 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and binding of proflavine diazides as functional intercalators for directed assembly on DNA†
Shahrbanou MoradpourHafshejania, Joseph H. Hedleyab, Alexandra O. Haighac, Andrew R. Pike*a and Eimer M. Tuite*a
aChemical Nanoscience Laboratory, School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: andrew.pike@ncl.ac.uk; eimer.tuite@ncl.ac.uk
bQuantuMDx, International Centre for Life, Times Square, Newcastle upon Tyne, NE1 4EP, UK
cChurchill Community College, Sixth Form, Churchill Street, Wallsend, NE28 7TN, UK
First published on 12th July 2013
Abstract
Proflavine diazide (PD) with amido-azide substituents on the amine groups and its N-methylated analogue (MePD) bind strongly to DNA by nearest-neighbour intercalation with little sequence selectivity, presenting reactive azide groups in the major groove. PD is neutral in aqueous solution but experiences binding-coupled protonation on interaction with DNA with an apparent pKa shift of 2.5 units. MePD can be click modified in situ on DNA with alkyne-functionalised thienyl-pyrrole as a precursor for conducting polymer synthesis, and remains intercalated after reaction with the substituents aligned in the groove.
Introduction
The use of DNA as an architectural material was revolutionized by the seminal work of Seeman and coworkers in constructing 2D and 3D DNA nanostructures,1 which resulted in an explosion of research in this field. The exquisite specificity of nucleic acid recognition, together with chemical stability, has made DNA the polymer of choice for the construction of increasingly intricate nano-architectures.2 The DNA duplex is an attractive candidate as a 1D template or scaffold for assembly of functional materials via chemical reactions, coordination chemistry, or non-covalent association.3Assembly of conducting polymers, e.g. polyaniline,4 polypyrrole and polythienylpyrrole5,6 on DNA has been a key area of interest for development of molecular wires. Control of polymerization can be achieved by tethering monomers to DNA on the bases, sugars, or modified backbones.7 Herein, we present first generation molecules for an alternative strategy which uses unmodified DNA as a scaffold to facilitate the linear assembly of functional materials. This strategy uses small molecules (ligands) that bind strongly to DNA with specific recognition modes (e.g. intercalation or groove binding)8 to present reactive substituents in one DNA groove. Hence, the duplex becomes an adaptable scaffold without the requirement for chemical modification of DNA. Proflavine is the framework we have chosen initially for development of functional intercalators.9 The strong and well-characterized intercalative binding of acridines has led to their development as anticancer drugs,10 and these properties also make them attractive candidates for anchoring supramolecular architectures to a DNA scaffold.
Previously, 9-aminoacridine has been used as an intercalative ligand to assemble a copper catalyst on DNA for asymmetric synthesis,11 and proflavine has been modified with platinum complexes for improved therapeutics.12 Searcey and coworkers produced a library of substituted acridine intercalators using click chemistry in solution,13 one of which drove formation of Holliday junctions.13b 9-Aminoacridine azide was used for in situ click with an alkene peptide, where the reactants were pre-assembled.14a More recently, Balasubramanian and coworkers have used in situ click substitution of well-known tetraplex binders to identify drugs that bind selectively to G4 motifs.14b Also, minor groove binding azido-ligands have been used for assembly of functional molecules on AT-rich DNA.14c In this paper we report the synthesis of novel proflavine derivatives with amido-azide substituents that intercalate DNA and undergo in situ click reactions15 with molecules such as alkyne-substituted thienylpyrrole (TP). In this paper, we fully characterize their binding to DNA and in situ click reaction, and in related work5f we have shown that conducting poly(TP)n nanowires can be formed from the resultant assembly.
Results
Synthesis and absorbance of diazido-proflavines
Modification of proflavine (1, Pf)‡ with azide groups produces a ligand that presents “click” functionalities along DNA after intercalation. Initially, the exocyclic amines were converted to azides but that product was relatively unreactive. Thus, an extended amide linker was introduced to increase accessibility of the azides, and overcome the reduced reactivity of Pf exocyclic amines after intercalation with DNA16 (Scheme 1).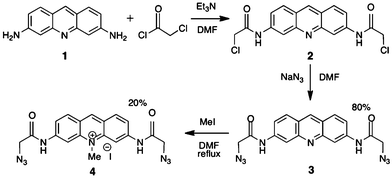 | ||
| Scheme 1 Synthesis of azide-modified proflavines. | ||
Product 3 (proflavine diazide, PD) was obtained in good yield but proved poorly soluble in water, although it dissolved in polar solvents and in DNA solution. Consequently, methylated proflavine diazide (4, MePD) was synthesized, in which the ring nitrogen is quaternized to give a cationic species at pH 7. For direct comparison of 1, 3 and 4, all experiments were carried out in aqueous buffered solution containing 1% DMSO by volume. This allowed dissolution of PD to tens of millimolar and did not perturb the DNA conformation as judged by circular dichroism.
Aqueous solutions of Pf, PD, and MePD are yellow-orange due to absorbance in the 300–500 nm region (Fig. 1). The modified acridines absorb at higher energy with smaller extinction coefficients than Pf, as previously reported for spin-labelled Pf.17 All compounds also have significant absorbance in the 260 nm DNA region. Protonation on the ring nitrogen of Pf and PD is calculated to lower the energy of the absorption (supplementary information†), consistent with observed shifts to longer wavelength of the absorption maxima at low pH (Table 1).
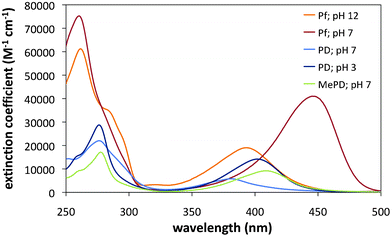 | ||
| Fig. 1 Absorption spectra of the proflavine dyes in aqueous solution. | ||
| Species | pH | λmax/nm | εmax/M−1 cm−1 | ΔE/eV |
|---|---|---|---|---|
| a λmax and εmax, measured; ΔE, calculated. | ||||
| Pf | 12 | 393 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 | 3.51 |
| PfH+ | 7 | 445 | 41000 | 3.29 |
| PD | 7 | 381 | 5800 | 3.62 |
| PDH+ | 3 | 403 | 13200 | 3.27 |
| MePD+ | 7 | 409 | 9200 | 3.35 |
pKas for Pf in aqueous solution are reported at 0.3 and 9.5,18 and our data (supplementary information†) concur. At pH 7 the dominant monocation has an S0 → S1 absorption maximum at 445 nm with an extinction coefficient of 41000 M−1 cm−1.19 At high pH, deprotonation of the ring nitrogen produces the neutral form absorbing at higher energy (λmax = 393 nm) with a smaller extinction efficient. The spectra of the di- and tri-cationic forms (pH < 0), for further protonation on the exocyclic nitrogens, are reported to have maxima at 350–360 nm and extinction coefficients comparable to the neutral species.18,20 MePD is also monocationic at pH 7 with a pKa of 9.6 for deprotonation. The poor aqueous solubility of PD is consistent with a dominant neutral form at pH 7, which renders the compound very hydrophobic. As pH drops from 9 to 1, the PD spectrum changes substantially with an isosbestic point at 365 nm (Fig. 2a). A pKa is observed at pH ∼4.4, and is assigned to protonation of the ring nitrogen.
![Effect of pH on the absorption spectrum of PD. (a) spectra and calculated partial charges, and (b, inset) titration, [PD] = 54 μM.](/image/article/2013/RA/c3ra43090a/c3ra43090a-f2.gif) | ||
| Fig. 2 Effect of pH on the absorption spectrum of PD. (a) spectra and calculated partial charges, and (b, inset) titration, [PD] = 54 μM. | ||
Partial charges calculated for the neutral and monocation forms of PD, Pf, and MePD (supplementary information†) explain the change of pKa when Pf carries amido-azide substituents. For neutral Pf, the ring nitrogen carries a high negative partial charge with partial positive charges on the exocyclic amines, and the charge density changes significantly after protonation of the ring nitrogen. For neutral PD, the negative charge on the ring nitrogen is much smaller due to significant negative charge density on the amide linker and azide nitrogens. After protonation of the ring nitrogen, the negative charges remain localized on the amido-azide substituents. Cationic MePD has a similar partial charge distribution to PD monocation.
Interaction of diazido-proflavines with DNA
For PD, a dramatically different behaviour is observed (Fig. 3). A 35 nm red-shift is accompanied by a hyperchromism of >50%. Although this apparently indicates a different mode of binding, it is actually consistent with intercalation where binding is coupled to protonation. The maximum absorption of the DNA-bound dye at 416 nm represents a 13 nm shift from the maximum of PDH+. Additionally, the large absorbance increase compared to the PD spectrum represents a hypochromic change compared to the spectrum of free PDH+. Thus, the changes in PD absorbance on addition of DNA can be rationally interpreted in terms of the dominant DNA-bound species being the monocationic PDH+.
![Effect of binding to calf thymus(CT)-DNA on the absorption spectrum of PD. The spectrum without DNA is shown in dark red. P/D is the DNA (nucleotide phosphate) to dye ratio. [PD] = 56 μM.](/image/article/2013/RA/c3ra43090a/c3ra43090a-f3.gif) | ||
| Fig. 3 Effect of binding to calf thymus(CT)-DNA on the absorption spectrum of PD. The spectrum without DNA is shown in dark red. P/D is the DNA (nucleotide phosphate) to dye ratio. [PD] = 56 μM. | ||
Coupled protonation and DNA binding has been reported previously for neutral dibenzoacridine22a as well as other intercalators,22b minor groove binders22c and proteins.22d It is also known that the apparent pKa of cytosine is raised significantly from ∼4.5 when it protonates on formation of CGC-triplexes, with a 3–5 unit increase reported for internal positions.23 Moreover theory predicts that the DNA minor groove is more acidic than the surrounding solvent,24a with experiments indicating a drop of up to 2 units.24b This is likely a result of the high negative potential in the minor groove caused by electrostatic focussing.25 Thus, the increase of apparent pKa of PD on binding to DNA is not without precedent.
Absorption changes were analysed by the Scatchard method to obtain binding constants and apparent site sizes as shown in Table 2. Proflavine binds very strongly to CT-DNA and only a lower limit for the association constant could be determined at low ionic strength. As the salt concentration was raised, binding weakened and an association constant was readily determined with 500 mM NaCl added to buffer. The binding constant in 5 mM phosphate was estimated theoretically using Record/Manning theory, which states that K for a monocationic intercalator varies with added inert monocation according to eqn (1), with B = 0.24, Z = 1, and Ψ = 0.8226 or 1.27
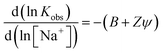 | (1) |
A binding constant of K ≈ 1.5 × 107 M−1 was predicted at 7.5 mM Na+, which is higher than the experimental value, and represents an upper limit at low ionic strength. The binding constants measured for Pf have similar magnitudes to previously reported values.28,29 PD and MePD also show high affinities for DNA, with site sizes that are close to nearest-neighbour, although their binding constants are lower than that for Pf. Nonetheless, at low ionic strength, both PD and MePD are quantitatively bound to DNA at high ratios of DNA basepair to dye concentrations, expressed as P/D ([nucleotide phosphate]/[dye]).
![CD spectra in the visible region showing induced CD of the dyes in the presence of calf thymus (CT)-DNA. [Pf] = 10 μM, P/D = 50; [PD] = 50 μM, P/D = 20; [MePD] = 50 μM, P/D = 10. P/D is the DNA (nucleotide phosphate) to dye ratio. Data are smoothed (see supplementary information for raw data).](/image/article/2013/RA/c3ra43090a/c3ra43090a-f4.gif) | ||
| Fig. 4 CD spectra in the visible region showing induced CD of the dyes in the presence of calf thymus (CT)-DNA. [Pf] = 10 μM, P/D = 50; [PD] = 50 μM, P/D = 20; [MePD] = 50 μM, P/D = 10. P/D is the DNA (nucleotide phosphate) to dye ratio. Data are smoothed (see supplementary information† for raw data). | ||
The ICD for Pf resembles that reported previously for similar binding ratios at low ionic strength in native DNA, [poly(dA–dT)]2, and [poly(dG–dC)]2.29,32–34 The non-conservative splitting pattern has been attributed to degenerate vibronic exciton coupling between intercalated and externally bound dyes.28c,34 External binding is found to be minimal at high salt concentrations but to occur to some extent at low salt concentrations even at the high P/D ratios used in this work.34,35 The deconvoluted CD spectrum of pure intercalated Pf is reported to be positive but that of acridine orange, which is tetramethylated on the exocyclic nitrogens, is negative.34 This was attributed to different intercalation geometries of the two dyes, since the transition moment involved is the same long-axis polarized π–π* transition for each.34–36 It was suggested that H-bonds between Pf and the DNA backbone, as observed in crystal structures, are responsible for that difference since the magnitude of the Pf ICD was sensitive to increasing ionic strength. Negative ICD spectra for PD and MePD suggest that these dyes might have intercalation orientations more similar to acridine orange than Pf; indeed, small slides or twists of the dye in the intercalation pocket can cause a sign inversion.37 The absence of splitting in the ICD spectra of PD and MePD suggests that external binding is less important for these dyes than for Pf, perhaps because the increased bulk of the side chains hinders association of additional dyes in the groove as observed when Pf external binding is blocked by glycosylation of the major groove in T2-DNA.32
| LDr(λ) = LD (λ)/Aiso(λ) = 1.5 S (3 〈cos2α〉 − 1) | (2) |
The LD signal of DNA is negative in the 200–350 nm absorption region, where the strongly absorbing transition moments (TM) are polarized in the planes of the basepairs. Since the helix axis of aligned DNA is oriented parallel to the flow direction, negative LD indicates that the basepairs are oriented more perpendicular than parallel to the helical axis, as expected for B-form DNA. For all three acridines, the LD signals in the visible spectrum are also negative (supplementary information†). This implies that the TMs responsible for visible absorption, which are polarized in the acridine aromatic planes,34,36 also have an average orientation >54° to the helix axis.
For structural interpretation of LD, reduced linear dichroism (LDr) spectra were computed (Fig. 5) using eqn (2). In general, the LDr signals at 260 nm for DNA with and without dye report on changes in base orientation induced by dye binding, although overlap of strong dye absorption with DNA at 260 nm precludes quantitative analysis. The ratio of LDr signals in the visible and UV regions allows calculation of the angle between the dye and base pair planes. An important caveat is that the latter comparison is valid only if all the dye absorption arises from bound material, since free dye contributes to isotropic absorption but not to LD. Therefore, spectra were measured under conditions (low salt and high P/D) that favour complete binding. For our samples, there was good correspondence between the isotropic absorption and LD spectra, showing close to 100% of dye is bound.
![Reduced linear dichroism (LDr) spectra of acridine dyes with calf thymus (CT)-DNA. P/D = 50; [DNA] = 1 mM. Shear gradient = 1900 s−1 (600 rpm). P/D is the DNA (nucleotide phosphate) to dye ratio. The free DNA spectrum is shown in grey.](/image/article/2013/RA/c3ra43090a/c3ra43090a-f5.gif) | ||
| Fig. 5 Reduced linear dichroism (LDr) spectra of acridine dyes with calf thymus (CT)-DNA. P/D = 50; [DNA] = 1 mM. Shear gradient = 1900 s−1 (600 rpm). P/D is the DNA (nucleotide phosphate) to dye ratio. The free DNA spectrum is shown in grey. | ||
Negative LDr signals in the dye visible absorption bands (Fig. 5) are consistent with the chromophore long axes lying approximately parallel to the base pair planes. For Pf, this agrees with previous electric LD results which showed the ring system was parallel to the base planes for DNA with various base compositions.38 Pf shows greater magnitude LDr in the visible than in the UV. For PD and MePD, the visible LDr magnitude is lower than that in the UV. Previous studies have inferred an effective value of 80–86° for the orientation of the basepairs to the helix axis.29,39 Nevertheless, significantly more negative LDr in the dye than the DNA band, as observed for Pf, has also been reported for other intercalators such as methylene blue.37c,d Previous spectral analysis36 of Pf and related dyes assigned the 465 nm absorption solely to a long-axis polarized transition, and 263 nm absorption predominantly to long-axis polarized transitions with a small contribution from a short-axis polarized transition. Similar assignments are likely for PD and MePD. Thus different values of LDr(vis)/LDr(UV) suggest that PD and MePD adopt slightly different intercalation geometries than Pf, as also inferred from CD spectra. Wedging intercalation from the major groove, due to the bulky substituents impeding full insertion between the basepairs, would result in smaller LDr for the dye since the chromophore would sample a range of orientation, as observed for piperazinecarbonyloxyalkyl derivatives of anthracene and pyrene.40
Linear dichroism spectroscopy shows that the ring systems of PD and MePD remain intercalated in DNA after their azide groups undergo click reactions (Scheme 2) with 5-pentenyl-thienyl-pyrrole (pTP). On the other hand, 6 formed in solution did not bind strongly to DNA. Thus, an in situ click reaction is necessary to assemble such a molecule on DNA.
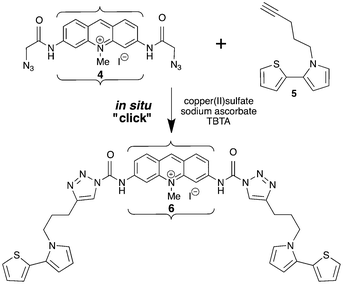 | ||
| Scheme 2 In situ “click” reaction of MePD (4) and pTP (5) to generate MePD-pTP (6). Brackets represent the intercalation site in DNA. Arrows represent presumed main transition moment directions. | ||
At high dye loading (Fig. 6), the LDr signal at 425 nm in the MePD absorption band remains negative after the click reaction. A new negative signal is observed in the 300–350 nm region, where MePD has no contribution, which corresponds to absorption of the pTP substituent (supplementary information†). The absorbing transition moment of the clicked TP chromophore thus has an average orientation of <54° to the helix axis. If we set the DNA basepair (260 nm) orientation at 90° to the helix axis, the calculated angles are 74° for MePD (425 nm) and 63° for pTP (350 nm). By contrast, at low dye loading (supplementary information†), the LDr signal at 300–350 nm is positive whilst the signal in the PD absorption band at 430 nm remains negative, and the angle for pTP substituents in this case is calculated as 52°. Thus, the proximity of nearest neighbour intercalated PD forces the TP residues into an orientation somewhat more parallel to the basepairs. Nonetheless, the TP residues can polymerise into long conducting poly(TP)n nanowires from this orientation, as we have demonstrated elsewhere.5f
![Reduced linear dichroism (LDr) spectrum of in situ generated MePD-pTP (6) bound to calf thymus (CT)-DNA compared with DNA-bound MePD.5f P/D = 50; [DNA] = 1 mM. P/D is the DNA (nucleotide phosphate) to dye ratio. Shear gradient = 3170 s−1 (1000 rpm).](/image/article/2013/RA/c3ra43090a/c3ra43090a-f6.gif) | ||
| Fig. 6 Reduced linear dichroism (LDr) spectrum of in situ generated MePD-pTP (6) bound to calf thymus (CT)-DNA compared with DNA-bound MePD.5f P/D = 50; [DNA] = 1 mM. P/D is the DNA (nucleotide phosphate) to dye ratio. Shear gradient = 3170 s−1 (1000 rpm). | ||
![Relative intrinsic viscosity of calf thymus (CT)-DNA on addition of acridine dyes. [DNA] = 300 μM; 25 °C.](/image/article/2013/RA/c3ra43090a/c3ra43090a-f7.gif) | ||
| Fig. 7 Relative intrinsic viscosity of calf thymus (CT)-DNA on addition of acridine dyes. [DNA] = 300 μM; 25 °C. | ||
PD and MePD are also quantitatively bound, and although their slopes (0.99 and 0.50, respectively) are lower than for Pf, they are consistent with intercalation. The slope for PD is substantially lower than for Pf or MePD, and its low value implies either that some bound PD is externally associated, or that binding causes a reduction in persistence length that counteracts the increase in contour length due to intercalation. However, the lack of splitting in CD spectroscopy indicates that external binding is not important for PD and MePD, certainly not to the extent of 50% being externally bound.
Minor groove binders cause little change in viscosity45 but partial intercalators decrease DNA viscosity by bending through wedging,46 whilst covalent binding of cisplatin decreases DNA viscosity through static bending.47 These bending interactions decrease the viscosity of rod-like DNA by shortening the axial length. In long DNA, the bending effect is translated to a decrease of persistence length, which reduces the relative viscosity according to eqn (3). Since MePD and PD still increase viscosity, albeit less so than Pf, they cannot be considered to behave as true partial intercalators that bind by wedging open the basepairs toward one groove. Nonetheless, the results suggest that they adopt an intercalation geometry that reduces DNA persistence length.
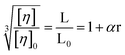 | (3) |
![Variation of emission intensity of acridine dyes on addition of calf thymus (CT)-DNA and alternating homopolynucleotides. P/D is the DNA (nucleotide phosphate) to dye ratio. [dye] = 5 μM.](/image/article/2013/RA/c3ra43090a/c3ra43090a-f8.gif) | ||
| Fig. 8 Variation of emission intensity of acridine dyes on addition of calf thymus (CT)-DNA and alternating homopolynucleotides. P/D is the DNA (nucleotide phosphate) to dye ratio. [dye] = 5 μM. | ||
Both PD and MePD+ are quenched by adenine as well as guanine in polynucleotides, indicating that their singlet states are more readily reduced than 1Pf. Ground state reduction is predicted to occur more readily than for Pf due to lower electron densities on their aromatic ring systems, and their (0,0) transitions are at higher energy than that of Pf. Taken together, these features rationalize why the excited singlet states of PD and MePD are significantly more oxidizing than that of Pf. Emission titrations indicate that PD and MePD show little selectivity in binding to [poly(dA–dT)]2, [poly(dG–dC)]2 or mixed sequence CT-DNA. Pf binds most strongly to [poly(dG–dC)]2 under our conditions, other studies report little selectivity at higher ionic strength.29 However, recent molecular modelling50 suggests that Pf should indeed bind more strongly to [poly(dG–dC)]2 than [poly(dA–dT)]2 due to greater π–π stacking in the former case. We interpret our observations in terms of the smaller substituents on Pf allowing it to intercalate deeply into the base pair pockets, thus benefiting from enhanced π–π stacking in the GC pockets. By contrast, if PD and MePD cannot intercalate as deeply as Pf from the major groove due to their bulky substituents, they will experience similar stacking interactions in all types of base pair pockets.
Discussion
Linear dichroism and viscometry results demonstrate that Pf, PD and MePD intercalate between the DNA base pairs. The small induced CD signals are also consistent with intercalative binding. Other results such as absorption, emission, and thermal denaturation titrations reflect the relative binding affinities of the three compounds. Proflavine was chosen as the framework molecule for our functional intercalators since it known to be an avid intercalator. A recent PDB deposition (3FT6)51a shows a crystal structure of proflavine intercalated at terminal CG/CG steps in a hexamer duplex (supplementary information†). The heterocyclic system is deeply intercalated and the exocyclic amino groups are both oriented towards the major groove but do not protrude. Symmetric intercalation with the dye long axis parallel to the base pair long axis allows the amines to form H-bonds with the phosphate and sugar groups of the backbone. This crystal structure confirms an earlier nmr structure with a tetramer [d(CCGG)]2,51b and crystal structures with RNA51c and DNA51d,e CG dinucleotide minihelices. By comparison, N,N′-tetramethyl proflavine (or acridine orange, AO) and N-dimethyl proflavine are intercalated asymmerically in dinucleotide minihelix crystals with ∼10° twist from the symmetric geometry.52 These dyes are slightly displaced towards the major groove in the basepair pocket, with one dimethyl amine group protruding and the other amine in a position similar to Pf. This small change of conformation is enough to cause a change in ICD signal from positive for Pf to negative for AO.34 Given the negative ICD spectra for PD and MePD bound to DNA, a reasonable hypothesis is that they adopt an intercalation geometry similar to AO, displaced towards the major groove, but likely with both their bulky azide substituents protruding. The wide major groove should readily accommodate these substituents and present them for reaction with molecules approaching from bulk solution.Such variation of intercalation geometry also rationalises differences in LD and viscosimetry for different dyes. Partial intercalators with small aromatic groups strongly reduce the viscosity of DNA.46a However, although PD and MePD give lower slopes than Pf, they nevertheless markedly increase DNA viscosity. Likewise, the LDr signals for these compounds are still strongly negative, although smaller than for Pf. Taken together, these observations suggest that PD and MePD bind by intercalation with slightly different geometries and dynamics than Pf, since their bulky substituents probably prevent the aromatic tricycle from embedding deeply in the intercalation pocket. Instead, they may be displaced towards the edges of the basepairs closest to the major groove which could give rise to dynamic or static bending of DNA, as seen for the partial intercalator [Ru(phen)3]2+46b,53 or for covalently bound cisplatin.47 Smaller LDr signals for the modified dyes are consistent with this postulate, since partially intercalated dyes can explore a range of orientation angles other than perpendicular to the helix axis.40 Consequently, PD and MePD show little sequence selectivity which is advantageous for their general application as versatile intercalative anchors for directed assembly on a DNA scaffold.
Conclusions
Proflavine, Pf, can be readily modified with azide groups on its exocyclic amines via an amide linker. The resultant compound, PD, is neutral but methylation on the ring nitrogen gives a cationic dye, MePD. PD is poorly soluble in water, but becomes protonated on binding to DNA, resulting in an apparent pKa shift of >3 units. Like proflavine, PD and MePD intercalate DNA, as evidenced by linear dichroism and viscometry. The Pf exocyclic amines reside in the major groove of DNA, and it is likely that the modified dyes adopt a similar binding geometry. However, their larger substituents appear to hinder deep intercalation, implying displacement towards the edge of the major groove, as reported for acridine orange, so that the basepairs become slightly wedged apart to reduce the DNA persistence length. DNA-bound PD and MePD undergo in situ click functionalization with pTP, and remain intercalated after reaction thus placing the monomers in the major groove ready for polymerization to form a conducting chain. In summary, PD and MePD are good candidates for application as an intercalative anchor for assembly of supramolecular structures on a DNA scaffold since they bind strongly with little sequence selectivity, and remain intercalated after click reactions with bulky functional groups.Experimental
All chemicals and solvents were of the highest grade available from Sigma-Aldrich. Proflavine hemisulfate salt hydrate was used as received, and proflavine derivatives were synthesized (Scheme 1) and purified as described in the supplementary information.† To ensure complete dissolution of dyes, solids were initially dissolved in 100% DMSO, and diluted with 5 mM phosphate buffer with the required pH to give samples in 5 mM phosphate with 1% DMSO. Polynucleotides [poly(dG–dC)]2 and [poly(dA–dT)]2, and high molecular weight calf-thymus DNA were from Sigma. All nucleic acids were dialyzed extensively against pure water before use to remove excess salts, and were stored in 5 mM sodium phosphate buffer (pH 6.9). All experiments were carried out in 5 mM phosphate buffer/1% DMSO (v/v) at 21 °C, unless otherwise stated, which allowed dissolution of PD to tens of millimolar. Although 10% DMSO can distort binding of ligands,54 1% DMSO did not perturb the DNA conformation as judged by circular dichroism, and binding results for Pf were consistent with those reported in the absence of DMSO. The concentrations of all materials were determined by UV/vis absorption spectroscopy using the following extinction coefficients, determined analytically for PD and MePD and obtained from the literature for the other materials. Pf (445 nm) 41000 M−1 cm−1;19 PD (381 nm) 6600 M−1 cm−1; MePD (409 nm) 9200 M−1 cm−1; CT-DNA (260 nm) 6600 M−1 cm−1; [poly(dA–dT)]2 (262 nm) 6700 M−1 cm−1; [poly(dG–dC)]2 (254 nm) 8400 M−1 cm−1. Nucleic acid concentrations are given per nucleotide. Buffer of required pH was prepared by adjusting the pH of a 5 mM phosphate (pH 6.9) solution using small aliquots of concentrated phosphoric acid or sodium hydroxide.UV/vis spectra and thermal denaturation were measuring with a Cary 100 Bio UV-visible spectrophotometer, and all data are normalized to a 1 cm pathlength. Titrations with calf thymus DNA solution were performed by adding aliquots of concentrated DNA to a constant concentration of ligand. Corrected fluorescence emission and excitation spectra were measured with a SPEX FluoroMax spectrophotometer. CD spectra were measured on a Jasco J-810 spectropolarimeter, and data were normalized to a 1 cm pathlength. The data are presented, as collected, in mdeg; these data can be converted to absorbance units through division by 32980 mdeg. LD spectra were measured on an Applied Photophysics Chirascan CD spectropolarimeter, adapted to produce linearly polarized light. Orientation of the intercalator nucleic acid samples was achieved in a flow Couette cell with an outer rotating cylinder and an inner cylinder of 3 cm diameter. The experimental path length was 1 mm, and data are normalized to a 1 cm path length.
Molecular modelling was performed with Spartan 04 (Wavefunction) using the semi-empirical PM3 method and density functional (DFT) method B3LYP/6-31G* to calculate potential densities and HOMO and LUMO energies of Pf, PD, MePD and their protonated forms.
A Cannon-Manning extra low charge size 75 semi-micro viscometer, immersed in a water bath thermostated at 25 °C, was used to measure the relative intrinsic viscosity55 of dilute solutions of CT-DNA. The DNA concentration and the viscometer volume (300 mL) were kept constant for a series of added dye concentrations. The flow time for water was 177 s, and for solutions containing DNA was >245 s. Measurements were carried out in triplicate and gave standard deviations of <±1 s. For long DNA, if the persistence length does not change on intercalation, a plot of the cube root of the relative intrinsic viscosity against binding ratio yields a slope of 1.4 (supplementary information†).56
Acknowledgements
We express our gratitude to the Nuffield Foundation for supporting a high school summer studentship for AOH, to COST D35 for networking opportunities, and to Newcastle University School of Chemistry for financial support.References
- (a) J. Chen and N. C. Seeman, Nature, 1991, 350, 631 CrossRef CAS PubMed; (b) E. Winfree, F. Liu, L. A. Wenzler and N. C. Seeman, Nature, 1998, 394, 539 CrossRef CAS PubMed.
- (a) N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed; (b) P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed; (c) A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763 CrossRef CAS PubMed; (d) T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stultz, Chem. Soc. Rev., 2011, 40, 138–148 RSC.
- (a) E. Meggers, P. L. Holland, W. B. Tolman, F. E. Romesberg and P. G. Schultz, J. Am. Chem. Soc., 2000, 122, 10714 CrossRef CAS; (b) G. H. Clever, K. Kaul and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 6226 CrossRef CAS PubMed; (c) S. Liu, G. H. Clever, Y. Takezawa, M. Kaneko, K. Tanaka, X. Guo and M. Shionoyo, Angew. Chem., Int. Ed., 2011, 50, 8886 CrossRef CAS PubMed; (d) I. Bouamaied, T. Nguyen, T. Ruhl and E. Stultz, Org. Biomol. Chem., 2008, 6, 3888 RSC; (e) F. Menacher and H.-A. Wagenknecht, Photochem. Photobiol. Sci., 2011, 10, 1275 RSC.
- (a) P. Nickels, W. U. Dittmer, S. Beyer, J. P. Kotthaus and F. C. Simmel, Nanotechnology, 2004, 15, 1524 CrossRef CAS; (b) B. Datta, G. B. Schuster, A. McCook, S. C. Harvey and K. Kakrzewska, J. Am. Chem. Soc., 2006, 128, 14428 CrossRef CAS PubMed.
- (a) A. R. Pike, S. N. Patole, N. C. Murray, T. Ilyas, B. A. Connolly, B. R. Horrocks and A. Houlton, Adv. Mater., 2003, 15, 254 CrossRef CAS; (b) S. A. F. Al-Said, R. Hassanien, J. Hannant, M. A. Galindo, S. Pruneanu, A. R. Pike, A. Houlton and B. Horrocks, Electrochem. Commun., 2009, 11, 550 CrossRef; (c) J. Hannant, J. H. Hedley, J. Pate, A. Walli, S. A. F. Al-Said, M. A. Galindo, B. A. Connolly, B. R. Horrocks, A. Houlton and A. R. Pike, Chem. Commun., 2010, 46, 5870 RSC; (d) M. A. Galindo, J. Hannant, R. W. Harrington, W. Clegg, B. R. Horrocks, A. R. Pike and A. Houlton, Org. Biomol. Chem., 2011, 9, 1555 RSC; (e) A. Mishchenko, M. Abdualla, A. Rudnev, Y. Fu, A. R. Pike and T. Wandlowski, Chem. Commun., 2011, 47, 9807 RSC; (f) S. MoradpourHafshejani, S. M. D. Watson, E. Tuite and A. R. Pike, Chem. Sci., 2013 Search PubMed , under review; (g) D. Erts, U. Malinovskis, I. Muiznieks and E. Tuite, Thin Solid Films, 2008, 516, 8969 CrossRef CAS.
- (a) W. Chen, G. Güler, E. Kuruvilla, G. B. Schuster, H.-C. Chiu and E. Riedo, Macromolecules, 2010, 43, 4032 CrossRef CAS; (b) Z. Ma, W. Chen and G. B. Schuster, Chem. Mater., 2012, 24, 3916–3922 CrossRef CAS; (c) W. Chen and G. B. Schuster, J. Am. Chem. Soc., 2012, 134, 840–843 CrossRef CAS PubMed.
- (a) A. Houlton, A. R. Pike, M. A. Galindo and B. R. Horrocks, Chem. Commun., 2009, 1797 RSC; (b) Rajesh, T. Ahuja and D. Kumar, Sens. Actuators, B, 2009, 136, 275 CrossRef CAS.
- Nucleic Acids in Chemistry and Biology, ed. G. M. Blackburn, M. J. Gait, D. Loakes and D. M. Williams, RSC Publishing, Cambridge, 3rd edn, 2006 Search PubMed.
- (a) L. S. Lerman, J. Mol. Biol., 1961, 3, 18 CrossRef CAS PubMed; (b) V. Luzzati, F. Masson and L. S. Lerman, J. Mol. Biol., 1961, 3, 634 CrossRef CAS PubMed; (c) W. D. Sasikala and A. Mukherjee, Phys. Chem. Chem. Phys., 2013, 15, 6446 RSC.
- W. A. Denny, Curr. Med. Chem., 2002, 9, 1655 CrossRef CAS PubMed.
- (a) G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230 CrossRef CAS PubMed; (b) A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083 RSC.
- (a) E. Ceci, R. Cini, A. Karaulov, M. B. Hursthouse, L. Maresca and G. Natile, J. Chem. Soc., Dalton Trans., 1993, 2491 RSC; (b) S. Biagini, A. Bianchi, T. Biver, A. Boggioni, I. V. Nikolayenko, F. Secco and M. Venturini, J. Inorg. Biochem., 2011, 105, 558 CrossRef CAS PubMed.
- (a) L. A. Howell, A. Howman, M. A. O'Connell, A. Mueller and M. Searcey, Bioorg. Med. Chem. Lett., 2009, 19, 5880 CrossRef CAS PubMed; (b) L. A. Howell, Z. A. E. Walker, R. Bowater, M. O'Connell and M. Searcey, Chem. Commun., 2011, 47, 8262 RSC.
- (a) S. Imoto, T. Hirohama and F. Nagatsugi, Bioorg. Med. Chem. Lett., 2008, 18, 5560 CrossRef PubMed; (b) M. Di Antonio, G. Biffi, A. Mariani, E.-A. Raiber, R. Rodriguez and S. Balasubramanian, Angew. Chem., Int. Ed., 2012, 51, 11073 CrossRef CAS PubMed; (c) M. I. Sánchez, O. Vázquez, J. Martínez-Costas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2012, 3, 2383 RSC.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS PubMed; (c) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC; (d) A. H. El-Sagheer and T. Brown, Acc. Chem. Res., 2012, 45, 1258 CrossRef CAS PubMed.
- L. S. Lerman, J. Mol. Biol., 1964, 10, 367 CrossRef CAS PubMed.
- K. Yamaoka, S. Noji and M. Yoshida, Bull. Chem. Soc. Jpn., 1981, 54, 31 CrossRef CAS.
- S. G. Schulman, D. V. Naik, A. C. Capomacchia and T. Roy, J. Pharm. Sci., 1975, 64, 982 CrossRef CAS PubMed.
- (a) N. F. Gersch and D. O. Jordan, J. Mol. Biol., 1965, 13, 138 CrossRef CAS PubMed; (b) G. R. Haugen and W. H. Melhuish, Trans. Faraday Soc., 1964, 60, 386 RSC.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 373 CrossRef CAS.
- (a) A. R. Peacocke and J. N. H. Skerrett, Trans. Faraday Soc., 1956, 52, 261 RSC; (b) E. Tuite and J. M. Kelly, Biopolymers, 1995, 35, 419 CrossRef CAS.
- (a) J. Booth and E. Boyland, Biochim. Biophys. Acta, 1953, 12, 75 CrossRef CAS PubMed; (b) F. Pierard, A. Del Guerzo, A. Kirsch-De Mesmaeker, M. Demeunynck and J. Lhomme, Phys. Chem. Chem. Phys., 2001, 3, 2911 RSC; (c) B. Njuyen, J. Stanek and W. D. Wilson, Biophys. J., 2006, 90, 1319 CrossRef PubMed; (d) F. M. Dullweber, T. Stubbs, J. Musil, J. Sturzebecher and G. Klebe, J. Mol. Biol., 2001, 313, 593 CrossRef CAS PubMed.
- (a) W. D. Wilson, H. P. Hopkins, S. Mizan, D. D. Hamilton and G. Zon, J. Am. Chem. Soc., 1994, 116, 3607 CrossRef CAS; (b) J. L. Asensio, A. N. Lane, J. Dhesi, S. Bergqvist and T. Brown, J. Mol. Biol., 1998, 275, 811 CrossRef CAS PubMed.
- (a) G. Lamm and G. R. Pack, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9033 CrossRef CAS PubMed; (b) S. Hanlon, L. Wong and G. R. Pack, Biophys. J., 1997, 72, 291 CrossRef CAS PubMed.
- R. Rohs, A. M. West, A. Sosinsky, P. Liu, R. S. Mann and B. Honig, Nature, 2009, 461, 1248 CrossRef CAS PubMed.
- (a) M. T. Record, C. F. Anderson and T. M. Lohman, Q. Rev. Biophys., 1978, 11, 103 CrossRef CAS PubMed; (b) W. D. Wilson and I. G. Lopp, Biopolymers, 1984, 23, 3025 Search PubMed.
- (a) G. S. Manning, Quart. Rev. Biophys., 1978, 11, 179 CrossRef CAS; (b) R. A. G. Friedmann and G. S. Manning, Biopolymers, 1984, 23, 2671 CrossRef PubMed.
- (a) J. C. Thomes, G. Weill and M. Daune, Biopolymers, 1969, 8, 647 CrossRef CAS PubMed; (b) F. Quadrifoglio, V. Crescenzi and V. Giancotti, Biophys. Chem., 1974, 1, 319 CrossRef CAS PubMed; (c) R. W. Armstrong, T. Kurucsev and U. P. Strauss, J. Am. Chem. Soc., 1970, 92, 3174 CrossRef CAS PubMed.
- B. García, J. M. Leal, R. Ruiz, T. Biver, F. Secco and M. Venturini, J. Phys. Chem. B, 2010, 114, 8555 CrossRef PubMed.
- B. Nordén, A. Rodger and T. Dafforn, Linear Dichroism and Circular Dichroism: A Textbook on Polarized Light Spectroscopy, RSC Publishing, Cambridge, 2010 Search PubMed.
- R. Lyng, T. Härd and B. Nordén, Biopolymers, 1987, 26, 1327 CrossRef CAS PubMed.
- H. J. Li and D. M. Crothers, Biopolymers, 1969, 8, 217 CrossRef CAS.
- M. Kamiya, Biochim. Biophys. Acta, 1979, 562, 70 CrossRef CAS.
- D. Fornasiero and T. Kurucsev, J. Phys. Chem., 1981, 85, 613 CrossRef CAS.
- H. J. Li and D. M. Crothers, J. Mol. Biol., 1969, 39, 461 CrossRef CAS PubMed.
- Y. Matsuoka and K. Yamaoka, Bull. Chem. Soc. Jpn., 1979, 52, 3163 CrossRef CAS.
- (a) R. Lyng, A. Rodger and B. Nordén, Biopolymers, 1991, 31, 1709 CrossRef CAS PubMed; (b) R. Lyng, A. Rodger and B. Nordén, Biopolymers, 1992, 32, 1201 CrossRef CAS PubMed; (c) E. Tuite and B. Nordén, J. Am. Chem. Soc., 1994, 116, 7548 CrossRef CAS; (d) E. Tuite and B. Nordén, Chem. Comm., 1995, 53–54 RSC.
- J. Ramstein, C. Houssier and M. Leng, Biochim. Biophys. Acta, 1973, 335, 54 CrossRef.
- (a) Y. Matsuoka and B. Nordén, Biopolymers, 1982, 21, 2433 CrossRef CAS PubMed; (b) P. J. Chou and J. Johnson, J. Am. Chem. Soc., 1993, 115, 1205 CrossRef CAS.
- H.-C. Becker and B. Nordén, J. Am. Chem. Soc., 2000, 122, 8344 CrossRef CAS.
- G. Cohen and H. K. Eisenberg, Biopolymers, 1969, 8, 45 CrossRef CAS.
- J. Ramstein, M. Dourlent and M. Leng, Biochem. Biophys. Res. Comm., 1972, 47, 874 CrossRef CAS PubMed.
- J. Ramstein, M. Ehrenberg and R. Rigler, Biochemistry, 1980, 19, 3938 CrossRef CAS PubMed.
- K. Günther, M. Mertig and R. Seidel, Nucl. Acids Res., 2010, 38, 6526 CrossRef PubMed.
- D. Suh and J. B. Chaires, Bioorg. Med. Chem., 1995, 3, 723 CrossRef CAS PubMed.
- (a) L. Kapiak and E. J. Gabbay, J. Am. Chem. Soc., 1975, 97, 403 CrossRef; (b) S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319 CrossRef CAS PubMed.
- J.-L. Butour and J.-P. Macquet, Biochim. Biophys. Acta, 1981, 653, 305 CrossRef CAS.
- (a) E. Tuite, J. M. Kelly, G. S. Beddard and G. D. Reid, Chem. Phys. Lett., 1994, 226, 517 CrossRef CAS; (b) G. D. Reid, D. J. Whittaker, M. A. Day, C. M. Creely, E. M. Tuite, J. M. Kelly and G. S. Beddard, J. Am. Chem. Soc., 2001, 123, 6953 CrossRef CAS PubMed.
- Y. Kubota and R. F. Steiner, Biophys. Chem., 1977, 6, 279 CrossRef CAS PubMed.
- R. Ruiz, B. García, G. Ruisi, A. Silvestri and G. Barone, J. Mol. Struct: THEOCHEM, 2009, 915, 86 CrossRef CAS.
- (a) T. Maehigashi, O. Persil, N. V. Hud and L. D. Williams, RCSB Protein Databank ID 3FT6 DOI:10.2210/pdb3ft6/pdb; (b) D. J. Patel and L. L. Canuel, Proc. Natl. Acad. Sci. USA, 1977, 74, 2624 CrossRef CAS PubMed; (c) S. Neidle, A. Achari, G. L. Taylor, H. M. Berman, H. L. Carrell, J. P. Glusker and W. C. Stallings, Nature, 1977, 269, 304 CrossRef CAS PubMed; (d) H.-S. Shieh, H. M. Berman, M. Dabrow and S. Neidle, Nucl. Acids Res., 1980, 8, 85 CrossRef CAS PubMed; (e) B. Schneider, S. L. Ginell and H. M. Berman, Biophys. J., 1992, 63, 1572 CrossRef CAS PubMed.
- (a) A. H.-J. Wang, G. J. Quigley and A. Rich, Nucl. Acids Res., 1979, 6, 3879 CrossRef CAS PubMed; (b) B. S. Reddy, T. P. Seshadri, T. D. Sakore and H. M. Sobell, J. Mol. Biol., 1979, 135, 787 CrossRef CAS PubMed; (c) T. D. Sakore, K. K. Bhandary and H. M. Sobell, J. Biomol. Struct. Dyn., 1984, 1, 1219 CrossRef CAS PubMed.
- (a) P. Lincoln and B. Nordén, J. Phys. Chem. B, 1998, 102, 9583 CrossRef CAS; (b) A. Reymer and B. Nordén, Chem Comm., 2012, 48, 4941 RSC.
- (a) H.-K. Kim, P. Lincoln, B. Nordén and E. Tuite, Chem. Commun., 1997, 2375 RSC; (b) A. W. McKinley, P. Lincoln and E. M. Tuite, Dalton Trans., 2013, 42, 4081 RSC.
- E. L. Gilroy, M. R. Hicks, D. J. Smith and A. Rodger, Analyst, 2011, 136, 4159 RSC.
- H. Yamakawa and M. Fujii, Macromolecules, 1974, 7, 128 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, spectra, calculations, physical data. See DOI: 10.1039/c3ra43090a |
| ‡ Abbreviations: CD, circular dichroism; CT-DNA, calf thymus DNA; PD, proflavine diazide; Pf, proflavine; LD, linear dichroism; LDr, reduced linear dichroism; MePD, methylated proflavine diazide; P/D, nucleotide phosphate/dye ratio; [poly(dA–dT)]2, poly(deoxyadenylic-thymidylic) acid; [poly(dG–dC)]2, poly(deoxyguanylic-cytidylic) acid; TM, transition moment. |
| This journal is © The Royal Society of Chemistry 2013 |