 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concurrent formation of furan-2,5- and furan-2,4-dicarboxylic acid: unexpected aspects of the Henkel reaction†
Shanmugam
Thiyagarajan
a,
Aliaksei
Pukin
a,
Jacco
van Haveren
a,
Martin
Lutz
b and
Daan S.
van Es
*a
aFood & Bio-based Research, Wageningen University and Research Centre, P.O. Box 17, 6700 AA Wageningen, The Netherlands. E-mail: daan.vanes@wur.nl; shanmugam.thiyagarajan@wur.nl; Fax: +31 317 483011; Tel: +31 317 481160
bBijvoet Center for Biomolecular Research, Crystal and Structural Chemistry, Utrecht University, Padualaan 8, 3584, Utrecht, The Netherlands
First published on 25th June 2013
Abstract
The concurrent formation of furan-2,5- and furan-2,4-dicarboxylic acid under solvent free conditions via a disproportionation reaction is described. By reacting potassium-2-furoate at 260 °C in the presence of 22 mol% of (Lewis acidic) catalysts like CdI2 or ZnCl2, potassium-2-furoate is disproportionated to furan and furandicarboxylic acids. Besides furan and furan-2,5-dicarboxylic acid (2,5-FDCA) as the main products, furan-2,4-dicarboxylic acid (2,4-FDCA) is also formed as a by-product. Experimental evidence has been obtained that, under the reaction conditions applied, 2,5-FDCA and 2,4-FDCA are formed by separate reaction pathways. Selectivity towards the different FDCA isomers is affected by the type of catalyst used. Single-crystal X-ray analysis shows that 2,4-FDCA has a more ‘linear’ character compared to 2,5-FDCA and hence is structurally more comparable to terephthalic acid (TA), making it an interesting monomer for synthetic polyesters.
Introduction
In recent years, the potential replacement of fossil resources by inexpensive and renewable starting materials such as starch, cellulose, proteins and vegetable oils is increasingly being explored, with the aim to develop a more sustainable bio-based economy.1 The application of renewable feedstocks for the synthesis of a variety of bio-based building blocks for performance polymers is also a growing subject of interest.2 2,5-FDCA is one among the top twelve bio-based building blocks derived from biomass,3 and is widely advocated as a potential substitute for terephthalic acid (TA), the main constituent of synthetic polyesters such as polyethylene terephthalate (PET) and polybutylene terephthalate (PBT). Recent reports shows that, polyethylene furanoate (PEF) and polybutylene furanoate (PBF) derived from 2,5-FDCA have properties comparable to their terephthalate analogues.4 For instance, Avantium reported superior gas barrier properties of PEF towards CO2 and O2 compared to PET.5Currently, the majority of academic and industrial efforts in the development of 2,5-FDCA are concentrated on routes starting from food-grade carbohydrates, in particular fructose, via the 5-(hydroxymethyl)-2-furaldehyde (HMF) route.6 Since competition with food production is undesirable for ethical as well as economical reasons, we are focusing on the use of non-edible feed stocks, such as lignocellulose, agro residues, and aqueous biomass (e.g. algae) for the production of chemicals and building blocks.
The first reported synthesis of 2,5-FDCA goes back almost one and a half centuries to a paper by Fittig and Heinzelman in 1876 who obtained 2,5-FDCA by reacting mucic (galactaric) acid with hydrobromic acid (Scheme 1, route 2).7 Since isolated yields are moderate and galactaric acid is not (yet) readily available, thus far little attention has been paid to this route. This may change if aldaric acids become more widely available, resulting from pectin or alginate streams produced by bio-refineries.8 Currently, the by-far most widely explored synthetic route to 2,5-FDCA is based on the catalytic oxidation of 5-(hydroxymethyl)-2-furaldehyde (HMF),9 which in turn can be obtained from the acid-catalyzed cyclodehydration of C6-sugars like glucose and fructose (Scheme 1, route 1).6,10Fructose is the preferred C6 sugar, because it results in substantially higher yields of HMF than glucose. The only reported process that efficiently converts glucose into HMF requires Cr-based catalysts in combination with ionic liquids, which is undesirable from a sustainability point of view.11Fructose can be economically obtained by acid catalyzed hydrolysis of sucrose or inulin, or by hydrolysis of starch and potentially cellulose followed by a selective partial isomerization of glucose to fructose (Scheme 1, route 1).12 The use of sucrose or starch is however highly undesirable given their primary use as food component.
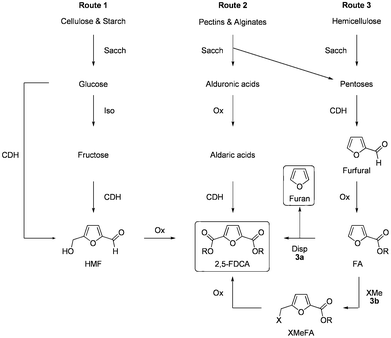 | ||
| Scheme 1 Reported routes to 2,5-FDCA starting from different carbohydrate sources: Sacch = saccharification, CDH = cyclodehydration, Iso = isomerization, Ox = oxidation, XMe = chloromethylation (X = Cl) or hydroxymethylation (X = OH). | ||
Over decades, considerable attention has been paid to develop technology to produce HMF in high yields with efficient and cost-effective methods,6,10c,13 as HMF is not only a potentially low cost precursor to 2,5-FDCA, but can also serve as a renewable platform chemical. Furthermore, HMF is a precursor of next generation biofuels such as 2,5-dimethylfuran (DMF), 5-ethoxymethylfurfural (EMF), ethyl levulinate (EL), and χ-valerolactone (gVL).13
Till now no large scale industrial commercial process for the production of HMF is operational, although various companies have announced plans for pilot-scale facilities. The relatively poor stability of HMF under acidic conditions (required for cyclodehydration reactions) often leads to the formation of undesired by-products like levulinic acid (LA), formic acid (FA), and insolubles such as furan resins and humins. These by-products severely complicate down-stream processing and reduce the yield of purified HMF.6,14 In order to address these issues, companies like Avantium are developing alternative fructose and glucose dehydration technologies in organic solvent like methanol yielding more stable HMF-ethers and HMF-esters as products.15
In addition to routes 1 and 2, a third route based on the use of hemicellulose and its C5 sugar components is also possible (Scheme 1, route 3). Given that hemicellulose is a non-edible (and not easily fermentable) residue from lignocellulosic biorefineries as well as the agro-food industry, it is an attractive feedstock for the production of chemicals. Analogous to the cyclodehydration of C6 sugars to HMF, cyclodehydration of C5 sugars (e.g.xylose and arabinose) yields 2-furaldehyde or furfural. Whereas HMF is still in a developmental stage, furfural is already produced commercially on a 400–500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ton scale, using agro-residues like bagasse as feedstock.16 (Catalytic) Oxidation of furfural gives 2-furoic acid (FA), or the corresponding ester, depending on the reaction conditions in high yields, however, subsequent introduction of a carbon substituent at C5 is non-trivial (Scheme 1, route 3b). Older reports on hydroxymethylation using p-formaldehyde in sulphuric acid or chloromethylation via the Blanc reaction gave only low to moderate yields of 5-hydroxymethyl- or 5-chloromethyl-2-furoic acid respectively.17 (N.B. Another drawback of the Blanc reaction is that the product is an extremely potent allergen.)18 Subsequently, oxidation is required in order to transform this intermediate into product 2,5-FDCA.19
000 ton scale, using agro-residues like bagasse as feedstock.16 (Catalytic) Oxidation of furfural gives 2-furoic acid (FA), or the corresponding ester, depending on the reaction conditions in high yields, however, subsequent introduction of a carbon substituent at C5 is non-trivial (Scheme 1, route 3b). Older reports on hydroxymethylation using p-formaldehyde in sulphuric acid or chloromethylation via the Blanc reaction gave only low to moderate yields of 5-hydroxymethyl- or 5-chloromethyl-2-furoic acid respectively.17 (N.B. Another drawback of the Blanc reaction is that the product is an extremely potent allergen.)18 Subsequently, oxidation is required in order to transform this intermediate into product 2,5-FDCA.19
A more attractive synthetic approach for the conversion of 2-furoic acid into 2,5-FDCA is based on the well-known Henkel reaction (also called Raecke/Henkel process) (Scheme 1, route 3a). This process is based on the thermal disproportionation of alkaline salts of aromatic carboxylates to give the symmetrical aromatic dicarboxylate and the unsubstituted aromatic compound. Raecke and co-workers were the first to examine this type of reaction already in 1952 to Henkel & Co.20 This resulted in a commercial process for the production of TA and benzene as by-product starting from potassium benzoate (Scheme 2).21 In general, Henkel-type reactions are carried out in the presence of cadmium or other metal (e.g. zinc) salts as catalysts, under CO2 or N2 atmosphere at temperatures between 350 °C and 500 °C along with high static pressures.20,22
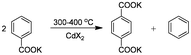 | ||
| Scheme 2 Synthesis of terephthalic acid (TA) via disproportionation of potassium benzoate. | ||
The Henkel-type disproportionation of aromatic carboxylate salts is however not limited to benzene derivatives but was also shown to proceed with heteroaromatic compounds like the carboxylate salts of furan, thiophene, pyrrole, and pyridine (Scheme 3).20
 | ||
| Scheme 3 Synthesis of heteroaromatic dicarboxylic acidvia disproportionation reaction. | ||
Raecke et al. also studied the influence of catalysts. When the reaction was carried out in the absence of catalyst the yield was very low, while in the presence of Lewis acid such as CdF2, CdCl2, CdI2, or ZnCl2, both conversion and selectivity were found to be significantly improved.20 Later, Andrisano et al.22 reported a detailed investigation on the reaction conditions with respect to type of catalyst, catalyst concentration, temperature, time and atmosphere (N2/CO2). These authors reported yields of up to 70–80% of dipotassium 2,5-furandicarboxylate by reacting potassium 2-furoate (K-2F) at 250–300 °C in the presence of CdI2 under a nitrogen atmosphere.22 Industrial production of TA via the Henkel reaction was later on abandoned in favor of the more cost effective catalytic oxidation of xylene (since the commercial value of benzoic acid is higher than that of p-xylene, while the reverse is true for benzenevs. TA).
However, the economics in the case of 2-furoic acid differ from that of benzoic acid. While the price of 2-furoic acid can be comparable with that of HMF, the co-product furan can be the chemical intermediate for high value products like tetrahydrofuran (THF) and 1,4-butanediol, whose prices exceed those of TA.23
Since, the development of economically viable routes to polymer building blocks like 2,5-FDCA from non-edible biomass is one of our main research interests,2a,2b,4d,24 we decided to reinvestigate the Henkel route in order to evaluate its industrial viability. Till recently, the last report on this reaction was by Ratusky et al. in 1971 who investigated the transcarboxylation reactions of heterocyclic compounds like pyridine, thiophene, furan and pyrrole carboxylates.25 Very recently, however, in parallel to our own developments, Pan et al. reported the use of the Henkel reaction as part of the strategy to obtain fully biobased polybutylene 2,5-furanoate (PBF).23 In contrast to all the reports mentioned here, we found that the Henkel reaction of potassium 2-furoate (K-2F) is much less selective than previously assumed. Here, we report the novel co-production of 2,5-FDCA and 2,4-FDCA by thermal disproportionation of potassium 2-furoate(K-2F).
Results and discussion
In their 1963 paper, Andrisano and Angeloni reported the disproportionation of K-2F into 2,5-FDCA using cadmium (mostly CdI2) and zinc (mostly ZnCl2) based catalysts.22 After a reaction time of 4 h at 250 °C typical yields of 20–67% of 2,5-FDCA were reported. The article describes a general procedure for the isolation of the 2,5-FDCA but does not give any detail whether the conversion of K-2F is incomplete or other byproducts besides furan and 2,5-FDCA are being formed. The authors mention one example in which, in the presence of 100 atm of CO2, 80% yield of 2,5-FDCA is obtained. Very recently, Pan et al. described the Henkel disproportionation of K-2F to furan and 2,5-FDCA with a selectivity of 86% at 61% conversion using ZnCl2 as catalyst.23 While these authors reported the conversion and formation of 2,5-FDCA based on HPLC analysis, information on isolated yields and the purification of 2,5-FDCA are lacking. Monomer purity is a crucial parameter in step-growth polymer synthesis, and therefore non-trivial.In order to explore the versatility and limitations of the Henkel reaction for the synthesis of polymer-grade furanic building blocks, our primary aim was to reproduce the results initially reported by Andrisano, and subsequently investigate alternative catalysts systems. The starting material K-2F can be efficiently synthesized by catalytic oxidation of furfural.26 When the oxidation is performed under aqueous conditions using KOH, in the presence of e.g. supported Pt or Au catalysts, K-2F can be obtained directly.23,27 The Henkel-type reactions are usually carried out between 300 to 500 °C. For benzene derived aromatics, the preferred temperature range is between 400 °C to 500 °C. Due to the lower thermal stability heteroaromatic rings, K-2F should be treated at lower temperatures (<400 °C) as previously suggested by Raecke and Andrisano.20,22
In a first set of experiments K-2F was mixed with the catalyst CdI2 and ground well under nitrogen atmosphere. The finely powdered mixture was then transferred into a round-bottom flask and inserted into a Kugelrohr glass oven and was slowly rotated for 5.5 h at elevated temperatures. As the reaction progresses, furan is formed as co-product, which was collected in a cooled (acetone/dry-ice) receiver bulb. After completion of the reaction the crude reaction mixture was dissolved in water, whereupon after acidification with conc. HCl, 2,5-FDCA precipitates out in high purity (>98% from NMR and GC analyses) (Scheme 4). Temperature variation (200–280 °C) (Table 1, entries 1–4) showed that while moderate conversion (32%) was obtained at 235 °C, high conversion (92%) could be obtained by increasing the temperature to 260 °C. Further increasing the temperature is detrimental, since at 280 °C significant decarboxylation of the products occurs, resulting in very poor FDCA yields.
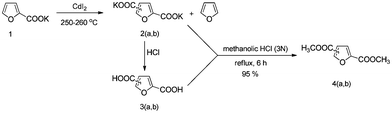 | ||
| Scheme 4 Schematic representation of the synthesis of 2,5-FDCA 3a, 2,4-FDCA 3b and its corresponding dimethyl esters 4a,b. | ||
| Entry | Catalysta | Time (h) | Temp.b (°C) | K-2F conversion (%) | FDCAs formationc (%) | Selectivityd | |
|---|---|---|---|---|---|---|---|
| 2,5-FDCA (%) | 2,4-FDCA (%) | ||||||
| a 22 mol% catalyst (10 mol% of catalyst gave poor yields, increasing the catalyst loading to two-fold, 22 mol% gave sufficient yields). b All reactions were performed in a Kugelrohr glass oven. c Total percentage of furandicarboxylic acids formed, calculated from NMR. d Individual selectivity of furandicarboxylic acids. | |||||||
| 1 | CdI2 | 5.5 | 200 | 0 | 0 | 0 | 0 |
| 2 | CdI2 | 5.5 | 235 | 32 | 30 | 80 | 20 |
| 3 | CdI2 | 5.5 | 260 | 92 | 91 | 70 | 30 |
| 4 | CdI2 | 5.5 | 280 | 88 | 60 | 71 | 29 |
| 5 | ZnI2 | 5.5 | 260 | 48 | 46 | 86 | 14 |
| 6 | ZnCl2 | 5.5 | 260 | 39 | 37 | 84 | 16 |
Since the use of Cd salts is undesirable from an ecological and toxicological point of view, alternative catalysts for the furan–Henkel reaction are required for this process to become industrially feasible. Pan et al. showed that ZnCl2 was the only effective dichloride metal salt of the extensive range of metal chloride salts they tested. Exchanging CdI2 for ZnI2 (on a 1:1 molar basis) at 260 °C results in a significant drop in K-2F conversion. Subsequent exchange of iodide for chlorine results in an apparent further drop (Table 1, entries 5–6). Nevertheless, in all the cases 2,5-FDCA can be effectively isolated from the reaction mixture with high purity (98% from GC and NMR analyses). Moreover, we also screened a wide range of catalysts like FeCl2, FeCl3, FeI2, MgI2, CoCl2, MnCl2 and ZrCl4, which were not reported by Pan et al. However, these catalysts were not effective for the Henkel reaction under the typical conditions.
NMR analysis of the crude product from the CdI2 catalyzed reactions showed, besides the expected product (the di-potassium salt of 2,5-FDCA 2a) and residual starting material K-2F, an additional unknown aromatic compound. The 1H NMR spectrum shows one proton signal corresponding to 2,5-FDCA 2a and 2 proton signals corresponding to the unknown compound (Fig. 1). GC-MS and HRMS analysis indicated comparable molecular ion mass values and fragmentation patterns for both compounds. Based on these observations we concluded that the unknown compound should be an asymmetrical isomer of 2,5-FDCA 2a. The only possibilities are either 2,4-FDCA or 2,3-FDCA. However, the lack of coupling between both proton signals (singlets) is a strong indication for the 2,4-isomer. Furthermore, 2,4-FDCA synthesized according to a known literature procedure28 gave an identical NMR spectra, confirming our assumptions.
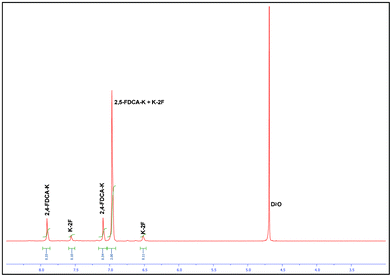 | ||
| Fig. 1 The 1H NMR spectrum of disproportionated crude reaction mixture. | ||
Since 2,4-FDCA has never been reported (and identified) as a by-product in the Henkel reaction of K-2F, including the recent paper by Pan et al., this came as a surprise. Although Ratusky reported the formation of furan dicarboxylic acids at 380 °C starting from K-2F and CdI2 as catalyst, no evidence is given on the isomeric composition.25 Furthermore, the reported reaction temperatures are over 100 °C higher compared to our case. Ratusky also observed partial isomerization of 2,5-FDCA to other isomers at the same high temperatures, comparable to benzenedicarboxylic acid salts.20,29
The presence of significant amounts of 2,4-FDCA (up to 30% of total FDCA yield) is intriguing, and raises the question why this has not been observed by others before. The answer could lie in the work-up procedure, which is based on dissolution of the crude reaction mixture in water, followed by (selective) precipitation of 2,5-FDCA by acidification. NMR analysis of the acidic water layer indicated the presence of 2,4-FDCA 3b and trace amounts of 2,5-FDCA 3a. The aqueous solubility of 3b is apparently significantly higher than that of the scarcely soluble 2,5-isomer 3a, which could prevented previous authors from observing the 2,4-isomer 3b.20,22,23
Evaporation of the aqueous layer afforded the majority of 2,4-FDCA 3b and 2,5-FDCA 3a. Residual 3a was removed from this mixture by soxhlet extraction using acetone followed by chloroform affording 3b in 85% purity. In order to improve the separation/purification procedure, a crude reaction mixture was refluxed in methanolic HCl, giving the corresponding dimethyl esters. GC-MS and NMR analysis showed beside the presence of dimethyl 2,5- and 2,4-FDCA also small amounts of dimethyl 3,4-FDCA and 2,3-FDCA (<5% based on 1H NMR and GC-MS analyses). This mixture was subsequently separated by column chromatography (silicagel, ethylacetate/petroleum ether, 6/94), giving dimethyl 2,5-FDCA 4a and dimethyl 2,4-FDCA 4b in high purity (≥ 99 + % from GC and NMR analyses) (Scheme 4, Fig. 2).
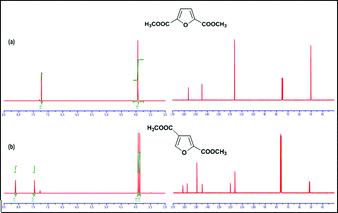 | ||
| Fig. 2 1H and 13C NMR spectra of isolated (a) dimethyl 2,5-furandicarboxylate4a and (b) dimethyl 2,4-furandicarboxylate4b in CDCl3. | ||
In order to gain insight into the reaction mechanism and the factors governing selectivity, a number of reference reactions as well as reactions with alternatively substituted furoate salts were performed under typical reaction conditions, using CdI2 as catalyst. Surprisingly, changing the position of the carboxylic acid group from the 2- to the 3-position resulted in a complete lack of reactivity (Table 2, entry 1). Introduction of a methyl substituent on either the 3- or the 5 position of K-2F also resulted in a complete lack of reactivity (Table 2, entry 2 & 3). These results indicate a strong influence of electronic factors in the Henkel reaction of furans. The previously suggested carbanion character of the reactive intermediate will be destabilised by the presence of electron releasing substituents like methyl groups. Steric factors appears less prominent, given the fact that both the 3- and 5-methyl 2-furoate were found to be unreactive. High negative charge density is also more likely to be stabilised at C2 due to the neighbouring oxygen group, than at C3.
| Entrya | Substrateb | Conversionc (%) | Resultd (NMR) | Entrya | Substrateb | Stabilityf | Resultd (NMR) |
|---|---|---|---|---|---|---|---|
| a All reactions were performed in a Kugelrohr glass oven, using CdI2 as catalyst (22 mol %) at 260 °C for 5.5 h. b The potassium salts of the corresponding (di) acid were prepared by drop-wise addition of KOH in ethanol to a solution of the free acid in ethanol at room temperature. The suspension was stirred further for 12 h, filtered and dried in vacuum oven at 50 °C. c The conversion was determined by mass balance after the end of reaction and also followed by 1H NMR analyses. d The crude product was analysed by 1H NMR. e No product was observed in 1H NMR and the starting materials were recovered. f The dipotassium salts of the furandicarboxylic acids were mixed with the catalyst and heated at 260 °C for 5.5 h and subsequently analysed by 1H NMR. g The starting material was recovered. | |||||||
| 1 |

|
— | —e | 4 |
|
stable/no weight loss | no changeg |
| 2 |

|
— | —e | 5 |

|
stable/no weight loss | no changeg |
| 3 |
|
— | —e | 6 |

|
stable/no weight loss | no changeg |
The thermal stabilities of the dipotassium salts of the three FDCA isomers (2,5-, 2,4- and 3,4-FDCA) were also investigated under typical reaction conditions. No significant weight loss was found after reaction, indicating that decarboxylation did not occur to any significant extent. 1H NMR analysis of the crude mixtures after reaction showed that no detectable isomerisation had occurred at 260 °C (Table 2, entry 4–6). Furthermore, the TGA analyses show that these FDCA isomers (2,5-, 2,4- and 3,4-FDCA) were stable under the typical reaction temperatures. In contrast, others have reported dynamic interconversion of (hetero)aromatic diacids under Henkel reaction conditions at much higher temperatures.25 Our results suggest that under more mild conditions the observed FDCA isomers are formed via independent reaction pathways instead of by interconversion.
The selectivity of the reaction towards 2,5-FDCA or 2,4-FDCA formation appears to depend on both the reaction temperature and the type of catalyst (Table 1).
Whereas comparison of selectivity is only valid at comparable levels of conversion, time variation experiments (vide infra) showed that maximum conversion is attained relatively quickly, making it next to impossible to meet this requirement. With the CdI2catalyst selectivity towards 2,4-FDCA 2b is increased by 50% when the reaction temperature is increased from 235 °C to 250 °C (and upward) indicating that formation of the 2,5-isomer has a lower activation energy. The metal component of the catalyst also has a pronounced effect on selectivity. Both zinc salts showed a 50% drop in selectivity towards 2,4-FDCA 2b compared CdI2 at the same temperature, albeit at lower conversion.
The effect of reaction time on conversion and product distribution was investigated by performing multiple reactions with varying time intervals ranging from 10 min. to 12 h. NMR analysis of the crude reaction mixtures indicates that conversion starts after approx. 20 min. at 260 °C, and reaches a maximum within less than 1 h yielding 90% of the theoretical yield of mixtures of potassium salts of 2,5-FDCA 2a and 2,4-FDCA 2b (see graph in supporting information).
This is in contrast to the results obtained by Andrisano,22 and Pan et al.23 where the reaction apparently took 3–4 h to reach completion, yielding only 2,5-FDCA. NMR spectra of the reactions performed from 2 h till 12 h showed no characteristic changes in the spectrum or the 2,4-FDCA/2,5-FDCA ratio (Fig. 3), indicating that the disproportionation is a very rapid reaction.
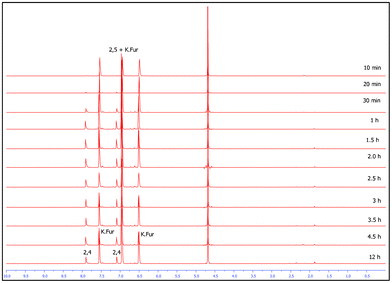 | ||
| Fig. 3 1H NMR spectra (in D2O) of crude products recorded at different time intervals. | ||
Recrystallization of purified dimethyl 2,4-FDCA 4b yielded crystals that allowed for structural characterisation by single crystal X-ray diffraction (Fig. 4).
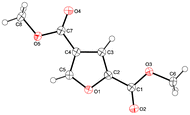 | ||
| Fig. 4 Molecular structure of 4b in the crystal. Displacement ellipsoids are drawn at the 50% probability level. | ||
When comparing the structural parameters of 2,4-FDCA with those of 2,5-FDCA and TA several critical aspects with regard to their (potential) application as polymer building block come to mind.27 Although 2,5-FDCA is widely advocated as potential alternative for TA in e.g. polyesters, their structural differences can have a profound influence on polymer properties. The higher order of symmetry of TA, together with the linearity, generally benefit chain stiffness and rate of crystallisation. At first glance 2,5-FDCA (γ 129°) is more comparable to isophthalic acid (IPA, γ 120°) than to TA (γ 180°) (Table 3). Yet on average 2,5-FDCA polyesters are more comparable to the TA analogues.4e,30 The distance between the carboxylic acid groups in 2,4-FDCA is close to that in the 2,5-isomer, yet this parameter is more crucial for application in polyamides (for lining up hydrogen bonding) than in polyesters. The angle γ (projected angle between carboxylic acid groups) is however closer to that of TA, making the molecule more linear (Table 3, Fig. 5). The lack of symmetry on the other hand can also have a profound effect on crystal packing and rate of crystallisation. Investigations on the effect of incorporating 2,4-FDCA in step-growth polymers are currently in progress.
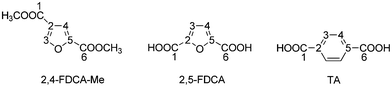 | ||
| Fig. 5 Atom labelling scheme for 2,4-FDCA-Me, 2,5-FDCA and TA. | ||
Overall, reports on the synthesis of 2,4-FDCA 3b are very scarce in the literature. Several authors,17c,31 described the synthesis of 2,4-FDCA from methyl coumalate. Although in principle also bio-based, this compound is more expensive when compared to the present precursor 2-furoic acid. Pearl et al.32 described the use of vanillin to synthesize 2,4-FDCA 3b, however in very low isolated yield (<3%). During the preparation of this manuscript, we came across the recent report by Citron et al.33 describing the synthesis of dimethyl 2,4-furandicarboxylate in 3-steps starting from furan-3-carbaldehyde. The strategy involves low temperature lithiation followed by carboxymethylation using methyl chloroformate, and subsequent oxidation using sodium chlorite giving the target compound in an overall yield of 16%. Hence, we can conclude that our current methodology for obtaining high purity 2,4-FDCA by disproportionation of K-2F is one of the most straightforward and effective routes to date.
Conclusions
The Henkel reaction of potassium-2-furoate (based on agricultural residues) is an efficient alternative method for obtaining furan dicarboxylic acids (FDCA), in total (unoptimised) isolated yields of up to 90%. In contrast to the scarce literature reports, besides the 2,5-isomer 3a the 2,4-isomer 3b is also formed. Selectivity towards either of the major FDCA isomers can be tuned by reaction temperature and type of catalyst used. Both isomers can be efficiently separated and purified after esterification of the crude product. The structure of 2,4-FDCA dimethyl ester 4b was confirmed by single crystal X-ray diffraction which showed that 2,4-FDCA has unique structural properties. The effect of incorporation of the 2,4-FDCA isomer in step-growth polymers is currently under investigation.Experimental section
Materials
2-furoic acid (98% Sigma-Aldrich), 3-furoic acid (98% Sigma-Aldrich), 5-methyl 2-furoic acid (97% Sigma-Aldrich), 3-methyl 2-furoic aicd (97% Sigma-Aldrich), dimethyl 3,4-furandicarboxylate (98% Sigma-Aldrich), cadmium iodide (99% Sigma-Aldrich), zinc chloride (99.99% Sigma-Aldrich), zinc iodide (99.99% Sigma-Aldrich), hydrochloric acid (reagent grade, 37%, Sigma-Aldrich), potassium hydroxide (reagent grade, 90%, flakes, Sigma-Aldrich), acetone (Merck, p.a.), methanolic HCl (3N, Sigma-Aldrich), chloroform (Merck, p.a.), methanol (Merck, p.a.), petroleum ether (Acros Organics, 40–60 °C), ethyl acetate (Acros Organics, 99 + %), silicagel (Alfa Aesar, 230–400 mesh), chloroform-D (99.8 atom % D Aldrich), deuterium oxide (99.9 atom % D Aldrich), magnesium sulfate (Acros Organics, 99% extra pure, dried, contains 3 to 4 moles of water were used as received, unless stated otherwise.Methods
Fourier transform infrared (FT-IR) spectra were obtained on a Varian Scimitar 1000 FT-IR spectrometer equipped with a Pike MIRacle ATR Diamond/ZnSe single reflection plate and a DTSG-detector. The measurement resolution was set at 4 cm−1, and the spectra were collected in the range 4000–650 cm−1 with 32 co-added scans. NMR spectra were recorded on a Bruker Avance III spectrometer operating at 400.17 MHz (1H) and 100.62 MHz (13C). Gas chromatography was performed on an Interscience Focus GC equipped with an AS 3000 series auto sampler. Injection volume 1 μL. Injector temperature 275 °C. Split ratio 1:33. Column flow (at 275 °C) 50 mL min−1 helium. GC column: Restek Rxi-5 ms, 30 m × 0.25 mm × 0.25 μm. GC program (2,5-FDA.mth); hold 2 min at 70 °C, ramp 10 °C min−1, final temperature 300 °C, hold 2 min. Total run time 27 min. Detector; FID at 300 °C. GC-MS analysis was performed on an Interscience TraceGC Ultra GC with AS3000 II autosampler (He carrier gas, flow 1 ml min−1, split flow 20 ml min−1; Restek GC column Rxi-5 ms 30 m × 0.25 mm × 0.25 μm; GC program hold 3 min at 50 °C, ramp 7.5 °C min−1, final temperature 330 °C) connected to a Interscience TraceDSQ II XL quadrupole mass selective detector (EI, mass range 35–500 Dalton, 150 ms sample speed). Electrospray Ionisation (ESI) mass spectrometry was carried out using a Bruker microTOF-Q instrument in positive ion mode (capillary potential of 4500 V). Melting points were measured on a Thermal Fisher Scientific IA 9000 Series digital melting point apparatus. Differential Scanning Calorimetry (DSC) measurements were conducted on a Perkin Elmer Diamond series DSC. The temperature range used was 20 °C up to 250 °C at a heating rate of 10 °C min−1. Short path distillations and disproportionation reactions were carried out using a Buchi Glass Oven B-585 (Kugelrohr). The thermal stability of dimethyl furan-2,5- and 2,4-dicarboxylate was determined by thermogravimetric analysis (TGA) with a STA 6000 (Simultaneous Thermal Analyser) from PerkinElmer Instrument. The samples were heated from 30 to 600 °C at a heating rate of 10 °C min−1 under a nitrogen flow of 40 mL min−1.
X-ray crystal structure determination of dimethyl furan 2,4-dicarboxylate 4b
C8H8O5, Fw = 184.14, colourless needle, 0.75 × 0.26 × 0.14 mm3, monoclinic, C2/c (no. 15), a = 15.2706(19), b = 11.0750(13), c = 11.6658(14) Å, β = 122.294(3)°, V = 1667.8(3) Å3, Z = 8, Dx = 1.467 g cm−3, μ = 0.12 mm-1. 14085 Reflections were measured on a Bruker Kappa ApexII diffractometer with sealed tube and Triumph monochromator (λ = 0.71073 Å) at a temperature of 150(2) K up to a resolution of (sin θ/λ)max = 0.61 Å−1. Intensity data were integrated with the SAINT software.34 Absorption correction and scaling was performed based on multiple measured reflections with SADABS (correction range 0.58–0.75).35 1563 Reflections were unique (Rint = 0.044), of which 1315 were observed [I > 2σ(I)]. The structure was solved by Direct Methods using the program SIR-97.36 Least-squares refinement was performed with SHELXL-97 against F2 of all reflections.37 Non-hydrogen atoms were refined freely with anisotropic displacement parameters. Hydrogen atoms were located in difference Fourier maps and refined with a riding model. 120 Parameters were refined with no restraints. R1/wR2 [I > 2σ(I)]: 0.0411/0.1096. R1/wR2 [all refl.]: 0.0516/0.1200. S = 1.048. Residual electron density between 0.24 and 0.22 e Å−3. Geometry calculations and checking for higher symmetry was performed with the PLATON program.38Synthesis of furan-2,5-dicarboxylic acid 3a and furan-2,4-dicarboxylic acid 3b
Potassium 2-furoate 1 (10.0 g, 0.066 mol) and CdI2 (5.3 g, 0.014 mol) were manually ground well in a mortar and pestle under nitrogen atmosphere and charged into a round-bottom flask and inserted into the Kugelrohr glass oven. The setup was slowly rotated at 260 °C under a continuous (low) flow of nitrogen. During the course of the reaction, the furan formed was collected in the cooling flask which was pre-cooled at −78 °C using a dry-ice/acetone bath, yielding furan quantitatively (95% of the theoretical amount). After 5.5 h, the mixture had turned into black colored solid lumps. The reaction was stopped and allowed to cool down to room temperature over 1 h. The black mass was subsequently dissolved in demineralized water (100 mL). Residual insoluble black material was filtered off and the pale yellow filtrate was acidified using 12 N HCl until the pH attains 1. Precipitated 2,5-FDCA was filtered off using a G-3 glass filter. The product was subsequently dried in vacuum oven at 40 °C over Sicapent for 12 h affording an off-white powdered material.Yield 3a: 6.47 g, 62.2%; purity: >95% (from GC and NMR analyses); m.p. > 250 °C (lit.39 m.p. 342 °C); 1H NMR (400 MHz, DMSO-d6, δ (ppm)): 7.27 (s, 2H); 13C NMR (100 MHz, DMSO-d6, δ (ppm)): 158.94, 147.07, 118.43; IR (neat, υ cm−1): 3147 (C–H), 3125 (C–H), 2878 (br, OH), 1667 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1571, 1523, 1417, 1268, 1224, 1187, 1163 and 1041; GCMS (EI): found 156.0513. C6H4O5 requires 156.0059.
O), 1571, 1523, 1417, 1268, 1224, 1187, 1163 and 1041; GCMS (EI): found 156.0513. C6H4O5 requires 156.0059.
The pale yellow aqueous filtrate was evaporated to dryness under reduced pressure using a rotary film evaporator. The crude product of 2.9 g, was subjected to soxhlet extraction using acetone for 6 h at reflux temperature. The residue was evaporated to dryness under reduced pressure affording a mixture of predominantly 2,4-FDCA 3b and trace amounts of 2,5-FDCA 3a. The crude mixtures of diacids were subjected again to soxhlet extraction using chloroform for 3 h at reflux temperature. The residue was evaporated to dryness under reduced pressure using a rotatory film evaporator affording 2,4-FDCA 3b with 85% purity.
Yield 3b: 2.8 g, 27%; m.p. 266 °C (lit.32 m.p. 264 °C); 1H NMR (400 MHz, DMSO-d6, δ (ppm)): 8.43 (s, 1H), 7.26 (s, 1H); 13C NMR (100 MHz, DMSO-d6, δ (ppm)): 170.91, 166.11, 149.77, 147.58, 125.67, 114.32; IR (neat, υ cm−1): 3150 (C–H), 3120 (C–H), 2880 (br, OH), 1683 (CO), 1580, 1520, 1418, 1262, 1191, 1165, 1036; GCMS (EI): found 156.0674. C6H4O5 requires 156.0059.
Synthesis of dimethyl furan-2,5-dicarboxylate 4a and dimethyl furan-2,4-dicarboxylate 4b
5.0 g of a crude reaction mixture containing potassium salts of 2-furoic acid, 2,5-FDCA, 2,4-FDCA and CdI2, were refluxed in methanolic HCl (1.2 M) (50 mL) at 75 °C for 6 h. After completion, the reaction mixture was cooled down to room temperature and the insoluble metal salts were filtered off. The deep yellow colored residue was evaporated under reduced pressure using a rotatory film evaporator. The resulting yellow viscous oily product was dissolved in chloroform (100 mL) and washed with water (3 × 50 mL), dried over magnesium sulfate, filtered and evaporated to dryness affording the dimethyl esters of 2,5-FDCA and 2,4-FDCA and 2-methyl furoate. The crude products were separated by column chromatography over silica gel using a mixture of ethyl acetate and petroleum ether (6:94). The dimethyl 2,5-furandicarboxylate 4a and dimethyl 2,4-furandicarboxlyate 4b were obtained in high purity (>95%, from NMR and GC analyses). The diesters were further purified by recrystallization from methanol.
Dimethyl furan-2,5-dicarboxylate 4a
Yield, 3.7 g, 67%; mp 110 °C (lit.25 mp 107–111 °C); 1H NMR (400 MHz, CDCl3, δ (ppm)): 7.21 (s, 2H), 3.93 (s, 6H); 13C NMR (100 MHz, CDCl3, δ (ppm)): 158.34 (CO), 146.61, 118.38, 52.29; IR (neat, υ cm−1): 3118 (C–H), 3021 (C–H), 2963 (C–H), 1721 (CO), 1583, 1516, 1432, 1379, 1272, 1237, 1191, 1161, 1133, 1030; GCMS (EI): found 184.0488. C8H8O5 requires 184.0372.
Dimethyl furan-2,4-dicarboxylate 4b
Yield, 1.6 g, 28%; mp 109 °C (lit.31 mp 109–110 °C); 1H NMR (400 MHz, CDCl3, δ (ppm)): 8.10 (s, 1H), 7.46 (s, 1H), 3.91 (s, 3H), 3.86 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3, δ (ppm)): 162.28 (CO), 158.47 (CO), 150.08, 145.48, 120.97, 117.02, 52.23, 51.92 ppm; IR (neat, υ cm−1): 3138 (C–H), 3015 (C–H), 2962 (C–H), 1719 (CO), 1585, 1519, 1436, 1394, 1270, 1195, 1138, 1081; HRMS (ESI): MH+, found 185.0442. C8H9O5 requires 185.0450.
Acknowledgements
Dr G. Frissen, W. Teunissen and Elwin Janssen are gratefully acknowledged for NMR, GC-MS and HR-MS measurements respectively. This work was (partly) financed by Braskem S/A. Dr A.T. Morita, Dr R. Coimbra and Dr P.A. Coutinho are thanked for fruitful discussions.References
- (a) P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; (b) H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC; (c) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC; (d) A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- (a) S. Thiyagarajan, L. Gootjes, W. Vogelzang, J. Van Haveren, M. Lutz and D. S. Van Es, ChemSusChem, 2011, 4, 1823–1829 CrossRef CAS; (b) J. Wu, P. Eduard, S. Thiyagarajan, J. Van Haveren, D. S. Van Es, C. E. Koning, M. Lutz and C. Fonseca Guerra, ChemSusChem, 2011, 4, 599–603 CrossRef CAS; (c) S. Thiyagarajan, L. Gootjes, W. Vogelzang, J. Wu, J. Van Haveren and D. S. Van Es, Tetrahedron, 2011, 67, 383–389 CrossRef CAS; (d) A. Gandini and M. N. Belgacem, Monomers, Polymers and Composites from Renewable Resources, Ed., Elsevier, Amsterdam, 2008 Search PubMed; (e) F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- (a) M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS; (b) A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 295–298 CrossRef CAS; (c) A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407–6422 RSC; (d) D. S. van Es, J. Renew. Mater., 2013, 1, 61–72 CAS; (e) J. Zhu, J. Cai, W. Xie, P.-H. Chen, M. Gazzano, M. Scandola and R. A. Gross, Macromolecules, 2013, 46, 796–804 CrossRef CAS; (f) R. J. I. Knoop, W. Vogelzang, J. van Haveren and D. S. van Es, J. Polym. Sci. Part A: Polym. Chem., 2013 DOI:10.1002/pola.26833.
- E. d. Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, vol. 1105, pp. 1–13 Search PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- R. Fittig and H. Heinzelman, Ber. Dtsch. Chem. Ges., 1876, 9, 1198 Search PubMed.
- (a) D. S. van Es, F. van der Klis, J. van Haveren, A. E. Frissen, H. W. C. Raaijmakers and G. P. F. M. van Engelen, Europ. Pat., EP1263081, 2012 Search PubMed; (b) F. van der Klis, A. E. Frissen, J. van Haveren and D. S. van Es, ChemSusChem, 2013 DOI:10.1002/cssc.201300367.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- (a) A. P. G. K. a. H. V. H. E. Vandam, Starch/Staerke, 1986, 38, 95 CrossRef; (b) M. J. Antal Jr, T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71–85 CrossRef; (c) B. F. M. Kuster, Starch/Staerke, 1990, 42, 314–321 CrossRef CAS.
- (a) V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef CAS; (b) A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS; (c) Y. Zhang, E. A. Pidko and E. J. M. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS; (d) E. A. Pidko, V. Degirmenci, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2010, 49, 2530–2534 CrossRef CAS.
- (a) A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC; (b) M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2010, 111, 397–417 CrossRef; (c) X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 385, 1–13 CrossRef CAS.
- S. Dutta, S. De and B. Saha, ChemPlusChem, 77, 259–272 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 13, 340–349 RSC.
- (a) Gruter, Gerardus Johannes Maria and F. Dautzenberg, Europ. Pat., EP 1834950A1, 2006 Search PubMed; (b) Gruter, Gerardus Johannes Maria and F. Dautzenberg, Europ. Pat., EP 1834951A1, 2006 Search PubMed; (c) Alexandra J. Sanborn and Stephen J. Howard, US Patent, US2009/0156841A1, 2008 Search PubMed.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its Many by-products, Elsevier, The Netherlands, 2000 Search PubMed.
- (a) Germany Pat., 1955 Search PubMed; (b) R. D. Sanderson, D. F. Schneider and I. Schreuder, J. Appl. Polym. Sci., 1994, 53, 1785–1793 CrossRef CAS; (c) Y. Hachihama, T. Shono and K. Hyono, Technol. Rep. Osaka Univ., 1958, 8, 475–480 CAS.
- V. O. Moldenhauer, W. Irion, R. Pfluger, H. Doser, D. Mastaglio and H. Marwitz, Liebigs. Ann. Chem., 1953, 580 Search PubMed . Band.
- O. Moldenhauer, G. Trautmann, W. Irion, R. Pfluger, H. Döser, D. Mastaglio and H. Marwitz, Justus Liebigs Ann. Chem., 1953, 580, 169–190 CrossRef CAS.
- (a) B. Raecke, Angew. Chem., 1958, 70, 1–5 CrossRef CAS; (b) B. Raecke, 1952, DE 958920.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010 Search PubMed.
- R. Andrisano and A. S. Angeloni, Ann. Chim., 1963, 53, 1658–1664 CAS.
- T. Pan, J. Deng, Q. Xu, Y. Zuo, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 47–50 CrossRef CAS.
- J. Le Nôtre, J. van Haveren and D. S. van Es, ChemSusChem, 2013, 6, 693–700 CrossRef.
- J. Ratusky, Collect. Czech. Chem. Commun., 1971, 36, 2831–2845 CrossRef CAS.
- (a) Y. Yang, C.-W. Hu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 405–410 CrossRef CAS; (b) E. S. Kim, S. Liu, M. M. Abu-Omar and N. S. Mosier, Energy Fuels, 2012, 26, 1298–1304 CrossRef CAS.
- R. J. Harrisson and M. Moyle, Org. Synth., 1963, Coll. Vol. 4, 493 Search PubMed.
- I. W. Ashworth, M. C. Bowden, B. Dembofsky, D. Levin, W. Moss, E. Robinson, N. Szczur and J. Virica, Org. Process Res. Dev., 2002, 7, 74–81 CrossRef.
- V. V. S. Revankar and L. K. Doraiswamy, Ind. Eng. Chem. Res., 1992, 31, 781–786 CrossRef CAS.
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- F. Feist, Ber. Dtsch. Chem. Ges., 1901, 34, 1992–1996 CrossRef CAS.
- I. A. Pearl and J. S. Barton, J. Am. Chem. Soc., 1952, 74, 1357–1357 CrossRef CAS.
- C. A. Citron, P. Rabe and J. S. Dickschat, J. Nat. Prod., 2012, 75, 1765–1776 CrossRef CAS.
- Bruker, SAINT-Plus., Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, SADABS: Area-Detector Absorption Correction, Universität Göttingen, Germany, 1999 Search PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148–155 CrossRef.
- S. M. Payne and F. M. Kerton, Green Chem., 2010, 12, 1648–1653 RSC.
- E. Martuscelli and C. Pedone, Acta Crystallogr. Sect. B: Struct. Sci., 1968, 24, 175–179 CrossRef CAS.
- M. Bailey and C. J. Brown, Acta Crystallogr., 1967, 22, 387–391 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMRspectra of potassium 2-furoate, 2,5-furandicarboxylic acid and 2,4-furandicarboxylic acid, Relative ratio of 2,5-FDCA and 2,4-FDCA formation over time according to 1H-NMR. CCDC 907968. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra42457j |
| This journal is © The Royal Society of Chemistry 2013 |