 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold dissolution: towards understanding of noble metal corrosion
Serhiy
Cherevko
*,
Angel A.
Topalov
,
Aleksandar R.
Zeradjanin
,
Ioannis
Katsounaros
and
Karl J. J.
Mayrhofer
*
Department of Interface Chemistry and Surface Engineering, Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Straße 1, 40237 Düsseldorf, Germany. E-mail: cherevko@mpie.de; mayrhofer@mpie.de; Fax: +49 (211) 6792-218; Tel: +49 (211) 6792-160
First published on 8th July 2013
Abstract
The electrochemical dissolution of gold is an intricate topic and even though it has been studied for more than 50 years, its understanding remains rather limited. In the current work, we obtain unique information on gold dissolution by using a setup composed of a micro-electrochemical scanning flow cell (SFC) and inductively coupled plasma mass spectrometry (ICP-MS). Thus, comprehensive online gold dissolution profiles during the initial stage of oxidation as a function of the potential, time and pH are presented. A microscopic model explaining the experimental findings is proposed. According to this model, two dissolution mechanisms take place in two different potential regions: at low anodic potentials the dissolution is driven by the place-exchange between metal and adsorbed hydroxyl/oxygen ions, while at higher potentials the oxygen evolution reaction taking place on the surface of gold oxide initiates concomitant gold loss.
1. Introduction
Gold is a commonly used material in electrochemistry. Due to its chemical inertness and resistance to oxide formation within the stability potential window of water, gold is a material of choice for many electrode reactions.1 In general, gold is considered as an ideally inert electrode in these studies, even though it is known that it is susceptible to dissolution in the presence of complexing agents like CN−, Cl− or Br−,2–4 which often restricts its application to solutions free from those species. However, also in the absence of such complexing agents Au can still dissolve at positive potentials, an effect that is frequently neglected. Despite its importance, our knowledge on the fundamentals of gold dissolution is based on few works only. More than a century ago, in 1907, Campbell observed gold dissolution on the anode and its deposition on a platinum cathode, when a voltage of 10 V was applied in a two-electrode cell.5 The authors, however did not find Au in solution, which is probably due to the insufficient detection limits of the methods used at that time. Most likely due to such limitations, there was no significant progress for decades, so that only in late 1960s and early 1970s with the rapid development of new electrochemical equipment and techniques the issue was revisited. Early voltammetric results on the oxidation/reduction of noble metals and particularly on gold revealed a charge imbalance between oxidation and reduction,6 which was attributed to the anodic dissolution of metals.7 In particular, Rand and Woods found that during the gold electrode cycling between +0.6 and +1.54 VRHE in 1 M H2SO4, the anodic charge was by 3.8 ± 0.2 μC cm−2 higher than the cathodic one, and they attributed this to the dissolution of gold during the positive-going sweep. Post analysis of the electrolyte performed by atomic absorption spectroscopy after 1040 cycles, confirmed the voltammetric data since the amount of dissolved gold corresponded (using the Faraday's law) to a similar charge of 3.6 ± 0.4 μC cm−2. In addition, extensive work on the investigation of the anodic dissolution of certain noble metals, including gold, was done at the Karpov Institute of Physical Chemistry in Moscow by Kolotyrkin, Chemodanov and others, by means of highly sensitive radiometric determination of corrosion products in electrolyte.8,9 On the other side, Cadle and Bruckenstein showed that gold dissolves also during the reduction of gold oxide using a rotating ring-disk electrode (RRDE) setup.10 In their experiments, gold was polarized at an anodic potential for 5 min, and then a negative-going sweep was recorded, where dissolution was observed. Similarly, Vesztergom et al. have also shown the existence of cathodic dissolution of gold using an RRDE setup and a phase-shifted double cyclic voltammetry method.11Recently, Tsuru et al. applied a double electrode flow cell to study the dissolution of platinum,12,13 palladium14 and, particularly, gold.15 The amount of dissolved gold was calculated using the currents recorded on the collector electrode when the potential on the generator electrode was in the region 1.35–1.6 VSHE (during the positive-going scan) and 1.4–1.1 VSHE (during the negative-going scan). The authors observed that gold dissolves during both directions of the sweep, and the integration of the absolute currents recorded on the collector electrode yielded a charge of 4.2 μC cm−2 per cycle between +0.4 and +1.6 VSHE.
While most of the previous works were driven solely by scientific curiosity and aimed at a fundamental understanding of the gold dissolution process, nowadays our interest is extended and related to practical applications as well. The lack of knowledge on the electrochemical gold oxidation and dissolution may retard the expansion and exploitation of new industrial applications. Gold was already proposed for several catalytic and sensor applications such as alcohol oxidation,16 especially in combination with platinum and palladium in alkaline media,17–19 CO oxidation20–22 and glucose oxidation.23 Gold-platinum alloys are also considered as active and stable oxygen reduction reaction (ORR) catalysts in proton exchange membrane fuel cells (PEMFCs)24–31 or lithium-air batteries.32,33 Moreover, unlike platinum, such alloys are poor catalysts for methanol oxidation, making them good candidates for methanol-tolerant cathodes in direct methanol fuel cells (DMFCs).34
For low-temperature fuel cell applications, the potential at the cathode may reach values as high as +1.5 VRHE during start-up or shut-down of the cell.35–37 Under these conditions, several degradation mechanisms may take place38 including the dissolution of the noble metal39,40 This work includes a comprehensive study of the stability of gold under transient and steady state potential perturbations, using a novel setup in which a commercial inductively coupled plasma mass spectrometer (ICP-MS) is utilized to detect the amount of gold dissolved during electrochemical experiments in a scanning flow cell (SFC). The current study complements previous works from our group on the rhodium,41 platinum39 and gold42 dissolution, aiming for a better understanding of the electrochemical dissolution of gold and noble metals in general, and eventually for the design of more stable catalysts.
2. Experimental
Before each experiment, the electrode (foil or disk) was polished with 1 and 0.3 μm alumina suspensions on a polishing cloth (Struers, MD Mol) followed by a short sonication in water, extensive washing in ultrapure water (PureLab Plus system, Elga, 18 MΩ, TOC < 3 ppb) and drying in a flow of argon. All potentials in this work are presented with respect to the reversible hydrogen electrode (RHE) potential, which was acquired against a Ag/AgCl electrode using a Pt disk electrode (5 mm diameter disk sealed in Teflon®, MaTeck, Germany) in the corresponding electrolyte after hydrogen saturation. The electrolytes (0.1 M H2SO4) were freshly prepared from superpure concentrated sulfuric acid (Merck, Germany) by dilution in ultrapure water. The potentiostat (Gamry Reference 600, USA), electrolyte, gas flow, and SFC components were automatically controlled using an in-house built LabVIEW software.43 Programmed measurement protocols were used to avoid any user interaction with the setup during measurements and thus increase reliability of the results.2.1. Experiments in the scanning flow cell coupled with online elemental analysis
A 0.1 mm thick 99.99% polycrystalline gold foil purchased from Aldrich or a gold disk (5 mm diameter, MaTeck, Germany) was used as a working electrode. The working electrode was placed on an xyz-positioning table and a tungsten needle was connected to the foil/disk to make an electrical contact. All experiments were performed in an in-house-built flow cell similar to the one reported in our previous paper.41 During measurements, the electrolyte was pumped to/from the cell by a standard ICP-MS (NexION 300X, Perkin Elmer) pump with constant flow rate of 140–150 μL min−1 through Tygon® tubing (380 μm inner diameter) from a reservoir of argon-saturated 0.1 M H2SO4 to the V-shaped channels of the polycarbonate cell and further downstream. The electrolyte was mixed with an internal standard (187Re, 7.5 μg L−1, mixing ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a Y-connector after the cell and was introduced into the ICP-MS. Calibration of the ICP-MS was made on each experiment day. The exposed area on the working electrode was ca. 1 × 10−2 cm2. Data are normalized to the exposed geometric area of the working electrode, unless otherwise stated. A homemade Ag/AgCl micro-reference electrode or a commercial saturated Ag/AgCl electrode (Metrohm, Germany) and a graphitic rod were used as the reference and the counter electrode, respectively. Electrochemical cleaning/activation of gold was performed by recording 30 cycles between 0 and +1.8 VRHE at 200 mV s−1 with simultaneous detection of gold dissolution. Then, the electrode was held at open circuit potential or a predefined potential in the double-layer region for 180–300 s to stabilize the dissolution signal on the background level. More details on the cell design, the setup and on the ICP-MS measurements can be found in the experimental part of a previous report.41
:1) in a Y-connector after the cell and was introduced into the ICP-MS. Calibration of the ICP-MS was made on each experiment day. The exposed area on the working electrode was ca. 1 × 10−2 cm2. Data are normalized to the exposed geometric area of the working electrode, unless otherwise stated. A homemade Ag/AgCl micro-reference electrode or a commercial saturated Ag/AgCl electrode (Metrohm, Germany) and a graphitic rod were used as the reference and the counter electrode, respectively. Electrochemical cleaning/activation of gold was performed by recording 30 cycles between 0 and +1.8 VRHE at 200 mV s−1 with simultaneous detection of gold dissolution. Then, the electrode was held at open circuit potential or a predefined potential in the double-layer region for 180–300 s to stabilize the dissolution signal on the background level. More details on the cell design, the setup and on the ICP-MS measurements can be found in the experimental part of a previous report.41
2.2. Experiments in a conventional three-electrode cell
A conventional three-electrode cell made of Teflon® with all electrode compartments separated by Nafion® membranes (Tschurl modification)44 was used in additional measurements. The working electrode was a gold disk of 5 mm in diameter (MaTeck, Germany) embedded in a Teflon® cylinder. The gold activation procedure was the same as described above for the SFC setup, with the only difference that dissolution products were not recorded. A saturated Ag/AgCl electrode and a graphite rod served as the reference and the counter electrode, respectively.3. Results
3.1. System validation
To make sure that the chosen electrode preparation procedure is appropriate, gold voltammograms in deaerated solution were recorded using both the SFC and a conventional three-electrode setup (see Fig. 1). The curves obtained with the two different techniques match each other well in the region of gold oxidation and oxide reduction. The small difference in the oxide formation region may be attributed to the difference in hydrodynamic conditions in the two setups, which may have an effect on the electrochemical cleaning step, e.g. presence/absence of re-deposition of dissolved gold. It should be noted that one may find different cyclic voltammogram (CV) profiles of gold/gold oxide system in various reports. Reasons of such diversity is the difference in electrode preparation (influence of grain size, exposed surface orientations, purity, etc.), electrode history (polishing and cleaning procedure), affinity of gold to poisoning by impurities, surface reconstruction during potential excursions, etc.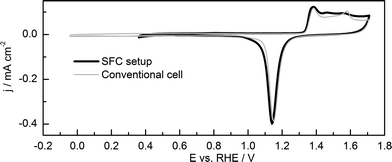 | ||
| Fig. 1 CVs taken on a gold foil electrode in the SFC setup (thick black line) and on a gold disk electrode in a conventional three-electrode cell setup (thin gray line) in 0.1 M H2SO4, showing double layer charging, formation of gold oxide and oxide reduction. Scan rate: 50 mV s−1. | ||
The voltammograms recorded in both setups display similar characteristics as described in literature;7,11 which can be explained following the most established mechanism of gold oxidation proposed by the Conway school.45–48 In the positive-going sweep, in the so-called double-layer region (i.e. below ca. +1.2 to +1.3 VRHE), the surface of gold is covered by adsorbed supporting electrolyte anions (e.g. HSO4− and/or SO42−), while the coverage with OHads/Oads species is relatively low. Beyond +1.2–1.3 VRHE the adsorbed anions of the supporting electrolyte are gradually displaced by OHads/Oads, and even Au–OH/O place-exchange, i.e. incorporation of O into the sub-surface layers, occurs. At even higher potentials and longer times, the formation of bulk phase oxide(s)/hydroxide(s) or hydrous oxide(s) takes place.49–51 It should be noted, that there is uncertainty about the exact nature of the adsorbed oxygenated species such as OHads/Oads or oxide(s)/hydrous oxide(s). For the sake of convenience, only the term oxide will be used in the text below, still bearing in mind that it may consist of several forms of oxides, hydroxides or hydrous oxides. Only when the nature of the oxide state is crucial to the discussion, a distinction between oxides will be emphasized, particularly between adsorbed and place-exchanged oxygen.
The CVs were recorded up to a potential close to the Burshtein minimum, corresponding to the onset of oxygen evolution reaction (OER).52 The negative potential limit in the measurements with the SFC setup was limited to the onset for the ORR, because in this setup (contrary to the conventional cell) residual oxygen was still present even after prolonged purging.41 The roughness factors (ratio of real to geometric surface area) of the two electrodes were estimated by integration of the oxide reduction peaks in the negative-going scan from +1.3 V to +0.95 V, and were ca. 1.4 (Au disk) and ca. 1.5 (Au foil). Here, the assumption was made that at the Burshtein minimum, the electrode is covered by one monolayer of adsorbed oxygen with a transferred charge of 400 μC cm−2.53 In all other measurements described hereinafter, the roughness factor was used as a fingerprint of the electrode preparation procedure, in order to ensure good reproducibility of the results.
3.2. Potentiodynamic dissolution of gold–the effect of upper potential limit
The cyclic voltammogram on gold in 0.1 M H2SO4 electrolyte together with the dissolution profile recorded online in Fig. 2a shows that there is a concurrency of the gold dissolution and gold oxidation. Due to the presence of planes with different crystal orientation as well as crystal defects of lower coordination number, however, the exact determination of an onset potential for the dissolution on a polycrystalline gold electrode during the positive-going sweep is not straightforward. Therefore, rather than discussing a single potential value it is better to consider a potential range for the onset of dissolution. At potentials ca. 100–150 mV below the onset potential for significant OHads/Oads adsorption (ca. +1.3 VRHE), some small, almost negligible amount of gold dissolves in parallel to a minor increase in the current at ca. +1.1 VRHE most likely due to oxidation of defects (data not shown). A correlation between magnitude of anodic current in this potential region and amount of surface defects was indeed observed by Kolb et al.54 Very recently Compton et al. have shown that due to higher density of surface defects gold nanoparticles show more significant oxidation currents compared to a bulk gold electrode.55 Thus, oxidation of nanoparticles at much lower potentials and, as a result, dissolution should be taken into account, for instance, in degradation studies on platinum nanoparticles during oxygen reduction.38 Alternatively, the possibility of OH/O adsorption within the surface pattern of adsorbed (bi-)sulfate ions was reported by Conway et al.48 Higher amounts of dissolved gold are detected when the upper potential limit (UPL) during cycling exceeds the onset for gold oxidation (E > ca. +1.3 VRHE, as highlighted in the inset in the Fig. 2a). Above this potential, gold dissolves both anodically and cathodically, as can be seen by the sharp increase of detected amount of gold in the positive-going sweep, and the peak in the dissolution signal upon reduction of the oxide (i.e. below ca. +1.2 VRHE), in the negative-going sweep (in this work we also use the term “cathodic dissolution”, bearing in mind that this is a process during cathodic reduction of gold oxide).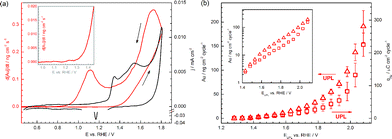 | ||
| Fig. 2 (a) Dissolution profile shown in line with cyclic voltammogram. The inset shows a magnified view of the area corresponding to the onset of dissolution. (b) Dependence of dissolved gold per cycle on the upper potential limits (UPL) of applied cycles. Open squares present data obtained from the experiments with increasing UPL and open triangles show results with decreasing UPL. The inset shows the same data presented in log scale. Electrolyte: 0.1 M H2SO4; scan rate: 10 mV s−1. | ||
In a previous work42 we showed that the reductive dissolution of gold dominates when the UPL is kept in a relatively low potential region (<ca. +1.6 VRHE), similarly to Pt39 and Rh.41 However, at higher potentials (>ca. +1.6 VRHE), anodic gold dissolution is the main cause of gold loss. Thus the rate of dissolution increases rapidly in the oxygen evolution region, and the total amount of gold dissolved during one cycle increases with the increase of the UPL, as can be seen in Fig. 2b. When the same electrode location is used for repetitive investigations using different UPLs, there is some ambiguity on whether there is any effect of the electrode history, since morphological changes may occur during consecutive cycles. To reject this hypothesis, an experimental series with decreasing UPL was performed. While generally the same trend for Au dissolution was obtained (see Fig. 2b and inset, where dissolution is shown in log scale), slightly higher amounts of Au dissolved at the same UPL especially in the high potential region. Interestingly, a similar difference in the exact dissolution amounts upon increasing and decreasing the UPL was also reported by Kolotyrkin et al. for platinum dissolution in an acetate electrolyte, which was attributed to the non-stationary nature of platinum dissolution at sufficiently low time scales.9
3.3. Potentiodynamic dissolution of gold–the effect of the scan rate
To study whether the time scale of the experiment besides the potential value itself affects gold dissolution, the effect of the scan rate using a fixed potential window (+0.4 to +1.8 VRHE) was studied and the results are presented in Fig. 3. Given that the time at which gold is prone to dissolution is shorter at higher scan rates, it is not surprising that an increase in the scan rate results in decreased quantities of dissolved gold per cycle. However, it is particularly interesting that such a decline is almost linear with the square root of the scan rate, as the slope of the log[Au] vs. log v diagram (Fig. 3b) is −0.56 ± 0.09 (R2 = 0.998). This may be an indication of a single diffusion-controlled Faradaic reaction of gold dissolution, similarly to what was proposed by Conway et al.2 for observations in the presence of complexing Cl− or Br− anions.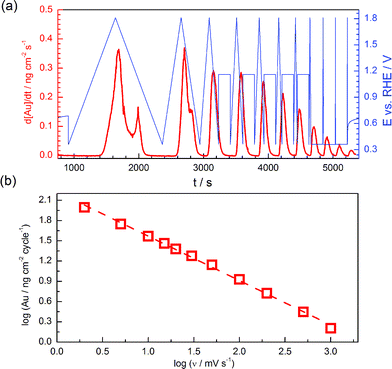 | ||
| Fig. 3 (a) Effect of scan rate on dissolution of gold during potential cycling between +0.4 and +1.8 VRHE. (a) Dissolution profile is presented together with potential transients (potential cycles followed by potential steps in case of high scan rates). (b) Dependence of gold dissolution amount on the scan rate presented on the log–log scale. Electrolyte: 0.1 M H2SO4. | ||
The simple relation found between dissolution quantity and scan rate, allows correlating and comparing literature data obtained with different scan rates. A re-calculation of the data of Rand and Woods7 and of Tsuru et al.15 to the main scan rate used in the current work (i.e. 10 mV s−1), yields a dissolution amount of ca. 5 and 4 ng cm−2 cycle−1, respectively, which are close to the value of 5–7 ng cm−2 cycle−1 observed here for similar UPLs. It should be noted that a decrease in the dissolution amount with scan rate was also observed on Rh and Pt,39,41 without, however, following such a simple dependence as for Au. This is most likely related to the fact that, contrary to gold, the dissolution during oxide reduction is the dominant dissolution process for those metals in the whole potential range.
3.4. Gold dissolution during formation/reduction of bulk oxide
It is generally accepted that gold forms bulk oxides at high anodic potentials (ca. above +1.8 to +2 VRHE) and that the formed phases are hydrous oxides with a varying, complex nature of hydration.49 In order to study the effect of bulk oxide formation and reduction on gold dissolution, a constant potential of +2.0 VRHE was applied for different times and always followed by a reduction ramp. The reduction peak shifts to more negative potentials for longer polarization times, indicating that the formation/reduction of such oxides is a highly irreversible process (Fig. 4a). Unlike the compact layer, whose thickness is limited by 2–3 layers only, bulk oxide may grow in virtually unlimited number of layers, depending on the potential and time of polarization.50,51 In our case, formation of a total of 4.5 MLs was achieved for an oxidation period of 105 s.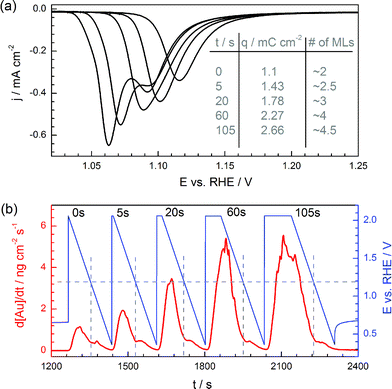 | ||
| Fig. 4 (a) Negative linear potential scans showing the reduction of gold oxides formed during anodization over various times. Charges obtained by integration of the cathodic peaks and corresponding number of gold oxide monolayers (assuming that one monolayer charge is 400 μC cm−2 and taking into account the roughness factor of the electrode of 1.45) are indicated in the table. (b) Corresponding dissolution profile, wherein the dotted lines correspond to the onset of gold oxide reduction. Scan rate during ramps: 10 mV s−1. | ||
While the anodic dissolution is enhanced at longer polarization times, the amount of gold dissolved during the reduction of the oxide does not show any significant increase. This suggests that the reduction of the complete bulk hydrous oxide is not associated with gold dissolution, but that the reduction of the compact oxide is dominating the cathodic gold dissolution. This is rather surprising, considering the surface roughening introduced by the formation of hydrous and porous bulk oxide.45,49,56
3.5. Potentiodynamic dissolution of gold–the effect of the re-deposition
Among the possible reasons for the decrease of the dissolved amount of Au with scan rate and for the independence from the formation/reduction of bulk oxide, one may consider a time effect and possible re-deposition of dissolved gold. To estimate the effect of re-deposition, additional experiments were carried out, where the cycle was interrupted during the backward scan and the potential was held at different values (ESTEP), as it is shown in Fig. 5a.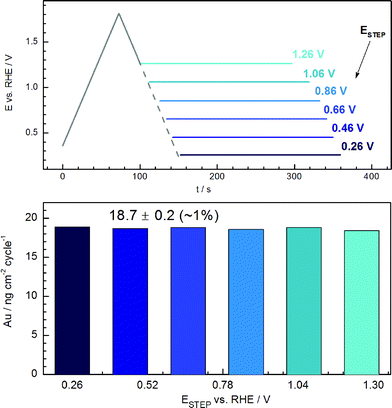 | ||
| Fig. 5 (a) Potential-time programs which consist of a positive-going sweep, an incomplete negative-going sweep and a potential step. (b) Dependence of the dissolved gold quantity on the lower potential limit of potential scans (step potentials ESTEP) shown in (a). Scan rate: 20 mV s−1. | ||
As we do not know the exact nature of the dissolved Au species and their activity at the vicinity of the electrode as a function of the potential and scan rate, it is impossible to calculate the deposition potential of the dissolved species. However, we can make the assumption that re-deposition can be possible only at potentials lower than those of the onset of gold oxidation. This sounds reasonable, if one considers that dissolved gold species do not deposit at potential of +0.8 VSHE15 (much lower than the maximum ESTEP of +1.26 VRHE applied in the current work). As one can see from the Fig. 1, at this potential, the surface is still covered by oxide, and re-deposition of gold on the oxide is not likely to happen. If re-deposition was taking place at the lower potentials in this experiment, one should expect a decreased amount of gold detected downstream. However, the amount of dissolved gold detected in the ICP-MS was almost the same (deviation is within 1%) regardless of the value of ESTEP. Thus, even if some re-deposition takes place microscopically, its effect must be negligible for the scan rates utilized in the current work. Based on this, the results presented in section 3.4 on the independence of cathodic dissolution on the bulk oxide formation cannot be attributed to re-deposition of dissolved gold.
3.6. Potentiostatic dissolution of gold
Fig. 6a presents dissolution profiles of gold during potential hold experiments at different potentials. At potentials below the onset of gold oxidation (ca. +1.3 VRHE) the dissolution rate remains below the detection limit. However, as soon as the potential is stepped to a value where gold oxidation takes place, continuous dissolution starts to appear. An initial dissolution peak right after the potential step is followed by a gradual decrease in the dissolution rate until an almost stable value is achieved after ca. 1000 s of polarization (Fig. 6a). In this respect, gold behaves differently to platinum and rhodium, the dissolution of which is suppressed to below the detection limit after the same timeframe.39,41 In order to obtain the amount of dissolved gold at different potentials, the integral over the area below the corresponding dissolution profiles was taken and plotted as a function of the applied potential (inset in Fig. 6a). Taking into account that one monolayer of gold corresponds to ca. 410 ng cm−2,42 the estimated number of dissolved layers gradually increases with the polarization potential, similarly to what was observed in experiments where the UPL was varied (inset on the Fig. 2b).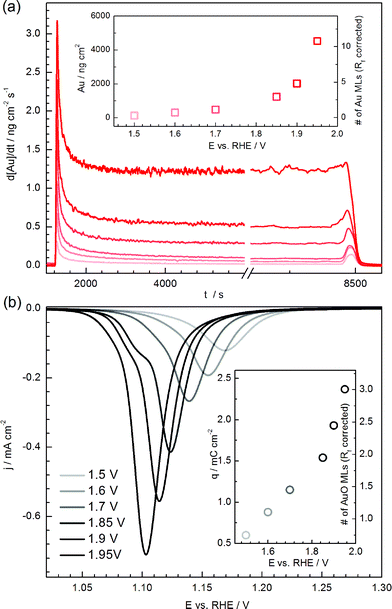 | ||
| Fig. 6 Potentiostatic dissolution of gold. (a) Dissolution of gold at different applied potentials (for potential values, see legend in 6b). For the sake of clearness, the initial section corresponding to the dissolution of gold during electrochemical cleaning is not shown. The total amount of dissolved gold and the corresponding number of lost gold monolayers against the step potential is shown in the inset. (b) Corresponding gold oxide reduction profiles following polarization at different potentials for 7200 s. The inset shows the dependence of the reduction charge and the number of gold oxide monolayers on the applied potential. | ||
The small peaks on the dissolution profiles at around 8500 s correspond to dissolution during cathodic ramps that were applied after the constant potential step, to get information on the amount of gold oxide(s) formed during each potential step. The corresponding voltammetric profiles are shown in Fig. 6b. Again, with increase of the polarization potential, more oxide forms on the surface of gold, while the reduction peak potentials shift to more negative values due to the formation of more stable oxides. The presence of at least two different oxide states is clearly seen by the small shoulder on the descending section of the peaks. The number of formed oxide layers increases from 0.75 (at +1.5 VRHE) to 3 monolayers (at +1.95 VRHE), indicating the qualitative analogy between the dissolution curve and the oxide isotherm in this region.
According to the classical theory of corrosion of metals, a constant dissolution rate should correspond to equal rates of oxide formation and metal dissolution.57 To verify this statement, the gold electrode was oxidized over different times and each step was followed by a cathodic ramp. Afterwards, CVs with 50 mV s−1 with UPL corresponding to the Burshtein minimum52 were taken to exclude any significant surface area change. The dissolution signal was recorded continuously as shown in Fig. 7a for three different applied polarization potentials. The dissolution rate changes with time for all studied potentials, and no complete steady state of dissolution is reached even after 3600 s of polarization. The amount of dissolved gold during each step (open symbols) and the dissolution rate at the end of each step (filled symbols) are plotted in Fig. 7b for E = 1.9 VRHE.
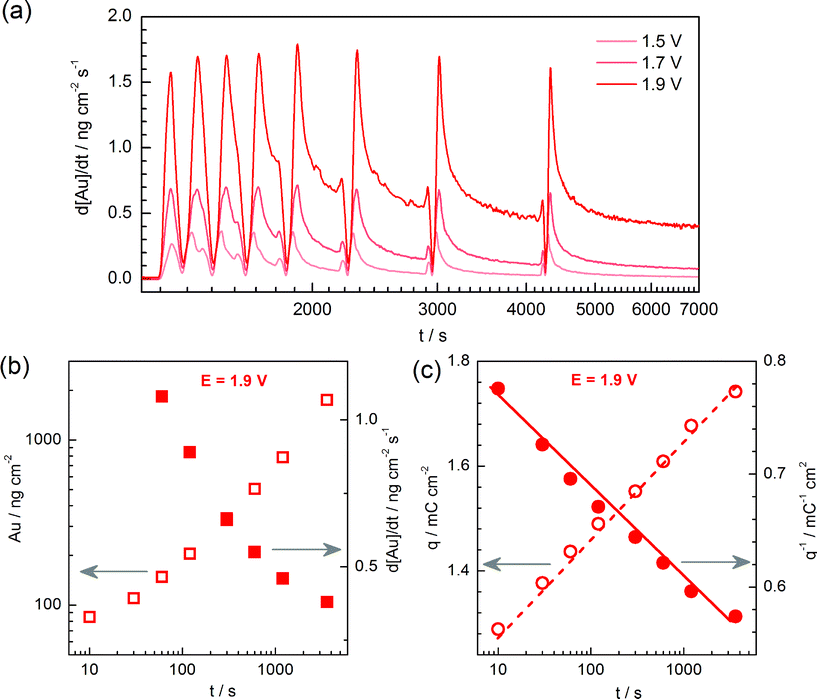 | ||
| Fig. 7 Time scale effect on gold dissolution and oxidation. (a) Dissolution profiles of gold vs. time under different polarization potential (see figure legend). (b) Corresponding dependence of the total amount of dissolved gold (open symbols) and rate of dissolution at the end of each step (filled symbols) on polarization time. (c) Charge associated to the oxidation of gold as a function of polarization time. For the sake of clearness, only data for E = 1.9 VRHE are presented in (b) and (c). | ||
The charge associated to the oxidation of gold, which is corresponding to oxide coverage, depends linearly on the logarithm of the polarization time, as shown in Fig. 7c (left axis). Conway et al. suggested that such a relationship is indicative of an activation-limited reaction, in which the Gibbs free energy of activation increases linearly with time.58 The growth of gold oxide however follows also the q−1vs. log(t) law (see Fig. 7c, right axis),50,59 indicating that the oxide growth is governed by the high-field-strength raised between metal and electrolyte fronts – known as the Cabrera–Mott model.60 The same tendency was also found for E = 1.5 VRHE and E = 1.7 VRHE (data are not shown). Thus the gold oxide growth may be explained by both kinetics laws – i.e. a direct logarithmic law for AuO thickness up to 2 MLs and an inverse logarithmic law when the oxide thickness becomes higher than 2–3 MLs,51 so that solid conclusions cannot be drawn. However, while the high field strength may only emerge when the two interfaces are separated by a relatively thin and insulating oxide, Conway's model is also applicable for the case of partially covered surfaces. Therefore, particularly in the studied potential range and experimental time scale where the oxide coverage is relatively low, the gold oxidation is likely to be governed by the electrochemical surface potential change with O/OH adsorption and Au–O/OH place-exchange. It should be additionally noted that for all applied potentials, a constant growth of gold oxide was found, however we did not find any significant increase in the electrode surface area (roughness factor) due to oxidation/dissolution. Thus, the obtained oxidation law is an intrinsic property of gold rather than an artifact due to surface area increase.
3.7. Effect of pH
The electrochemical oxidation and electrochemical dissolution of Au possess quite similar standard potentials, however can eventually be distinguished by the fact that protons are involved only in the former one, while the latter is governed by the concentration of gold ions only. The electrochemical oxidation of gold can be expressed as61| 2Au + 3H2O → Au2O2 + 6H+ + 6e−Eo = 1.457 + 0.0591 log[H+] | (1) |
| Au + 3H2O → Au(OH)2 + 3H+ + 3e−Eo = 1.511 + 0.0591 log[H+] | (2) |
| Au → Au3+ + 3e−Eo = 1.498 + 0.0197 log[Au3+] | (3) |
Assuming that the discharge of water and the adsorption of OH or O are fast processes and always in equilibrium for the scan rates employed in the current work, an effect of the pH should reveal whether dissolution is controlled by metal oxidation or by gold ionization.
Fig. 8 shows the variation of the CVs and the dissolution signals for different concentrations of sulfuric acid electrolyte. It should be noted, that the change of the pH was always accompanied with an unavoidable change in the concentration of (bi-)sulfate anions. The use of high concentrations of sulfate salts (such as Na2SO4) to keep the concentration of electrolyte anions the same for all measurements, is not feasible since a high concentration of alkali metal cations cause complications in the ICP-MS measurements. Nevertheless, it will be shown below that the impact of the proton and of the (bi-)sulfate concentration can be sufficiently decoupled.
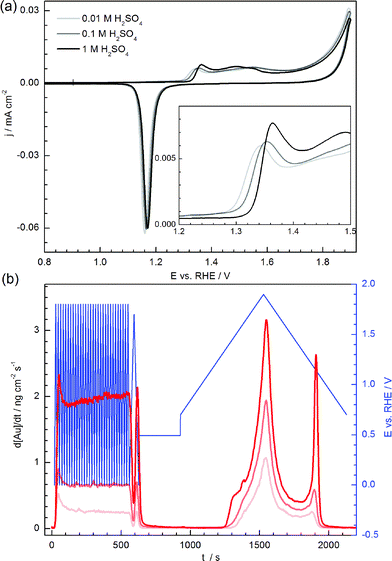 | ||
| Fig. 8 Effect of the electrolyte on gold dissolution during potential cycling. (a) CV curves recorded in three different electrolyte concentrations. The inset presents a magnified view of the onset of gold oxidation. (b) Dissolution of gold in three electrolytes during potentiodynamic conditions. | ||
When the CVs are plotted versus the pH-corrected RHE scale, they coincide relatively well (Fig. 8a), due to the fact that oxidation/reduction are indeed processes with protons involved in the corresponding rate-determining steps (RDS). The slight shift of the onset potential for gold oxidation to more positive values with increasing H2SO4 concentration is due to the competition between the adsorption of OH/O and (bi-)sulfate anions, i.e. a more positive potential is required for oxygenated species to displace (bi-)sulfate adsorbed species, as shown by Angerstein-Kozlowska et al.48
The dissolution signal together with the potential profile is shown in Fig. 8b. The signal corresponding to gold dissolution during the initial cleaning step is also included, to highlight that the dissolution signal increases almost exponentially with H+ concentration during these fast scans (200 mV s−1). The dissolution rate also grows with H+ concentration in the case of a slow scan rate of 2 mV s−1. Particularly, while the rate of anodic dissolution scales linearly with proton concentration, the rate of cathodic dissolution rather follows an exponential dependence, identical to that observed during the fast initial potential scans.
A similar effect of the dependence of gold dissolution on pH during anodic and cathodic process is seen for a prolonged constant potential step, followed by a cathodic ramp (Fig. 9a). The rate of anodic dissolution drops to an almost constant value after ca. 1000 s, with the ratio between dissolution rates in different electrolytes remaining stable. On the other hand, the dissolution rate during oxide reduction massively increases with pH decrease, as shown in the Fig. 9b, while the charge during oxide reduction (and thus the oxide coverage) is almost the same for all electrolytes (inset in Fig. 9a).
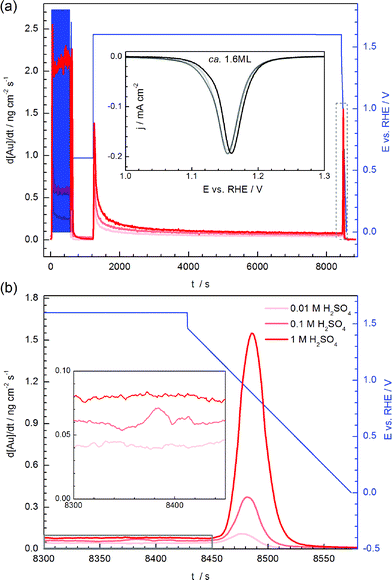 | ||
| Fig. 9 (a) Dissolution of gold in three electrolytes during potentiodynamic conditions. Corresponding reductive linear-sweep voltammograms are shown in the inset. (b) The magnified views of the dissolution profile at last minutes of the steady-state dissolution and during dissolution in the cathodic route. Higher magnification of the area highlighted with a gray box is shown in the inset. | ||
4. Discussion
Below, we will first summarize the most important experimental findings:(a) During a positive-going sweep, the onset of gold dissolution coincides with the onset of massive OH/O adsorption and, most likely, Au–OH/O place-exchange. During the negative going sweep, the rate of gold dissolution increases when gold oxide reduction starts;
(b) When the potential cycling at sweep rate of 10 mV s−1 does not exceed ca. +1.6 VRHE, dissolution during oxide reduction is the dominating dissolution process; for cycles with higher upper potential limits gold dissolves mainly anodically;
(c) The total amount of dissolved gold within a cycle decreases with increase in scan rate;
(d) Gold ions dissolve and diffuse away from the vicinity of the electrode, so that gold re-deposition is negligible within the time scale of the experiments;
(e) During anodic polarization experiments, a constant rate of gold dissolution is not achieved even after 7200 s at all studied potentials apart from E = +1.95 VRHE;
(f) The dissolved amount vs. potential graph follows roughly the same trend as the formed oxide amount vs. potential;
(g) The oxide growth kinetics can be described with both a q vs. log(t) and a q−1vs. log(t) dependence, while the dissolution rate decreases much slower (approximately it obeys log(Au) vs. log(t) law);
(h) The onset potential of dissolution, expressed in the RHE scale, is almost the same for electrolytes of different pH; small differences are attributed to the different concentration of counterions HSO4− and/or SO42−;
(i) The amount of dissolved gold during anodic and cathodic dissolution increases with increasing acidity; a change in the pH has a more pronounced effect on the dissolution during oxide reduction;
(j) The reduction of bulk hydrous gold oxide does not have a significant effect on gold dissolution.
The consistency between the onset of gold oxidation and transient dissolution in all experiments implies that both processes are closely linked to each other. Particularly, the observed shift of the dissolution onset potential with pH on the SHE scale reveals that one proton is involved in the rate determining step (RDS) of gold dissolution. Since the direct electrochemical dissolution does not involve protons (eqn (3)), gold dissolution at these potentials is governed by the kinetics of oxide formation. The increase in the dissolution rate with increasing acidity is then a sign of the shift of the water dissociation equilibrium, resulting in depressed amounts of hydroxyl ions in the vicinity of the electrode.
While the exact nature of the oxide still remains unresolved, the oxidation process of gold can tentatively be ascribed to OH/O adsorption and place-exchange between gold and oxygen ions, which commence in parallel.45,47,48 Interestingly, Au3+ ions, which are the thermodynamically stable species in acidic solution formed at sufficiently positive potentials according to the Pourbaix diagram,61 do not dissolve massively at such positive potentials due to passivation of the surface by gold oxide. During place-exchange, however, this passivity is probably disturbed either during surface reconstruction or during possible migration of gold ions from the gold matrix through the thin layer of gold oxide to the oxide/electrolyte interface. This leads to the exposure of gold ions to the electrolyte, which then experience a competition between two processes: (i) water discharge and consequent adsorption of OH/O that leads to re-passivation, and (ii) gold ion diffusion away from the electrode vicinity to the solution that leads to macroscopically observed dissolution. The extent of dissolution generally depends on the extent of the competition between these two processes, in particular for increasing acidity the re-passivation of the surface after place-exchange becomes less efficient. It is worth noting that the kinetics of the water discharge and OH/O adsorption is very facile in all cases, so that the observed rate of dissolution is overall relatively low.
Even though more work is required to derive an exact model for gold dissolution during transient conditions, it is still possible to consider a scheme that is consistent with the experimental observations. This tentative microscopic model (Scheme 1) of transient dissolution of gold includes processes taking place on the electrode during formation of oxide (a–c) and its reduction (c–e). As can be seen from the scheme, the main precursor of dissolved gold ion is the de-passivated ion formed anodically or cathodically as a result of place-exchange between O/OH and Au. It seems that the mechanism of the transient dissolution of gold at low potentials is similar to that of other noble metals which are prone to the formation of passivating oxides, i.e. Pt and, to less extent, Rh. Our observation that dissolution during the negative-going sweep is the main origin of dissolution, suggests that more exposed Au ions and thus more dissolution occur during the surface reduction on those metals.
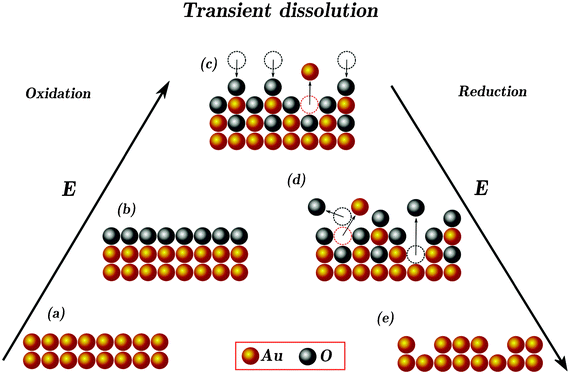 | ||
| Scheme 1 Proposed model of the transient dissolution of gold. For simplicity, gold in all valences is presented as “Au” and oxygenated species are shown as “O”. (a) Gold in the double-layer potential range. (b) Reversibly adsorbed O/OH ions on the surface of gold at potentials less than, or close to, ca. +1.3 V. Presence of O/OH ions on the surface protects gold from dissolution (passivation). Almost at the same potential place-exchange between O/OH and Au ions take place (de-passivation). After a simple act of the switch between the O/OH and Au ions, the two possibilities of the dissolution of Au and stabilization (re-passivation) are highlighted (c). During gold reduction, desorption of reversibly adsorbed O/OH causes de-passivation and gold dissolution (d). Reduced roughened surface is shown in (e). | ||
If the dissolution of gold was entirely driven by place-exchange in the entire potential region studied, one would expect a decrease in the dissolution rate approaching zero with time, regardless of the model the oxide growth follows. While this situation was indeed observed for platinum,39 gold dissolves almost at a constant rate at highly positive potentials. In addition, if dissolution was only due to place-exchange, one should expect proportionality between the charge associated with oxide formation and the amount of gold dissolving. However, for potentials above ca. +1.5 VRHE, it evolves from Fig. 7b and c that such a relationship does not exist (note that the dissolution amount is proportional to time in the log–log scale in Fig. 7b, but the charge corresponding to the formed oxide is linearly dependent to the time on the semi-log scale in Fig. 7c). The above observations indicate that there must be another process besides the place-exchange which contributes to additional gold corrosion at steady high potentials.
In a very recent work by the group of Koper62 it has been shown that the oxygen evolved during OER on gold is formed from oxygen ions in the oxide, the so-called “surface route” or “oxide route”.63 In this case one might expect a temporal local loss of passivation of the surface and thus an enhanced metal dissolution due to the same competition between (i) re-passivation and (ii) dissolution described above for the effect of place-exchange. In addition, the authors proposed that already at a potential as low as +1.54 VRHE some oxygen starts evolving as a result of the formation of Au2O3 from AuOOH, which is well in line with the observation for continuous gold dissolution starting already at that potential. The contribution becomes more dominating at higher potentials, when the OER rate is steadily increasing. It is worth mentioning that in contrast the enhanced stability of platinum in the OER region42 may thus be related to the fact that the reaction on platinum is going through a “solution route”.64
Since the formation of molecular oxygen on gold involves the participation of gold oxide at higher potentials, gold dissolution differs compared to other metals. This contribution of the OER in gold dissolution becomes more significant with increasing the potential, because the rate of the OER increases exponentially with potential. Eventually, anodic dissolution becomes the dominant dissolution process, while the cathodic dissolution is not significantly enhanced anymore. A model that can capture the nearly steady-state dissolution of gold during a constant oxygen evolution rate is sketched in Scheme 2.
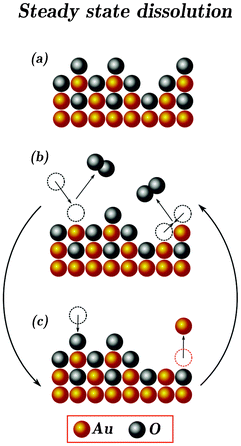 | ||
| Scheme 2 Proposed model of the steady state dissolution of gold. As in the Scheme 1, gold in all valences is presented as “Au” and oxygenated species are shown as “O”. (a) Oxidized passivated surface. Due to the logarithmic law of oxide growth, dissolution of gold is almost negligible at this stage after a relatively short period of time. (b) Oxygen evolution taking place on the surface of oxide-covered gold electrode (de-passivation). Formation of O2 from surface-surface and surface-solution oxygens is highlighted. Such de-passivation state of gold at high anodic potentials is not stable resulting in re-passivation due to the O/OH adsorption or dissolution. Both processes are presented in (c). | ||
Certainly, it needs to be emphasized that the elucidation of the exact dissolution mechanism is not straightforward, due to the uncertain and complex nature of the oxide species as well as the unresolved mechanism of noble metals passivation.65–68 Thus, further research efforts need to be directed towards a complete and combined investigation of gold oxidation and corrosion. Also other aspects, as for instance a possible effect of formation of hydroxyl radicals initiating and/or accelerating gold dissolution should be included.69–72 Nevertheless, the proposed models can capture all the experimentally observed aspects of gold dissolution described in this work. In summary, the local de-passivation of the surface during oxide formation/reduction as well as oxygen evolution is determining transient and steady-state dissolution of gold, respectively.
Acknowledgements
We thank the BMBF (Kz: 033RC1101A) for financial support and Andrea Mingers for experimental assistance.References
- F. Scholz, Electroanalytical Methods: Guide to Experiments and Applications, Springer, 2010 Search PubMed.
- M. Tian, W. G. Pell and B. E. Conway, Corros. Sci., 2008, 50, 2682–2690 CrossRef CAS.
- S. Ye, C. Ishibashi, K. Shimazu and K. Uosaki, J. Electrochem. Soc., 1998, 145, 1614–1623 CrossRef CAS.
- D. W. Kirk, F. R. Foulkes and W. F. Graydon, J. Electrochem. Soc., 1978, 125, 1436–1443 CrossRef CAS.
- F. H. Campbell, Trans. Faraday Soc., 1907, 3, 103–113 RSC.
- K. J. Vetter and D. Berndt, Zeitschrift für Elektrochemie, Berichte der Bunsengesellschaft für physikalische Chemie, 1958, 62, 378–386 Search PubMed.
- D. A. J. Rand and R. Woods, J. Electroanal. Chem., 1972, 35, 209–218 CrossRef CAS.
- Y. M. Kolotyrkin, Electrochim. Acta, 1973, 18, 593–606 CrossRef CAS.
- Y. M. Kolotyrkin, V. V. Losev and A. N. Chemodanov, Mater. Chem. Phys., 1988, 19, 1–95 CrossRef CAS.
- S. H. Cadle and S. Bruckenstein, Anal. Chem., 1974, 46, 16–20 CrossRef CAS.
- S. Vesztergom, M. Ujvári and G. G. Láng, Electrochem. Commun., 2011, 13, 378–381 CrossRef CAS.
- A. P. Yadav, A. Nishikata and T. Tsuru, J. Electrochem. Soc., 2009, 156, C253–C258 CrossRef CAS.
- B. R. Shrestha, A. P. Yadav, A. Nishikata and T. Tsuru, Electrochim. Acta, 2011, 56, 9714–9720 CrossRef CAS.
- B. R. Shrestha, A. Nishikata and T. Tsuru, Electrochim. Acta, 2012, 70, 42–49 CrossRef CAS.
- B. R. Shrestha, A. Nishikata and T. Tsuru, J. Electroanal. Chem., 2012, 665, 33–37 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Baumer, Science, 2010, 327, 319–322 CrossRef CAS.
- S. Cherevko, X. Xing and C.-H. Chung, Electrochim. Acta, 2011, 56, 5771–5775 CrossRef CAS.
- S. Cherevko, N. Kulyk and C.-H. Chung, Langmuir, 2012, 28, 3306–3315 CrossRef CAS.
- S. Cherevko, N. Kulyk and C.-H. Chung, Electrochim. Acta, 2012, 69, 190–196 CrossRef CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- C. T. Campbell, Science, 2004, 306, 234–235 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252–255 CrossRef CAS.
- S. Cherevko and C.-H. Chung, Sens. Actuators, B, 2009, 142, 216–223 CrossRef CAS.
- S. V. Selvaganesh, G. Selvarani, P. Sridhar, S. Pitchumani and A. K. Shukla, Phys. Chem. Chem. Phys., 2011, 13, 12623–12634 RSC.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS.
- Y. Zhang, Q. Huang, Z. Zou, J. Yang, W. Vogel and H. Yang, J. Phys. Chem. C, 2010, 114, 6860–6868 CAS.
- Y.-H. Fang and Z.-P. Liu, J. Phys. Chem. C, 2011, 115, 17508–17515 CAS.
- M. S. El-Deab and T. Ohsaka, J. Electroanal. Chem., 2003, 553, 107–115 CrossRef CAS.
- B. Du, O. Zaluzhna and Y. J. Tong, Phys. Chem. Chem. Phys., 2011, 13, 11568–11574 RSC.
- F. Ye, H. Liu, W. Hu, J. Zhong, Y. Chen, H. Cao and J. Yang, Dalton Trans., 2012, 41, 2898–2903 RSC.
- E. Higuchi, K. Hayashi, M. Chiku and H. Inoue, Electrocatalysis, 2012, 3, 274–283 CrossRef CAS.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y.-C. Lu, Z. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef CAS.
- J. B. Xu, T. S. Zhao, W. W. Yang and S. Y. Shen, Int. J. Hydrogen Energy, 2010, 35, 8699–8706 CrossRef CAS.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS.
- C. A. Reiser, L. Bregoli, T. W. Patterson, J. S. Yi, J. D. Yang, M. L. Perry and T. D. Jarvi, Electrochem. Solid-State Lett., 2005, 8, A273–A276 CrossRef CAS.
- J. C. Meier, C. Galeano, I. Katsounaros, A. A. Topalov, A. Kostka, F. Schüth and K. J. J. Mayrhofer, ACS Catal., 2012, 2, 832–843 CrossRef CAS.
- A. A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2012, 51, 12613–12615 CrossRef CAS.
- A. A. Topalov, S. Cherevko, A. Zeradjanin, J. Meier, I. Katsounaros and K. J. J. Mayrhofer, Submitted.
- S. O. Klemm, A. Karschin, A. K. Schuppert, A. A. Topalov, A. M. Mingers, I. Katsounaros and K. J. J. Mayrhofer, J. Electroanal. Chem., 2012, 677–680, 50–55 CrossRef CAS.
- S. Cherevko, A. A. Topalov, I. Katsounaros and K. J. J. Mayrhofer, Electrochem. Commun., 2013, 28, 44–46 CrossRef CAS.
- A. A. Topalov, I. Katsounaros, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2011, 82, 114103 CrossRef.
- K. J. J. Mayrhofer, S. J. Ashton, J. Kreuzer and M. Arenz, Int. J. Electrochem. Sci., 2009, 4, 1–8 CAS.
- B. E. Conway, Prog. Surf. Sci., 1995, 49, 331–452 CrossRef CAS.
- M. Tian, W. G. Pell and B. E. Conway, Electrochim. Acta, 2003, 48, 2675–2689 CrossRef CAS.
- H. Angerstein-Kozlowska, B. E. Conway, A. Hamelin and L. Stoicoviciu, Electrochim. Acta, 1986, 31, 1051–1061 CrossRef CAS.
- H. Angerstein-Kozlowska, B. E. Conway, A. Hamelin and L. Stoicoviciu, J. Electroanal. Chem., 1987, 228, 429–453 CrossRef CAS.
- D. Burke and M. E. G. Lyons, in Modern Aspects of Electrochemistry 28, ed. J. O. M. Bockris, B. E. Conway and R. E. White, Plenum Press, 1996 Search PubMed.
- G. Tremiliosi-Filho, L. H. Dall'Antonia and G. Jerkiewicz, J. Electroanal. Chem., 1997, 422, 149–159 CrossRef CAS.
- G. Tremiliosi-Filho, L. H. Dall'Antonia and G. Jerkiewicz, J. Electroanal. Chem., 2005, 578, 1–8 CrossRef CAS.
- A. A. Michri, A. G. Pshenichnikov and R. K. Burshtein, Elektrokhimiya, 1972, 8, 364–365 CAS.
- R. Woods, in Electroanalytical chemistry, ed. A. J. Bard, Marcel Dekker, INC., New York, 1976, vol. 9 Search PubMed.
- D. M. Kolb, Electrochim. Acta, 2000, 45, 2387–2402 CrossRef CAS.
- Y. Wang, E. Laborda, A. Crossley and R. G. Compton, Phys. Chem. Chem. Phys., 2013, 15, 3133–3136 RSC.
- L. D. Burke and P. F. Nugent, J. Electroanal. Chem., 1998, 444, 19–29 CrossRef CAS.
- K. J. Vetter and F. Gorn, Electrochim. Acta, 1973, 18, 321–326 CrossRef CAS.
- B. E. Conway, B. Barnett, H. Angerstein-Kozlowska and B. V. Tilak, J. Chem. Phys., 1990, 93, 8361–8373 CrossRef CAS.
- M. M. Lohrengel and J. W. Schultze, Electrochim. Acta, 1976, 21, 957–965 CrossRef CAS.
- N. Cabrera and N. F. Mott, Rep. Prog. Phys., 1949, 12, 163 CAS.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, National Association of Corrosion Engineers, 1974 Search PubMed.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2334–2343 RSC.
- M. Wohlfahrt-Mehrens and J. Heitbaum, J. Electroanal. Chem., 1987, 237, 251–260 CrossRef CAS.
- J. Willsau, O. Wolter and J. Heitbaum, J. Electroanal. Chem., 1985, 195, 299–306 CrossRef CAS.
- B. V. Ershler, Dokl. Akad. Nauk SSSR, 1942, 37, 258–261 Search PubMed.
- B. V. Ershler, Dokl. Akad. Nauk SSSR, 1942, 37, 262–264 Search PubMed.
- V. M. Byakov, M. L. Ezerskii, A. M. Skundin and G. V. Steinberg, Russ. J. Electrochem., 2009, 45, 2–10 CrossRef CAS.
- V. S. Bagotsky, Fundamentals of Electrochemistry, Wiley, 2005 Search PubMed.
- A. N. Chemodanov and I. M. July, Zashita metallov (Protection of metals), 1991, 27, 658–666 CAS.
- A. M. Nowicka, U. Hasse, M. Hermes and F. Scholz, Angew. Chem., Int. Ed., 2010, 49, 1061–1063 CrossRef CAS.
- A. M. Nowicka, U. Hasse, G. Sievers, M. Donten, Z. Stojek, S. Fletcher and F. Scholz, Angew. Chem., Int. Ed., 2010, 49, 3006–3009 CrossRef CAS.
- G. Sievers, U. Hasse and F. Scholz, J. Solid State Electrochem., 2011, 16, 1663–1673 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |