 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe growth of porous carbon fibres through in situ vapour deposition
Yi'en Zhou*a and
Liang Hongab
aDept of Chemical & Biomolecular Engineering, National University of Singapore, Singapore. E-mail: chezhouy@nus.edu.sg
bInstitute of Material Research Engineering, Singapore
First published on 16th August 2013
Abstract
This study investigates the transition of carbon flakes to porous carbon fibres (PCF) through vaporisation fragments of polyaromatic hydrocarbons (PAH) and their in situ condensation under the co-gas atmosphere of N2 and CO2 (v/v = 1), where CO2 plays a vital role in cutting out vaporizable PAH fragments. The polycrystalline PCF exhibit d-spacings as large as 17 Å within the mesoporous framework.
As a unique form of porous carbon, porous carbon fibres (PCF) are unlike either carbon nanotubes or granular activated carbons. PCF possess different contours, sizes and most importantly, polycrystalline structures with broad ranges of d-spacing values as well as a mesoporous carbon framework. Chemical vapour deposition method has traditionally been used to grow carbon nanotubes1 from CH4 or C2H2 and bamboo-like carbon nanotubes on copper foils from ethanol.2
It has been found that fibrous activated carbons exhibit better capabilities than their granular counterparts to entrap condensable vapours,3–6 owing to their structural traits of higher aspect ratios as well as interconnected micro- and meso-pores. Whilst long activated carbon fibres are normally produced from acrylic textile fibres and other polymer precursors through lengthy procedures,7 short and ultrathin fibres are formed by the electrospinning technique.8–13 In order to generate pores in a carbon matrix, carbon dioxide etching is a post carbonization treatment that engraves internal micropores without causing obvious alteration in contour to the fibres formed.14 Alternatively, the construction of various mesoporous carbon frameworks has been carried out through the design of polymer precursors, in which using templates is a typical strategy.15–17 Contrary to the direct transition from small organic molecules or polymers to fibrous carbons via carbonization and then CO2 etching, this work unveils an alternative transition from an activated carbon flake to a PCF structure undertaken under CO2 and a co-gas atmosphere only. Hence, this transition differs from the transformations of carbon allotropes18–21 because the former, in principle, involves convective mass transfer whereas the latter do not.
The initial carbon powder sample was prepared from 2-hydroxyethyl cellulose (HEC) by the method discussed elsewhere.22 The resulting carbonaceous powder was then activated at 700 °C under CO2 for 1 h (500 cm3 min−1) and cooled in an Ar purging stream. The activated carbon powder consisting of irregular and dense carbon flakes (Fig. 1a) was obtained thereafter through proper cleaning in water. The activated carbon powder was then heated in a tubular reactor to 800 °C under a specific gas stream (50 cm3 min−1) and this heat treatment (or incubation) lasted for 5 h (unless otherwise stated) to incubate PCF. The FESEM images (Fig. 1b and c) show clumps of the PCF developed under different incubation atmospheres resulting in different contours. The TEM image (Fig. 1d) was taken from a strand of the PCF harvested under N2–CO2. This fibre displays a spine-like structure assembled by sub-micron carbon wedges and interstitial voids. Although the complete mechanism of this transition has yet to be fully understood, experiments done to-date including those described in the following sections suggest that it takes place through the vaporization and in situ condensation of PAH fragments leading to the formation of polycrystalline carbon wedges, which subsequently assemble to form the fibres.
 | ||
| Fig. 1 (a) Activated carbon flake; (b) pure N2 5 h; (c) N2–CO2 5 h; (d) TEM image of fibre from sample 1 (c). | ||
The microstructures of the carbon products collated from the reactor were scrutinized by electron microscopy (JSM-6700F Field Emission Scanning Electron Microscope, JEOL) whilst the porous features of the prepared PCF samples were obtained from nitrogen isotherms at 77 K by Brunauer–Emmett–Teller (BET) surface area measurement (Autosorb-1, Quantachrome instruments). Prior to each measurement, the prepared PCF samples were degassed at 300 °C for 3 h. The surface area (ABET) was calculated from the linear region of the adsorption isotherm under the relative pressure (P/P0) from 0.05 to 0.3. Crystalline characteristics of the samples were analysed on a X-ray diffractometer (Brucker D8 Advance, Cu Kα radiation λ = 1.54 Å) and the pendant organic functional groups on carbon skeleton were characterized by Fourier transform infrared spectroscopy (FT-IR) using the KBr pellet window method. Finally, the H2S adsorption attributes of the PCF and the control samples were determined at 25 °C in a fixed bed microreactor (diameter: 10 mm; length: 200 mm) where 0.05 g of an adsorbent was packed for each measurement. A H2S (1100 ppm)–N2 balance stream was fed with a flow rate of 1000 cm3 h−1 at 1 atm. The H2S concentration in the outlet gas was detected by an electrochemical sensor (MOT500-H2S, Keernuo Electronics Technology). The change in H2S concentration in the outlet stream portrays the adsorption capacity of the testing sample. The breakthrough time is defined as the time that elapses before the bed exit-concentration of H2S reaches 1% of the inlet concentration.
The hypothesis of flake-to-fibre transition through vaporization of PAH was verified by using a specimen of graphite powder (Sigma Aldrich) in lieu of the carbon flakes to conduct the fibre growth experiment under identical conditions to those used to prepare the sample in Fig. 1c. Unlike the thick PCF growth in sample 1c, only a scarce number of fibres in larger diameters can be incubated from the graphite specimen because of the strong association of graphene sheets in graphite. The occurrence of the fibres supports the etching role of CO2 and the proposed sublimation/deposition mechanism. Fig. 2 provides a closer inspection of the micro-environment surrounding a short segment of an arbitrary fibre originating from graphite. Numerous small graphite chips display discernible rough shapes and emerge near the fibre segment on a graphite chunk. It is thus construed that these chips must be the remaining CO2-reactive graphite pieces from the incubation process. Besides this, the nick in the selected fibre segment shows the bulk morphology of the fibre that comprises tiny strands. These strands were regarded as the product of deposition of the stripped miniature graphene pieces. Accompanying the consecutive fusion of graphene pieces, the strands gradually merged together to form scaffolds, which ultimately connected with each other to form a fibre. As it would be far more difficult to strip graphenes than PAHs from their respective carbon sources under the incubation conditions, much fewer fibres could be eventually generated from graphite. Moreover, unlike what was shown in Fig. 1d, the dense fibre matrix might arise from continuous filling of tiny graphene pieces into niches throughout the course of fibre formation. We also observed that only trace quantities of fibre strands could be generated if CO2 was absent from the incubation atmosphere, validating the role of CO2 in skinning miniature graphenes.
 | ||
| Fig. 2 Magnified view of a single fibre grown from graphite particles attached to its surface under N2–CO2 incubation. | ||
With the exception of the above control experiment, the infrared spectra of the three incubated samples obtained from different treatment conditions (Fig. 3) show clearer aromatic ring stretching peaks (close to 1500 cm−1 and 1600 cm−1) compared to that of the activated carbon powder besides the overtone bands (1600–1800 cm−1). This is due to the occurrence of a fraction of smaller PAH rings with similar sizes22 as the compiling PCF. It is rational to deem that both CO2 and thermal etchings cut up PAH rings on the surface of flakes into smaller ones, which contribute to the construction of PCF. In particular, an extension of incubation from 5 to 18 h under N2–CO2 gave rise to a majority of smaller PAH species, which also bear phenolic groups according to the obvious O–H vibration band (3400 cm−1). Yet, the 18 h incubation left behind only a small amount of carbon residue, including negligible PCF. The reason for the depletion of PCF from this sample should be due to the fact that PCF is far reactive than the carbon flakes towards CO2. Hence, the growth of PCF and the in situ etching of them shifted to the latter side with the extension of treatment, leaving behind a collection of smaller PAH with the substituted –OH group. This functional group must initially come from CO2 through a complex conversion process since the activated carbon sample showed no oxy-group characteristics in its IR spectrum. It can also be deduced that the PAH with a high –OH substitution degree is immune to further etching by CO2 and converted to PCF. Therefore, the residue survived from the excessively extended incubation duration.
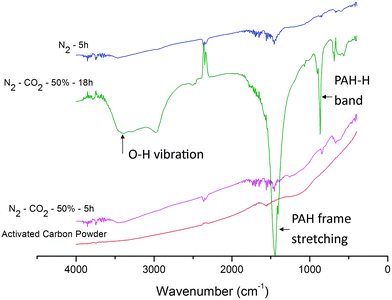 | ||
| Fig. 3 FT-IR spectra showing the change in organic functional groups due to activation and incubation. | ||
It is also noteworthy that the activation step had created some reactive sites in the carbon flakes before the incubation treatment. It is essential to the subsequent growth of PCF. This is because the incubation step applied a delicate flow rate of CO2 as described above in order to avert PCF formed from being destroyed by a higher CO2 flow. In addition, the use of N2 instead of CO2–N2 as the atmosphere for incubation also resulted in PCF as shown in Fig. 1c. This test shows as well the effect of the reactive sites inherited from the previous activation step. It is also clear that the PCF in Fig. 1b display straighter contours and a denser matrix compared to that in Fig. 1c. As per the observation, the thermal etching under N2 should produce PAH fragments with more regular structure and hence, a more closely packed PCF matrix as a logical conclusion, which is further justified subsequently by BET surface analysis.
The impact of incubation atmosphere on both the conversion and pore structures in the three co-gases (N2, He, or Ar) of CO2 demonstrated that lesser PCF were formed in CO2–He compared to CO2–N2 (Fig. 1c), whilst almost no PCF were obtained (with most of the initial carbon flakes remaining intact) when the transition was conducted in CO2–Ar. This fact suggests that the co-gas would have affected the activity of CO2. This observation may be explained using the pre-exponential factor A of bi-molecular collision model23 since it reflects collision effectiveness at a given temperature:
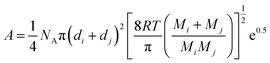
During the condensation process, a coaxial stacking of sub-micron carbon wedges that consists of the planar PAH molecules takes place (Fig. 1d). This hierarchy is verified by the X-ray diffraction results (Fig. 4). Against an amorphous structure displayed by the activated carbon flakes, the products from the incubation under the three different atmospheres (i.e. N2, CO2–N2, and CO2–He) display different polycrystalline structures as a result of multiple randomised stacking of PAH molecular fragments different in sizes and with oxy-groups attached. It is noted that the PCF incubated under N2 (or He)–CO2 displays a very broad coverage of d-spacing, ranging from 17.9 Å to a mere 0.91 Å. Thus, the PCF offer far greater distances between PAH sheets than normal graphitized carbon adsorbents that have the inter-graphene distance of 3.35 Å.25 As noted above, the residue of the 18 h incubation under N2–CO2 is prevalently amorphous phase due to the diminution of the majority of PCF. A conclusion can thus be drawn from the above X-ray characterization: it was the PCF rather than the activated carbon source that contributed to the crystalline features despite the difficulty in separating PCF from the carbon residue as both were entangled together. As such, the intensity of crystalline peaks could approximate the relative content of PCF in a sample. Indeed, similar conversions of the activated carbon flakes to PCF were attained under the He–CO2 and N2–CO2 streams, respectively.
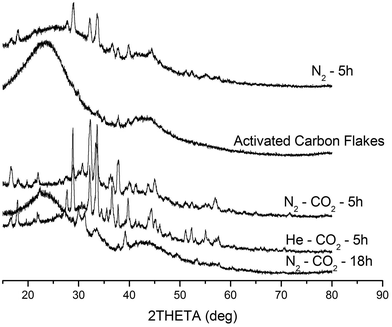 | ||
| Fig. 4 X-ray diffraction results. | ||
Although there was difficulty in separating the PCF from the flakes, close examination under electron microscopy shows that both the He–CO2 and N2–CO2 streams produced a conversion of ca. 40% to fibres by volume (40% yield) whilst conversion under Ar–CO2 stream produced no PCF (0% yield). A mass loss of ca. 20% was also observed in all samples regardless of the incubation atmosphere. In addition, as mentioned previously, CO2 plays a major role in this transition and the yield drops significantly to ca. 15% when CO2 was not included in the treatment during the incubation. The incubation duration, the identity of co-gas, and the flow rate of CO2 are the key factors affecting the yield of PCF at the selected incubation temperature, which have been elucidated in the preceding paragraphs.
As far as the porous structures evolved under different atmospheres are concerned, their BET isotherms and the specific surface properties are ostensibly greater than that of the activated carbon flakes specimen, which exhibits a feeble microporous structure with the most abundant pore size of 16 Å (0.24 cm3 g−1) and a specific surface area of 250 m2 g−1. When it is incubated under N2, the pure thermal etching triggers formation of mesoporosity (vm/vt = 0.66). It is in accordance to its lower 2θ XRD peaks together with an increase in surface area. Furthermore, a change of the incubation atmosphere to N2–CO2 permits CO2-etching and hence, resulting in an even greater harvest of PCF. This contributes towards the mesoporosity (vm/vt = 0.68), the total specific pore volume (0.81 cm3 g−1) and the surface area (1140 m2 g−1). As aforementioned, if the incubation was extended to 18 h under the same atmosphere, only the phenolic PAH remained and showed a low surface area of 131 m2 g−1 with a slightly increased mesoporosity (vm/vt = 0.73) but a vt of 0.10 cm3 g−1. This outcome supports the previous inference for the occurrence of small PAH with similar ring sizes on the basis of the FT-IR spectrum since these PAH rings favour closely packing. In addition, the use of He–CO2 as incubation atmosphere instead of N2–CO2 demonstrated the modest surface properties but a high mesoporosity (vm/vt = 0.80). Such effect can be presumed to be caused by the active collisions between CO2 by He molecules, which made CO2 could adsorb only those reactive carbon sites. With the CO2 etching role, these sites become micro-pores and more preferential to CO2 and hence the micro-pores were expanded to mesopores. The absorption of H2S to the three selected specimens showed different breakthrough durations on the basis of per gram adsorbent: 1.4 min (AC-flake); 14.2 min (CO2–N2), 23.4 min (CO2–He), which is in accord with the volume fraction of mesopores (vm/vt) (Fig. 5).
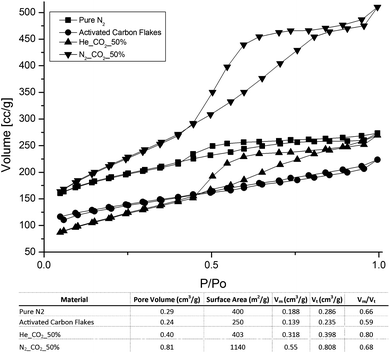 | ||
| Fig. 5 Isotherms of BET for various pore structures. | ||
In conclusion, we have observed and described the phenomenon that a specific type of activated carbon flakes undergoes a sublimation-like process to transform to fibrous structures under a CO2-containing binary atmosphere at 800 °C. This C-to-C transition leads to fibrous activated carbon with sizes between carbon nanotube and carbon fibre and a micro to meso porous structure. The co-gas of CO2 plays a vital role in shaping the evolution of the activated fibre and, in particular, its porous structure. This phenomenon is presumed to originate from the different molecular collision efficiencies and the kinetic diameters of gases. Finally, the H2S adsorption test was undertaken to validate the surface properties obtained from BET analysis.
The authors would like to thank the National University of Singapore (NUS) and Singapore National Research Foundation (NRF grant no.: R-279-000-261-281) for the support rendered in the course of this research work.
Notes and references
- H.-C. Lin, S.-T. Shiue, Y.-H. Cheng, T.-J. Yang, T.-C. Wu and H.-Y. Lin, Carbon, 2007, 45, 2004–2010 CrossRef CAS PubMed.
- J. T. Zhu, J. C. Jia, F. L. Kwong and D. H. L. Ng, Carbon, 2012, 50, 2504–2512 CrossRef CAS PubMed.
- E. A. Dawson, G. M. B. Parkes, P. A. Barnes, M. J. Chinn, L. A. Pears and C. J. Hindmarsh, Carbon, 2002, 40, 2897–2903 CrossRef CAS.
- M. Suzuki, Carbon, 1994, 32, 577–586 CrossRef CAS.
- Y. K. Zhan, C. Y. Nie, H. B. Li, L. K. Pan and Z. Sun, Electrochim. Acta, 2011, 56, 3164–3169 CrossRef CAS PubMed.
- J. Y. Zhou, Z. W. Wang, R. Zuo, Y. Zhou, X. M. Cao and K. Cheng, Asia-Pac. J. Chem. Eng., 2012, 7, S245–S252 CrossRef CAS.
- P. J. M. Carrott, J. M. V. Nabais, M. M. L. Ribeiro Carrott and J. A. Pajares, Carbon, 2001, 39, 1543–1555 CrossRef CAS.
- M. Aceituno-Medina, A. Lopez-Rubio, S. Mendoza and J. M. Lagaron, Food Hydrocolloids, 2013, 31, 289–298 CrossRef CAS PubMed.
- S. Agarwal, A. Greiner and J. H. Wendorff, Prog. Polym. Sci., 2013, 38, 963–991 CrossRef CAS PubMed.
- T. Maitra, S. Sharma, A. Srivastava, Y.-K. Cho, M. Madou and A. Sharma, Carbon, 2012, 50, 1753–1761 CrossRef CAS PubMed.
- Y. Qiu, J. Yu, T. Shi, X. Zhou, X. Bai and J. Y. Huang, J. Power Sources, 2011, 196, 9862–9867 CrossRef CAS PubMed.
- X. Song, Z. Wang, Z. Li and C. Wang, J. Colloid Interface Sci., 2008, 327, 388–392 CrossRef CAS PubMed.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- J. Alcañiz-Monge, D. Cazorla-Amorós, A. Linares-Solano, S. Yoshida and A. Oya, Carbon, 1994, 32, 1277–1283 CrossRef.
- D. Gunther, J. Beckmann, M. Schoneich, P. Schmidt and O. Klepel, Carbon, 2012, 50, 3096–3098 CrossRef PubMed.
- S. Lei, J. I. Miyamoto, T. Ohba, H. Kanoh and K. Kaneko, J. Phys. Chem. C, 2007, 111, 2459–2464 CAS.
- Y. Meng, D. Gu, F. Q. Zhang, Y. F. Shi, H. F. Yang, Z. Li, C. Z. Yu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- C. He, L. Z. Sun and J. Zhong, Journal of Superhard Materials, 2012, 34, 386–399 CrossRef.
- F. P. Bundy, W. A. Bassett, M. S. Weathers, R. J. Hemley, H. K. Mao and A. F. Goncharov, Carbon, 1996, 34, 141–153 CrossRef CAS.
- V. V. Brazhkin, A. G. Lyapin, S. V. Popova, S. C. Bayliss, T. D. Varfolomeeva, R. N. Voloshin, A. G. Gavrilyuk, M. V. Kondrin, V. V. Mukhamad'yarov, I. A. Troyan, S. V. Demishev, A. A. Pronin and N. E. Sluchanko, JETP Lett., 2002, 76, 681–692 CrossRef CAS.
- K. Yamada, Y. Tanabe and A. B. Sawaoka, Philos. Mag. A, 2000, 80, 1811–1828 CAS.
- M. Sun and L. Hong, Carbon, 2011, 49, 2173–2180 CrossRef CAS PubMed.
- P. W. Atkins, Atkins' physical chemistry, Oxford University Press, Oxford, New York, 2006 Search PubMed.
- G. Aguilar-Armenta, G. Hernandez-Ramirez, E. Flores-Loyola, A. Ugarte-Castaneda, R. Silva-Gonzalez, C. Tabares-Munoz, A. Jimenez-Lopez and E. Rodriguez-Castellon, J. Phys. Chem. B, 2001, 105, 1313–1319 CrossRef CAS.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier Science, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |