp = 224–226 °C, ref. 5a = 235 °C; [α]D −30.1 (c 0.53; pyr.), ref. 5a [α]D −41 (Pyr.); IR: νmax 3627, 3369, 3354, 3198, 3064, 2955, 2847, 2792, 1720, 1657, 1626, 1546, 1432, 1218, 1191, 1003, 755 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 0.86 (d, J = 6.7 Hz, 3H); 0.90 (d, J = 6.8 Hz, 3H); 0.94 (d, J = 6.5 Hz, 3H); 1.05 (d, J = 6.7 Hz, 3H); 1.40 (q, J = 12.4 Hz, 1H); 1.64–1.82 (m, 3H); 1.85–2.05 (m, 5H); 2.12 (dt, J1 = 13.5 Hz, J2 = 6.7 Hz; 1H); 2.22 (t, J = 12.2 Hz, 1H); 2.36 (s, 3H); 2.48–2.54 (m, 1H); 2.63 (bd, J = 12.2 Hz, 1H); 2.78–2.84 (m, 1H); 2.94–3.02 (m, 2H), 3.30 (dd, J1 = 14.7 Hz, J2 = 4.1 Hz, 1H); 3.35–3.41 (m, 2H); 3.72 (dd, J1 = 8.4 Hz, J2 = 5.9 Hz, 1H); 4.32 (dd, J1 = 7.6 Hz, J2 = 5.8 Hz, 1H); 6.74 (d, J = 7.02 Hz, 1H); 6.98 (bs, 1H); 7.02 (t, J = 7.9 Hz, 1H); 7.13 (d, J = 8.1 Hz; 1H); 7.57 (d, J = 1.58 Hz, 1H); 9.05 (s, 1H); 10.66 (d, J = 1.55 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 15.7, 16.6, 21.5, 22.2, 22.7, 24.3, 26.0, 26.6, 30.5, 33.0, 39.4, 41.8, 42.6, 43.0, 45.7, 52.4, 58.8, 63.5, 66.5, 90.15, 103.3, 108.9, 110.0, 111.7, 118.6, 122.1, 125.9, 132.16, 133.20, 164.9, 165.2, 177.1; HRMS (ESI): m/z calcd for C32H44N5O5: [M + H]+, 578.3337; found: 578.3337.:13%:12%); mp = decomposition at 250 °C, ref. 5a compound darkens at 250 °C and decomposes at 300 °C; [α]D −120 (c 0.1; pyr.), ref. 5a [α]D −122 (c 0.2; Pyr.); IR: νmax 3370, 3240, 3194, 3117, 3048, 2948, 2852, 2348, 1577, 1445, 1390, 1345, 1090, 1029, 1007, 764 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 1.34 (q, J = 12.9 Hz, 1H); 2.00 (dt, J1 = 10.9 Hz, J2 = 4.1 Hz, 1H); 2.17 (t, J = 11.5 Hz, 1H); 2.37 (s, 3H); 2.48–2.55 (m, 1H); 2.70–2.85 (m, 3H); 3.12 (dd, J1 = 11.0 Hz, J2 = 2.1 Hz, 1H); 3.29 (dd, J1 = 14.6 Hz, J2 = 4.1 Hz, 1H); 6.79 (d, J = 7.1 Hz, 1H); 6.98 (bs, 1H); 7.01 (t, J = 7.6 Hz, 1H); 7.13 (d, J = 8.1 Hz, 1H); 10.65 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 26.5, 30.5, 40.9, 42.6, 58.6, 66.6, 108.8, 110.0, 112.0, 118.6, 122.1, 125.9, 132.3, 133.2, 175.1; HRMS (ESI): m/z calcd for C16H19N2O2: [M + H]+, 271.1441; found: 271.1443.:1); mp = 189–190 °C; [α]D −66.1 (c 0.98; DCM); IR: νmax 3142, 3094, 3002, 2947, 2864, 2834, 2804, 1729, 1608, 1443, 1238, 1096, 1025, 745 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.56–1.66 (m, 1H); 2.21 (ddd, J1 = 11.1 Hz, J2 = 9.4 Hz, J3 = 4.3 Hz, 1H); 2.34–2,42 (m, 1H); 2.52 (s, 3H); 2.66–2.75 (m, 1H); 2.94–3.04 (m, 3H); 3.26–3.32 (m, 1H); 3.42 (dd, J1 = 14.7 Hz, J2 = 4.3 Hz, 1H); 3.75 (s, 3H); 6.89 (t, J = 1.6 Hz, 1H); 6.95–6.98 (m, 1H); 7.16–7.21 (m, 2H); 8.07 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 27.0, 30.7, 40.3, 41.6, 43.2, 51.9, 58.8, 66.8, 108.8, 111.9, 113.4, 118.0, 123.3, 126.2, 132.8, 133.5, 174.5; HRMS (ESI): m/z calcd for C17H21N2O2: [M + H]+, 285.1598; found: 285.1595.:2); mp = 173–175 °C, ref. 27 172 °C; [α]D −24 (c 1.33; DCM); IR: νmax 3402, 3261, 3144, 3093, 3003, 2946, 2861, 2801, 1728, 1601, 1548, 1517, 1435, 1248, 1234, 1199, 1109, 903, 732 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.63–1.72 (m, 1H); 2.74–2.92 (m, 6H); 2.96–3.02 (m, 1H); 3.05–3.12 (m, 1H); 3.46–3.52 (m, 1H); 3.74 (s, 3H); 6.88 (s, 1H); 6.94 (d, J = 6.7 Hz, 1H); 7.15–7.22 (m, 2H); 8.03 (bs, 1H). 13C NMR (CDCl3, 125 MHz) δ 29.6, 30.8, 41.3, 43.0, 48.4, 52.0, 59.6, 109.0, 111.8, 113.3, 117.8, 123.2, 126.6, 132.5, 133.6, 174.4; HRMS (ESI): m/z calcd for C16H19N2O2: [M + H]+, 271.1441; found: 271.1440.:12); mp = 146–149 °C, ref. 10 143–146 °C; [α]D −117.1 (c 1.0; DCM), ref. 10 [α]D −117.2; IR: νmax 3402, 3147, 2948, 2838, 1729, 1641, 1610, 1547, 1443, 1365, 1201, 751 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.63–1.73 (m, 1H); 2.49 (t, J = 11.6 Hz; 1H); 2.54 (td, J1 = 10.1 Hz, J2 = 4.8 Hz, 1H); 2.75 (bt, J = 13.0 Hz, 1H); 2.90–3.05 (m, 3H); 3.34 (d, J = 11 Hz, 1H); 3.38–3.49 (m, 2H); 3.60 (dd, J1 = 8.6 Hz, J2 = 5.8 Hz, 1H); 3.75 (s, 3H); 5.23 (d, J = 10.2 Hz, 1H); 5.29 (d, J = 17.1 Hz, 1H); 5.99 (m, 1H); 6.89 (s, 1H); 6.95 (d, J = 6.3 Hz, 1H); 7.15–7.21 (m, 2H); 8.07 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 26.7, 30.8, 40.5, 41.7, 51.9, 54.8, 56.5, 63.7, 108.8, 111.9, 113.4, 118.0, 118.6, 123.3, 126.2, 132.9, 133.4, 133.8, 174.7; HRMS (ESI): m/z calcd for C19H23N2O2: [M + H]+, 311.1754; found: 311.1753.:1:1); mp = 246–247 °C; [α]D −138.4 (c 0.72; DMSO); IR: νmax 3208, 2942, 2322, 1650, 1589, 1448, 1365, 1337, 1283, 952, 757 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 1.31–1.41 (m, 1H); 2.30 (t, J = 11.5 Hz, 1H); 2.29–2.37 (m, 1H); 2.56 (bt, J = 12.8 Hz, 1H); 2.66–2.75 (m, 1H); 2.78–2.86 (m, 2H); 3.16–3.21 (m, 1H); 3.27 (dd, J1 = 14.6 Hz, J2 = 7.4 Hz, 1H); 3.36 (dd, J1 = 14.6 Hz, J2 = 3.9 Hz, 1H); 3.51 (dd, J1 14.6 Hz, J2 = 5.5 Hz, 1H); 5.17 (d, J = 10.2 Hz, 1H); 5.26 (d, J = 17.0 Hz, 1H); 5.90–6.00 (m, 1H); 6.79 (d, J = 7.1 Hz, 1H); 6.98 (s, 1H); 7.02 (t, J = 7.6 Hz, 1H); 7.14 (d, J = 8.1 Hz, 1H); 10.65 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 26.2, 30.6, 39.9, 40.9, 54.7, 55.6, 63.5, 108.8, 110.1, 112.1, 117.9, 118.6, 122.1, 125.9, 132.4, 133.2, 134.7, 175.2; HRMS (ESI): m/z calcd for C18H21N2O2: [M + H]+, 297.1598; found: 297.1595.:12); mp = 201–203 °C, ref. 4a 200–202 °C; [α]D −63.0 (c 0.87; DCM); IR: νmax 3365, 3296, 3067, 2941, 2795, 2719, 1623, 1545, 1443, 1417, 1292, 911, 739 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.60–1.71 (m, 3H); 2.26 (s, 6H), 2.41–2.45 (m, 2H); 2.48 (t, J = 11.5 Hz, 1H); 2.49–2.56 (m, 1H); 2.62–2.68 (m, 1H); 2.68–2.75 (m, 1H), 2.79–2.86 (m, 1H); 2.94–3.02 (m, 1H); 3.21–3.26 (m, 1H); 3.36–3.43 (m, 3H); 3.46 (dd, J1 = 14.5 Hz, J2 = 4.1 Hz, 1H); 3.58 (dd, J1 = 14.4 Hz, J2 = 5.9 Hz, 1H); 5.19 (d, J = 10.1, 1H); 5.25 (dd, J1 = 17.1 Hz, J2 = 0.8 Hz, 1H); 5.92–6.03 (m, 1H); 6.87–6.93 (m, 2H); 7.13–7.20 (m, 2H); 7.64 (bs, 1H); 8.19 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ 25.5, 26.8, 31.1, 40.1, 40.7, 43.8, 45.6, 45.7, 55.8, 56.7, 59.4, 63.7, 108.8, 111.8, 113.2, 118.0, 118.7, 123.2, 126.2, 133.0, 133.4, 133.7, 173.7; HRMS (ESI): m/z calcd for C23H33N4O: [M + H]+, 381.2649; found: 381.2647.:1); mp = 176–178 °C; [α]D −126.2 (c 0.16; DMF); IR: νmax 3316, 3068, 3021, 2977, 2948, 2858, 2789, 1731, 1666, 1604, 1537, 1436, 1341, 1274, 1160, 1116, 1073, 929, 752, 635 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 1.15 (t, J = 7.2 Hz, 1H); 1.32–1.42 (m, 1H); 2.28–2.36 (m, 2H); 2.48–2.56 (m, 1H); 2.77–2.86 (m, 3H); 3.15–3.21 (m, 1H); 3.24–3.30 (m, 3H); 3.31–3.36 (m, 1H); 3.48–3.55 (m, 1H); 3.65 (s, 3H); 5.18 (d, J = 10.3 Hz, 1H); 5.27 (d, J = 17.0 Hz, 1H); 5.87–6.00 (m, 1H); 7.00 (d, J = 7.4 Hz, 1H); 7.19 (t, J = 7.7 Hz, 1H); 7.52 (d, J = 1.3 Hz, 1H); 7.78 (d, J = 8.2 Hz, 1H); 7.99 (t, J = 5.4 Hz, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 15.0, 25.7, 30.3, 34.9, 40.6, 51.6, 54.3, 55.4, 62.8, 112.9, 115.1, 115.9, 117.9, 118.1, 124.7, 127.1, 132.8, 133.1, 134.6, 151.8, 173.7; HRMS (ESI): m/z calcd for C22H28N3O3: [M + H]+: 382.2125; 382.2118.(a) Via saponification of 9a. In a round-bottom flask compound 9a (100 mg, 0.272 mmol) was dissolved in THF–H2O mixture (2
:1, 6 mL). To the mixture aqueous solution of NaOH (1.0 M, 0.6 mL) was added and reaction mixture was stirred for 1 h. After that aqueous HCl (1.0 M, 0.9 mL) was added reaction mixture was concentrated under reduced pressure. The residue on evaporation was purified by column chromatography (THF–H2O–AcOH = 98:1:1) to give titled compound as a yellowish solid (76.9 mg, 77%).(b) Via Pinnic oxidation of 15. To aldehyde 15 (1.84 g, 5.20 mmol), t-BuOH (18 mL) and THF (18 mL) were added and solution was stirred and cooled to ∼3 °C (ice/water bath). Then 2-methyl-2-butene (2.83 mL, 26.86 mmol) was added. To this a solution of NaH2PO4 (1.89 g, 15.7 mmol) and NaClO2 (1.42 g, 12.6 mmol, 80%) in H2O (18 mL) was added slowly. Reaction mixture was stirred for 1.5 h at ∼3 °C (ice/water bath) and then it was stopped with addition of Na2SO3 (2.02 g, 16.0 mmol). Precipitate formed was filtered off and filtrate was concentrated under reduced pressure. Residue on concentration was purified by column chromatography with two different mobile phases used sequentially. First mobile phase (DCM–MeOH = 10
:1) was used to separate yellow impurities and second mobile phase (THF–H2O–AcOH = 98:1:1) was used to elute product which upon evaporation and drying gave light yellowish solid (1.33 g, 68%). Rf: 0.57 (THF–H2O–AcOH = 98:1:1); mp = 155–158 °C (from EtOAc); [α]D −73.3 (c 1.19; DMSO); calc. for: C21H25N3O3·H2O: C, 65.44; H, 7.06; N, 10.90. Found: C, 65.03; H, 6.72; N, 10.58%; IR: νmax 3308, 2962, 2930, 2872, 2854, 1677, 1586, 1532, 1439, 1390, 1274, 1035, 753 cm−1. 1H NMR (DMSO-d6, 500 MHz) δ 1.15 (t, J = 7.1 Hz, 3H); 1.27–1.38 (m, 1H); 2.25–2.37 (m, 2H); 2.68 (t, J = 10.8 Hz, 1H); 2.76–2.85 (m, 2H); 3.17 (d, J = 11.1 Hz, 1H); 3.20–3.48 (m, 5H); 3.52 (dd, J1 = 14.6 Hz, J2 = 3.3 Hz; 1H); 5.17 (d, J = 10.1 Hz, 1H); 5.27 (d, J = 16.9 Hz, 1H); 5.90–6.00 (m, 1H); 7.01 (d, J = 7.2 Hz, 1H); 7.19 (t, J = 7.7 Hz, 1H); 7.52 (s, 1H); 7.87 (d, J = 8.0 Hz, 1H); 7.99 (bs, 1H), 12.50 (bs, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 15.0, 25.7, 30.5, 34.9, 39.8, 40.8, 54.6, 55.5, 62.9, 112.9, 115.2, 115.9, 117.8, 118.1, 124.7, 127.2, 133.0, 133.1, 134.7, 151.9, 175.1; HRMS (ESI): m/z calcd for C21H26N3O3: [M + H]+, 368.1969; found: 368.1967.:12); mp = 206–209 °C, ref. 8 204–206 °C; [α]D −74.4 (c 0.91, DMSO); IR: νmax 3240, 3056, 2923, 2847, 1868, 1797, 1644, 1612, 1552, 1469, 1445, 1342, 1035, 733 cm−1; 1H NMR (DMSO-d6, 500 MHz) δ 0.88–0.97 (m, 1H); 1.94 (t, J = 10.9 Hz, 1H); 2.27 (td, J1 = 10.2 Hz, J2 = 4.1 Hz, 1H); 2.52–2.59 (m, 2H); 2.73–2.80 (m, 1H); 3.07 (d, J = 9.9 Hz, 1H); 3.23 (dd, J1 = 14.6 Hz, J2 = 7.6 Hz, 1H); 3.26–3.31 (m, 1H); 3.36–3.43 (m, 3H); 3.49 (dd, J1 = 14.6 Hz, J2 = 5.6 Hz, 1H); 4.54 (t, J = 5.3 Hz, 1H); 5.15 (d, J = 10.4 Hz, 1H); 5.24 (d, J = 16.8 Hz, 1H); 5.95 (m, 1H); 6.77 (d, J = 7.1 Hz, 1H); 6.96 (bs, 1H); 7.00 (t, J = 7.6 Hz, 1H); 7.11 (d, J = 8.1 Hz, 1H); 10.60 (s, 1H). 13C NMR (DMSO-d6, 125 MHz) δ 26.4, 30.9, 38.3, 40.5, 56.1, 56.7, 64.1, 64.6, 108.6, 110.3, 112.0, 117.4, 118.4, 122.0, 126.0, 133.1, 135.1; HRMS (ESI): m/z calcd for C18H23N2O: [M + H]+, 283.1805; found: 283.1802.:12); mp = 225–227 °C (from MeOH); [α]D −69.0 (c 1.19; DCM); calc. for: C24H36N2OSi: C, 72.67; H, 9.15; N, 7.06. Found: C, 72.73; H, 9.39; N, 7.10%; IR: νmax 3096, 3013, 2949, 2929, 2899, 2856, 2813, 2784, 1738, 1645, 1618, 1549, 1518, 1462, 1357, 1217, 1098, 834, 772, 742 cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.08 (s, 3H); 0.09 (s, 3H); 0.93 (s, 9H); 1.09–1.18 (m, 1H); 2.02–2.16 (m, 2H); 2.49 (dt, J1 = 3.8 Hz, J2 = 10.9 Hz, 1H); 2.66 (d, J = 13.0 Hz, 1H); 2.75 (t, J = 12.7 Hz, 1H); 3.00 (bt, J = 9.1 Hz, 1H); 3.21 (d, J = 9.5 Hz, 1H); 3.38 (dd, J1 = 14.2 Hz, J2 = 7.5 Hz, 1H); 3.47 (dd, J1 = 14.6 Hz, J2 = 3.9 Hz, 1H); 3.51–3.56 (m, 1H); 3.58–3.63 (m, 2H); 5.20 (d, J = 10.1 Hz, 1H); 5.26 (d, J = 17.0 Hz, 1H); 5.95–6.06 (m, 1H); 6.89 (s, 1H); 6.92–6.95 (m, 1H); 7.15–7.20 (m, 2H), 8.01 (s, 1H). 13C NMR (CDCl3, 125 MHz) δ −5.2, −5.1, 1.2, 18.5, 26.1, 26.8, 29.8, 31.0, 38.7, 40.9, 56.8, 56.9, 64.3, 66.7, 108.58, 112.3, 113.3, 117.8, 118.2, 123.2, 126.4, 133.4, 134.0, 134.1; HRMS (ESI): m/z calcd for C24H37N2OSi: [M + H]+, 397.2675; found: 397.2673.:3) to give titled compound as pale yellow solid (16.94 g, 90%). Rf: 0.38 (EtOAc–heptane = 1:3); mp = 135–137 °C (from MeOH/H2O); [α]D −75.9 (c 1.40; DCM); calc. for: C27H41N3O2Si: C, 69.33; H, 8.84; N, 8.98. Found: C, 69.58; H, 9.11; N, 9.02%; IR: νmax 3377, 2952, 2925, 2853, 2810, 1737, 1661, 1605, 1535, 1460, 1449, 1438, 1291,1087, 837, 780, 754 cm−1; 1H NMR (CDCl3, 500 MHz) δ 0.07 (s, 3H); 0.08 (s, 3H); 0.91 (s, 9H); 1.05–1.13 (m, 1H); 1.28 (t, J = 7.2 Hz, 3H); 2.00–2.13 (m, 2H), 2.40 (td, J1 = 11.2 Hz, J2 = 3.9 Hz, 1H); 2.58–2.63 (m, 2H); 2.91 (bt, J = 9.1 Hz, 1H); 3.18 (d, J = 8.6 Hz, 1H); 3.31 (dd, J1 = 14.5 Hz, J2 = 7.6 Hz, 1H); 3.36 (dd, J1 = 15.0 Hz, J2 = 3.9 Hz, 1H); 3.46–3.64 (m, 5H); 5.19 (d, J = 10.2 Hz, 1H); 5.24 (d, J = 17.1 Hz, 1H); 5.66 (bs, 1H); 5.90–6.01 (m, 1H); 7.04 (d, J = 7.3 Hz, 1H); 7.20 (s, 1H); 7.27 (t, J = 7.8 Hz, 1H); 7.72 (d, J = 8.2 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ −5.23, −5.19, 15.1, 15.3, 18.4, 26.1, 26.5, 29.8, 30.9, 35.9, 38.5, 40.5, 56.6, 56.7, 63.8, 66.6, 111.7, 116.4, 116.5, 117.9, 118.4, 125.3, 128.4, 132.8, 133.8, 134.4, 152.5; HRMS (ESI): m/z calcd for C27H42N3O2Si: [M + H]+, 468.3041; found: 468.3040.:88), to give titled compound as a white solid (3.21 g, 94%). Rf: 0.38 (EtOH–EtOAc = 12:88); mp = 190–193 °C (from toluene); [α]D −92.5 (c 1.26; DMSO); calc. for: C21H27N3O2: C, 71.36; H, 7.70; N, 11.89. Found: C, 71.65; H, 8.02; N, 11.89%; IR: νmax 3424, 3303, 2932, 2913, 2856, 2812, 2784, 1664, 1603, 1533, 1439, 1243, 1142, 1039, 918, 812, 748 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.08–1.18 (m, 1H); 1.30 (t, J = 7.2 Hz, 3H); 1.80 (bs, 1H); 2.08–2.17 (m, 2H); 2.44 (td, J1 = 10.5 Hz, J2 = 4.0 Hz, 1H); 2.63–2.71 (m, 2H); 2.92–2.93 (m, 1H); 3.16–3.24 (m, 1H); 3.34–3.43 (m, 2H); 3.49–3.55 (m, 2H); 3.55–3.62 (m, 2H); 3.66 (dd, J1 = 10.8 Hz, J2 = 4.7 Hz, 1H); 5.21 (d, J = 10.2 Hz, 1H); 5.26 (d, J = 16.8 Hz, 1H); 5.50 (t, J = 4.7 Hz, 1H); 5.92–6.02 (m, 1H); 7.06 (d, J = 7.4 Hz, 1H); 7.20 (d, J = 1.3 Hz, 1H); 7.28 (t, J = 8.0 Hz, 1H); 7.69 (d, J = 8.2 Hz, 1H); 1H NMR (DMSO-d6, 500 MHz) δ 0.85–0.95 (m, 1H); 1.15 (t, J = 7.2 Hz, 3H); 1.95 (dd, J1 = 22.3 Hz, J2 = 11.2 Hz, 1H); 2.25 (dt, J1 = 10.3 Hz, J2 = 3.8 Hz, 1H); 2.46–2.53 (m, 1H), 2.56 (bd, J = 12.7 Hz, 1H); 2.71–2.79 (m, 1H); 3.07 (d, J = 10.2 Hz, 1H); 3.22 (dd, J1 = 14.6 Hz, J2 = 7.7 Hz; 1H); 3.25–3.32 (m, 3H); 3.32–3.42 (m, 3H); 3.50 (dd, J1 = 14.6 Hz, J2 = 5.3 Hz; 1H); 4.57 (t, J = 5.1 Hz, 1H); 5.16 (d, J = 10.3 Hz, 1H); 5.24 (d, J = 17.1 Hz, 1H); 5.89–5.99 (m, 1H); 6.98 (d, J = 7.3 Hz, 1H); 7.18 (t, J = 7.8 Hz, 1H); 7.50 (s, 1H); 7.85 (d, J = 8.2 Hz, 1H); 7.97 (t, J = 5.4 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 15.3, 26.5, 30.9, 36.0, 38.6, 40.5, 56.4, 56.8, 63.7, 66.5, 111.7, 116.3, 116.6, 118.4, 118.8, 125.4, 128.4, 132.7, 133.5, 134.2, 152.4; 13C NMR (DMSO-d6, 125 MHz) δ 15.0, 25.9, 30.8, 34.9, 38.2, 40.1, 56.1, 56.5, 63.6, 64.6, 112.7, 115.4, 115.9, 117.6, 118.0, 124.7, 127.3, 133.1, 133.6, 134.9, 151.9; HRMS (ESI): m/z calcd for C21H28N3O2: [M + H]+, 354.2176; found: 354.2177.(a) Via Pfitzner–Moffatt oxidation of 14. Alcohol 14 (3.00 g, 8.49 mmol) was dissolved in anhydrous DMSO (20 mL) and anhydrous DCM (20 mL) under N2 atmosphere. Solution was cooled to ∼3 °C (ice/water bath) and with stirring DCC (5.26 g, 25.5 mmol), anhydrous pyridine (1.38 mL, 17 mmol) and TFA (0.99 mL, 12.7 mmol) were added sequentially. Reaction mixture was stirred for 1.5 h at ∼3 °C (ice/water bath). Suspension formed was filtered and to the filtrate DCM (250 mL) was added and organic phase was washed with water (3 × 250 mL). Combined water phases were reextracted with DCM (250 mL). Then combined organic phases were dried over anhydrous Na2SO4, concentrated under reduced pressure and residue was purified by column chromatography (MeOH–EtOAc = 10
:90 and DCM–MeOH = 90:10) to give titled aldehyde as a white solid (2.60 g, 87%).(b) Via Parikh–Doering oxidation of 14. In an oven dried, round-bottom flask, alcohol 14 (50 mg, 0.141 mmol) under N2 atmosphere, was dissolved in anhydrous DMSO (2 mL) and to the solution anhydrous DCM (2 mL) was added. Et3N (0.197 mL, 1.41 mmol) was added and reaction mixture was cooled to ∼3 °C (ice/water bath). To this solution SO3·Py (67.4 mg, 0.423 mmol) dissolved in anhydrous DMSO (0.25 mL) was added dropwise and reaction mixture was stirred for 80 min. After that time reaction mixture was poured in to H2O (20 mL) and water phase was extracted with DCM (3 × 15 mL). Combined organic phases were dried over MgSO4 and concentrated under reduced pressure. Residue on concentration was purified by column chromatography (EtOH–EtOAc = 1
:9) to give aldehyde 15 as a white solid (33.7 mg, 68%). Rf: 0.56 (MeOH–EtOAc = 10:90); mp = 165–167 °C (from DCM/heptane); [α]D −70.0 (c 0.45; DCM); calc. for: C21H25N3O2: C, 71.77; H, 7.17; N, 11.96. Found: C, 71.61; H, 7.31; N, 11.84%; IR: νmax 3320, 2927, 2849, 1726, 1668, 1623, 1566, 1535, 1437, 1242, 1085, 892, 752, 640 cm−1; 1H NMR (CDCl3, 300 MHz) δ 1.30 (t, J = 7.2 Hz, 3H); 1.35–1.45 (m, 1H); 2.31 (t, J = 11.6 Hz, 1H); 2.42–2.50 (m, 1H); 2.65–2.75 (m, 1H); 2.85–3.05 (m, 3H); 3.30–3.44 (m, 3H); 3.45–3.63 (m, 3H); 5.19–5.32 (m, 2H); 5.59 (bt, J = 5.0 Hz, 1H); 5.90–6.00 (m, 1H); 7.07 (d, J = 7.4 Hz, 1H); 7.21 (d, J = 1.6 Hz, 1H); 7.30 (t, J = 7.6 Hz, 1H); 7.72 (d, J = 8.3 Hz, 1H); 9.76 (s, 1H). 13C NMR (CDCl3, 75.5 MHz) δ 15.3, 26.4, 27.9, 36.0, 40.2, 48.6, 52.5, 56.5, 63.5, 112.0, 115.9, 116.5, 118.2, 118.9, 125.4, 128.3, 132.8, 133.3, 133.4, 152.4, 202.6; HRMS (ESI): m/z calcd for C21H26N3O2: [M + H]+, 352.2020; found: 352.2019.:25:10:25); mp = 205–207 °C; [α]D −75.4 (c 0.83; DCM); IR: νmax 3294, 3066, 2934, 2861, 2765, 2725, 1663, 1639, 1535, 1439, 1346, 1212, 919, 751 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.28 (t, J = 7.2 Hz, 3H); 1.57 (m, 1H); 1.64–1.71 (m,2H); 2.26 (s, 6H); 2.35–2.46 (m, 4H); 2.51–2.58 (m, 1H); 2.58–2.64 (m, 1H); 2.72–2.78 (m, 1H); 2.79–2.86 (m, 1H); 3.15–3.20 (m, 1H); 3.27 (dd, J1 = 14.4 Hz, J2 = 7.7 Hz, 1H); 3.33 (dd, J1 = 14.9 Hz, J2 = 4.0 Hz, 1H); 3.36–3.40 (m, 2H); 3.46–3.54 (m, 3H); 5.17 (d, J = 10.2 Hz, 1H); 5.22 (d, J = 17.1 Hz, 1H); 5.79 (t, J = 5.4 Hz, 1H); 5.86–5.96 (m, 1H); 7.00 (d, J = 7.3 Hz, 1H); 7.22 (bs, 1H); 7.23–7.27 (m, 1H); 7.69 (bt, J = 4.6 Hz, 1H); 7.71 (d, J = 8.2 Hz, 1H). 13C NMR (CDCl3, 125 MHz) δ 15.3, 25.5, 26.4, 31.0, 35.9, 40.0, 40.3, 43.7, 45.5, 55.6, 56.5, 59.3, 63.1, 111.9, 116.0, 116.4, 118.1, 118.7, 125.3, 128.2, 132.8, 133.6, 152.5, 173.6; HRMS (ESI): m/z calcd for C26H38N5O2: [M + H]+, 452.3020; found: 452.3019.:90). First the desired product, impurity C, was eluted and evaporation of mobile phase gave titled compound as light yellowish foam (0.405 g, 58%). Part of the foam (55 mg) was further crystallized from toluene (0.2 mL) and heptane (3 mL) at −40 °C to give light yellowish solid (45 mg, 82%). Rf: 0.46 (Pyr.–Me2CO = 10%:90%); mp = 109–110 °C, ref. 4a 125–127 °C (from diisopropyl ether); [α]D −71.8 (c 0.19; DCM); IR: νmax 3334, 3274, 2971, 2971, 2936, 2867, 2828, 2781, 2245, 1668, 1525, 1438, 1340, 1525, 1438, 1340, 1271, 1138, 913, 729 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.18 (t, J = 7.2 Hz, 3H); 1.29 (t, J = 7.2 Hz, 3H); 1.73 (m, 1H); 1.83–1.91 (m, 2H); 2.28 (s, 6H); 2.34–2.44 (m, 2H); 2.49–2.58 (m, 2H); 2.63 (t, J = 12.9 Hz, 1H); 2.79 (d, J = 11.8 Hz, 1H); 2.95 (bt, J = 8.9 Hz, 1H); 3.16 (d, J = 10.8 Hz, 1H); 3.26–3.40 (m, 5H); 3.47–3.58 (m, 3H); 3.72–3.90 (m, 2H); 5.19 (d, J = 10.0 Hz, 1H); 5.25 (d, J = 17.1 Hz, 1H); 5.62 (bt, J = 5.3 Hz, 1H); 5.88–5.99 (m, 1H), 7.02 (d, J = 7.2 Hz, 1H); 7.26 (t, J = 7.7 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H); 9.45 (bs, 1H). 13C NMR (CDCl3, 125 MHz) δ 14.9, 15.3, 26.5, 29.8, 30.4, 31.3, 35.6, 36.0, 39.9, 42.3, 43.3, 44.9, 55.7, 56.2, 63.4, 111.8, 116.1, 116.6, 118.1, 118.7, 125.4, 128.3, 132.7, 133.5, 133.7, 152.4, 177.6; HRMS (ESI): m/z calcd for C29H43N6O3: [M + H]+, 523.3391; found: 523.3390.Continuing elution, regioisomer 16 was isolated and evaporation of mobile phase gave (67 mg, 10%) yellow oil. Rf: 0.29 (pyr.–Me2CO = 10:90); [α]D −40.2 (c 0.10; DCM); IR: νmax 3282, 2971, 2935, 2868, 2817, 2785, 2246, 1688, 1524, 1438, 1340, 1271, 1139, 917, 729 cm−1; 1H NMR (CDCl3, 500 MHz) δ 1.28 (t, J = 7.2 Hz, 3H); 1.30 (t, J = 6.7 Hz, 3H); 1.72–1.83 (m, 3H); 2.27 (s, 6H); 2.40 (m, 2H); 2.50–2.66 (m, 3H); 2.74 (d, J = 13.0 Hz, 1H); 2.92–2.99 (m, 1H); 3.15 (d, J = 13.4 Hz, 1H); 3.21 (d, J = 11.4 Hz, 1H); 3.29–3.36 (m, 3H); 3.39 (dd, J1 = 14.7 Hz, J2 = 3.7 Hz, 1H); 3.47–3.53 (m, 2H); 3.56 (dd, J1 = 15.2 Hz, J2 = 6.4 Hz, 1H); 3.92 (q, J = 7.0 Hz, 2H); 5.21 (d, J = 10.1 Hz, 1H); 5.26 (d, J = 17.0 Hz, 1H); 5.64 (bt, J = 5.4 Hz, 1H); 5.88–5.98 (m, 1H); 7.02 (d, J = 7.3 Hz, 1H); 7.23 (bs, 1H); 7.27 (t, J = 7.8 Hz, 1H); 7.72 (d, J = 8.3 Hz, 1H); 9.33 (bs, 1H). 13C NMR (CDCl3, 125 MHz) δ 15.3, 15.9, 26.5, 27.2, 29.4, 31.3, 35.9, 39.6, 40.1, 42.3, 45.3, 55.6, 56.4, 57.3, 63.4, 112.0, 115.9, 116.5, 118.2, 119.1 125.4, 128.2, 132.8, 133.1, 133.4, 152.4, 155.0, 177.9; HRMS (ESI): m/z calcd for C29H43N6O3: [M + H]+, 523.3391; found: 523.3385.
Acknowledgements
Financial support for this research was provided by Lek d.d., a Sandoz Company. We would like to acknowledge Roman Burja TLCM head for valuable help, Dr. Martin Črnugelj for NMR assistance and University of Ljubljana, Faculty of Chemistry and Chemical Technology for high resolution mass analyses and elemental microanalyses. Dr. Franc Požgan gratefully acknowledges The Ministry of Higher Education, Science and Technology of the Republic of Slovenia, the Slovenian Research Agency (P1-0230-0103) and EN→FIST Centre of Excellence for their partial financial support.Notes and references
- P. Del Dotto and U. Bonuccelli, Clin. Pharmacokinet., 2003, 42, 633 CrossRef CAS PubMed.
- ICH Q3A Guideline, Revision 2, 2008.
- European Pharmacopoeia, Monograph C, 7th edn (7.5), 2012, p. 1535.
- (a) E. Brambilla, E. di Salle, G. Briatico, S. Mantegani and A. Temperilli, Eur. J. Med. Chem., 1989, 24, 421 CrossRef CAS; (b) P. Salvati, A. M. Caravaggi, A. Temperilli, G. Bosisio, O. Sapini and E. di Salle, US Pat. 4526892, 1985, Chem. Abstr., 1985, 103, 160754.
- (a) A. Stoll and A. Hofmann, Helv. Chim. Acta, 1943, 26, 2070 CrossRef CAS; (b) A. Stoll, A. Hofmann and Th. Petrzilka, Helv. Chim. Acta, 1946, 29, 635 CrossRef CAS; (c) R. Ručman and N. Djordjevič, DE Pat. 2708042, 1976, Chem. Abstr., 1977, 88, 158466.
- G. Marzoni, W. L. Garbrecht, P. Fludzinski and M. L. Cohen, J. Med. Chem., 1987, 30, 1823 CrossRef CAS PubMed.
- J. V. Braun, K. Heider and E. Müller, Chem. Ber., 1918, 51, 273 CrossRef.
- (a) A. M. Crider, R. Grubb, K. A. Bachmann and A. K. Rawat, J. Pharm. Sci., 1981, 70, 1319 CrossRef CAS PubMed; (b) T. A. Montzka, J. D. Matiskella and R. A. Partyka, Tetrahedron Lett., 1974, 15, 1325 CrossRef; (c) Z. Časar and T. Mesar, WO Pat. 2009133097, 2009, Chem. Abstr., 2009, 151, 528967.
- E. C. Kornfeld and N. J. Bach, EP Pat. 3667, 1979, Chem. Abstr., 1979, 92, 181450.
- M. Hafner, M. Sulyok, R. Schuhmacher, C. Crews and R. Krska, World Mycotoxin J., 2008, 1, 67 CrossRef CAS.
- J. Beneš, A. Černý, V. Miller and S. Kudrnáč, Collect. Czech. Chem. Commun., 1983, 48, 1333 CrossRef.
- Amount of epimerisation in transformation of 8 to 9 was determined on the worked up, crude reaction mixture. Recording 1H NMR of the reaction mixture showed on 6:1 epimer ratio. Triplets of CH3CH2NCO at δ [ppm] 1.15 and 1.12 of 9a and 9b were used. See ESI.†.
- S. Ohno, Y. Adachi, M. Koumori, K. Mizukoshi, M. Nagasaka and K. Ichihara, Chem. Pharm. Bull., 1994, 42, 1463 CrossRef CAS PubMed.
- R. Gažák, V. Křen, P. Sedmera, D. Passarella, M. Novotna and B. Danieli, Tetrahedron, 2007, 63, 10466 CrossRef.
- Z.-X. Wang, Y. Q. Li, M. Kondamreddy and X. Cai, US Pat. 20080275240, 2008, Chem. Abstr., 2008, 149, 534450.
- (a) S. Mantegani, E. Brambilla, C. Caccia, E. di Salle, M. A. Cervini, R. A. McArthur, G. Traquandi and M. Varasi, Il Farmaco, 1998, 53, 65 CrossRef CAS PubMed; (b) L. Bernardi, S. Mantegani, A. Temperili and G. Traquandi, GB Pat. 2162173, 1986, Chem. Abstr., 1986, 105, 79216.
- K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1963, 85, 3027 Search PubMed . In general DCC was used for DMSO activation, albeit more expensive EDC·HCl was also employed. There were no differences in reaction yield between the two, although aldehyde prepared using EDC·HCl did not require so thorough purification with respect to the residual urea by-product.
- J. R. Parikh and W. v. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505 CrossRef CAS.
- (a) P. A. Stadler and R. K. A. Giger, in Natural Products and Drug Development, ed. P. L. Krogsgaard, S. C. Brøgger and H. Kofod, Munksgard, Copenhagen, 1984, pp. 463–484 Search PubMed; (b) P. M. Dewick, in Medicinal Natural Products, John Wiley and Sons, Ltd, 2nd edn, 2002, pp. 368–376 Search PubMed.
- S. Inuki, A. Iwata, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 2072 CrossRef CAS PubMed.
- (a) B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888 CrossRef CAS; (b) B. S. Bal, W. E. Childers, Jr and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS.
- (a) B. F. DeTar and R. Silverstein, J. Am. Chem. Soc., 1966, 88, 1013 CrossRef; (b) J. Podlech, Synthesis of Peptides and Peptidomimetics, in Houben-Weyl, Methods of Organic Chemistry, Additional and Supplementary Volume E22a to the 4th Edition, ed. M. Goodman, A. Felix, L. Moroder and C. Toniolo, Georg Thieme Verlag, Stutgart, New York, 2001, pp. 517–533 Search PubMed.
- (a) S. W. Ashford, K. E. Henegar, A. M. Anderson and P. G. M. Wuts, J. Org. Chem., 2002, 67, 7147 CrossRef CAS PubMed; (b) A. L. Gutman, G. Nisnevich, I. Rukhman, B. Tishin, A. Vilensky and B. Pertsikov, US Pat. 20020177709, 2002, Chem. Abstr., 2002, 138, 4728; (c) J. Galambos, L. Czibula, F. Sebõk, S. Bálint and F. Kassai, WO Pat. 2005085243, 2005, Chem. Abstr., 2005, 143, 286587.
- A solvent screening was made by others (see ref. 15) with the aim to improve the ratio of cabergoline (1) vs. its regioisomer in the reaction of cabergolinic acid A with EDC·HCl. The best results were achieved with halogenated solvents like DCM, DCE and chlorobenzene with the ratios of cabergoline (1) vs. regioisomer of 4.1, 4.1 and 4.7 respectively.
- Ratio of Cvs.15 was determined from 1H NMR spectrum of evaporated crude reaction mixture. Singlets of NMe2 at δ [ppm] 2.30 and 2.28 for C and 15 were used.
- I. Candiani, W. Cabri, F. Zarini, A. Bedeschi and S. Penco, Synlett, 1995, 605 CrossRef CAS.
- T. Fehr, P. A. Stadler and A. Hofmann, Helv. Chim. Acta, 1970, 53, 2197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all synthesised compounds. See DOI: 10.1039/c3ra43417f |
| This journal is © The Royal Society of Chemistry 2013 |