 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triborate and pentaborate salts of non-metal cations derived from N-substituted piperazines: synthesis, structural (XRD) and thermal properties†
Michael A. Beckett*a, Peter N. Hortonb, Michael B. Hursthouseb and James L. Timmisa
aSchool of Chemistry, Bangor University, Bangor, Gwynedd, LL57 2UW, UK. E-mail: m.a.beckett@bangor.ac.uk; Fax: +44(0)1248 382734; Tel: +44(0) 1248 37
bSchool of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK
First published on 17th June 2013
Abstract
The synthesis and characterization of a triborate salt, [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1), and four pentaborate salts, [H2N(CH2CH2)2NH][B5O6(OH)4] (2a), [MeHN(CH2CH2)2NH][B5O6(OH)4] (2b), [MeHN(CH2CH2)2NMe][B5O6(OH)4] (2c) and [Me2N(CH2CH2)2NMe2][B5O6(OH)4]2 (2d) are described. TGA and DSC analysis (in air, 25–1000 °C) indicate that triborate 1 decomposes to B2O3via a multistage process, with the first stage (<250 °C) being dehydration to condensed polymeric hexaborate of approximate composition: [H2N(CH2CH2)2NH2][B6O10]. The pentaborates (2a–2d) are thermally decomposed to B2O3via a 2 stage process involving polymeric [NMC][B5O8]. The anhydrous polyborates were amorphous. BET analysis of materials derived from the thermolysis of 1 at 250, 400, 600, and 1000 °C, were all non-porous (surface area <1.8 m2 g−1). A single-crystal X-ray diffraction study of 1 showed that it contains isolated triborate(1−) anions in a structure comprised of alternating cationic and anionic layers held together via extensive H-bonds. Single-crystal XRD structural studies on pentaborate salts 2c and 2d are also reported.
1. Introduction
There has been increased interest in recent years in non-metal cation (NMC) polyborate, notably pentaborate, salts as a result of their potential applications as porous1–5 or NLO6 piezoelectric7 or fluorescent8 materials. The chemistry of boric acid in aqueous solution is complicated with monomeric and various polymeric borate ions existing in equilibrium concentrations9 which are dependent upon B concentration, B/[OH−] ratio, and temperature. Despite this, pentaborate salts of NMCs are generally readily synthesized as crystalline solids from aqueous solution by the interaction of a free base, or its hydroxide salt, with B(OH)3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio.2,4,10 Structurally, NMC pentaborates are best described as supramolecular networks of H-bonded isolated [B5O6(OH)4]− anions, and the formation of this extended lattice is clearly a driving force in product formation. The NMCs occupy ‘cavities’ within the network, and usually (if possible) add further stabilizing H-bond cation-anion interactions.1,2,4 Occasionally, NMC polyborate salts other than pentaborates have been obtained from aqueous solution e.g. [B3O3(OH)4]−,11 [B4O5(OH)4]2−,12 [B7O9(OH)5]2− (2 isomers),13,14 [B8O10(OH)6]2−,2 [B9O12(OH)6]2−,15,16 [B14O20(OH)6]4−.17 More forcing solvothermal methods have led to NMC salts of isolated or condensed polyborate sheets, which are formally derived from isolated anions.18 We are currently undergoing a research programme investigating the structure directing effects of NMCs in borate chemistry4,14,19 and have recently isolated, by this method from aqueous solution,20 an unprecedented double salt of stoichiometry [H2en]2[B11O18(OH)]·7H2O which contains two isolated polyborate anions: [B4O5(OH)4]2− and [B7O9(OH)5]2−. As a continuation of this study we have investigated substituted piperazine based NMCs and now report the synthesis of an unusual triborate salt, as well as further examples of pentaborate salts. The structures of the anions present are illustrated in Fig. 1. The thermal properties of the triborate salt (TGA/DSC) has been investigated in detail and results compared with those of the pentaborate derivatives. Porosity (BET surface area measurements) of materials derived from the thermal decomposition of the triborate salt are also described. These results are compared with those from other known NMC polyborate salts.
:5 ratio.2,4,10 Structurally, NMC pentaborates are best described as supramolecular networks of H-bonded isolated [B5O6(OH)4]− anions, and the formation of this extended lattice is clearly a driving force in product formation. The NMCs occupy ‘cavities’ within the network, and usually (if possible) add further stabilizing H-bond cation-anion interactions.1,2,4 Occasionally, NMC polyborate salts other than pentaborates have been obtained from aqueous solution e.g. [B3O3(OH)4]−,11 [B4O5(OH)4]2−,12 [B7O9(OH)5]2− (2 isomers),13,14 [B8O10(OH)6]2−,2 [B9O12(OH)6]2−,15,16 [B14O20(OH)6]4−.17 More forcing solvothermal methods have led to NMC salts of isolated or condensed polyborate sheets, which are formally derived from isolated anions.18 We are currently undergoing a research programme investigating the structure directing effects of NMCs in borate chemistry4,14,19 and have recently isolated, by this method from aqueous solution,20 an unprecedented double salt of stoichiometry [H2en]2[B11O18(OH)]·7H2O which contains two isolated polyborate anions: [B4O5(OH)4]2− and [B7O9(OH)5]2−. As a continuation of this study we have investigated substituted piperazine based NMCs and now report the synthesis of an unusual triborate salt, as well as further examples of pentaborate salts. The structures of the anions present are illustrated in Fig. 1. The thermal properties of the triborate salt (TGA/DSC) has been investigated in detail and results compared with those of the pentaborate derivatives. Porosity (BET surface area measurements) of materials derived from the thermal decomposition of the triborate salt are also described. These results are compared with those from other known NMC polyborate salts.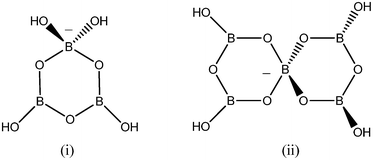 | ||
| Fig. 1 Schematic drawing of (i) triborate(1−) as observed in 1, and (ii) pentaborate(1−) as observed in 2a–2d. | ||
2. Results and discussion
2.1 Synthesis and characterization
The triborate salt [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1), and the pentaborate salts [H2N(CH2CH2)2NH][B5O6(OH)4] (2a), [MeHN(CH2CH2)2NH][B5O6(OH)4] (2b), and [MeHN(CH2CH2)2NMe][B5O6(OH)4] (2c) were conveniently prepared in H2O/MeOH solution, from B(OH)3 and (substituted) piperazines according to eqn (1) and (2).[Me2N(CH2CH2)2NMe2][B5O6(OH)4]2 (2d) was prepared by reacting a solution of [Me2N(CH2CH2)2NMe2][OH]2, which were obtained from ion exchange of the diiodide salt, with B(OH)3 as shown in eqn (3). Eqn (1) and (2) indicate that a piperazine:B(OH)3 ratio of 1:6 would be ideal for the preparation of 1, whereas a similar 1:5 ratio would be ideal for 2a. We obtained 1 in high yield from a 1:3 ratio reaction mixture in which half of the piperazine remained unreacted. We believe the function of the unreacted piperazine is to raise pH of the solution allowing the more basic triborate anion5,15 to crystallize.
| [HN(CH2CH2)2NH] + 6B(OH)3 → [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1) + 4H2O | (1) |
| [RN(CH2CH2)2NR′] + 5B(OH)3 → [RNH(CH2CH2)2NR′][B5O6(OH)4] + 5H2O (R = R′ = H, (2a); R = Me, R′ = H, (2b); R = R′= Me, (2c) | (2) |
| [Me2N(CH2CH2)2NMe2][OH]2 + 10B(OH)3 → [Me2N(CH2CH2)2NMe2][B5O6(OH)4]2 (2d) + 12H2O | (3) |
With the exception of 2d (obtained in 50%) products were readily obtained in high yield (86–97%) and were of good analytical purity by crystallization induced by removal of the solvents. Compounds 1 and 2a–d were characterized spectroscopically (NMR, IR) and X-ray diffraction (powder) techniques. Powder diffraction analysis showed that all samples were prepared crystalline, and crystals from 1, 2c and 2d were selected for a single-crystal structural determination (section 2.4). Unsubstituted piperazine salts usually contain dications, with monocationic salts occurring less frequently.2111B NMR spectra of salts 1 and 2a–d are all very similar, despite the different stoichiometry for 1, and showed peaks at +18 ppm, +13 ppm and +1 ppm. The similarity in these spectra may be explained by the existence of a complex series of equilibria existing in aqueous solution which link monomeric and oligomeric borate species.9 Similar spectra have been obtained from other pentaborate salts.4,14,191H and 13C NMR spectra, obtained in D2O solution, were in full accord with their structures, with signals associated with B–OH and NH protons producing a single broadened single at ∼4.7 ppm due to rapid exchange. The IR spectrum of 1 (Fig. 2) displayed strong B–O peaks at 1414 cm−1, 1305 cm−1, 985 cm−1 and 862 cm−1 which may be assigned by reference to Li et al.22 to νas(B(3)–O), νas(B(3)–O), νs(B(3)–O) and νs(B(4)–O), respectively. The strong peak at 1510 cm−1 may be an NH or OH bending vibration. The diagnostic23νs(B(3)–O)22 band at ∼925 cm−1 is apparent in the pentaborate samples 2a–d, but significantly is absent from 1.
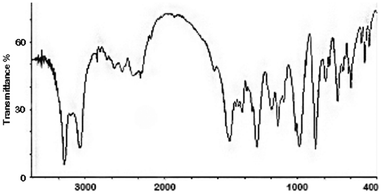 | ||
| Fig. 2 IR spectrum of the triborate salt for 1. | ||
2.2 Thermal properties of 1
The thermal properties of NMC pentaborate salts, which contain intricate 3D H-bonded networks (section 2.3), have been well investigated and the thermal behaviour of 2a–2d are consistent with earlier reports on pentaborate salts.1–5 The layered structure for 1 (see section 2.3) might suggest that its thermal properties would be different and therefore was worthy of further and more detailed investigation.Heating a sample of the triborate salt (1) in air resulted in thermal decomposition and the eventual formation of a glassy black solid. During this process the sample ‘chars’ and intumesces prior to collapsing down to the glass. This behaviour is similar to that observed for non-metal cation pentaborates.1–5 A thermal study of the one previously known NMC triborate, [HOCH2CMe2NH3][B3O3(OH)4],11 suggested dehydration (2 mole equivalent per triborate) by 120 °C to a viscous liquid by either condensation of anions or anion/cation esterification. The process for 1 was followed in more detail by DSC/TGA (Fig. 3). Closer inspection of the TGA curve shows that the decomposition is a multistep process, with a series of endothermic processes occurring below 275 °C, and two exothermic processes occurring at 400 °C and 600 °C. The weight loss below 275 °C is consistent with dehydration as shown in eqn (4). The final residue (55.4%) is in close agreement with that calculated (54.2%) for B2O3 according to eqn (5).
| [H2N(CH2CH2)2NH2][B3O3(OH)4]2 → [H2N(CH2CH2)2NH2][B6O10] + 4H2O | (4) |
| [H2N(CH2CH2)2NH2][B3O3(OH)4]2 → 3B2O3 + oxidation products | (5) |
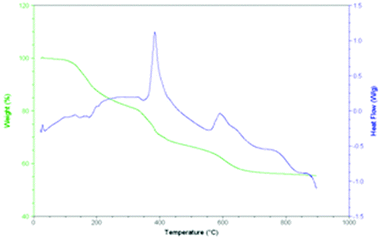 | ||
| Fig. 3 TGA/DSC trace for 1. | ||
It was of interest to examine the physical properties (crystallinity and porosity) of materials derived from the aerobic thermolysis of 1 and a furnace procedure was used to attempt to replicate the above TGA results on a small synthetic scale. Accordingly, samples (∼1.0 g) were heated to 275 °C (1a), 500 °C (1b), 600 °C (1c), or 1000 °C (1d) and weighed and characterized. Elemental analytical data of 1a were consistent with the dehydrated product (eqn (4)) and the residual weight associated with formation of 1d was in good agreement with the residual weight calculated expected for formation of B2O3. Elemental analysis data for 1b and 1c demonstrated residual presence of C, H and N with lower amounts present in 1c indicating that the oxidation process was still incomplete at this temperature/time. All thermally produced samples were amorphous by powder XRD studies. and SEM pictures of ground samples were all remarkably similar (exemplified for 1b in Fig. 4) and were consistent with glassy shards.
 | ||
| Fig. 4 SEM of 1b (sample 1 calcined at 500 °C, 24 h) having been ground in a mortar and pestle. | ||
The porosity of the thermally derived materials were determined by multipoint BET analysis.24 Essentially all samples were ‘non-porous’ with the isolated pentaborates having porosities of 0.12–0.85 m2 g−1, condensed pentaborates 0.04–0.86 m2 g−1, intumesced materials 0.12–1.59 m2 g−1, and the final glassy B2O3, 0.18 m2 g−1. The densities of 1b and 1c, determined as ‘tipped solids’, were extremely low (0.1, 0.2 g cm−3 respectively) and are consistent with the observation that the materials intumesced during oxidation to leave expanded materials. The low porosity of the final glassy black solid (∼B2O3) was expected, but the porosity of the intumesced materials were lower than expected and indicated macroporous aerogel-like structures, in which gaseous decomposition products were trapped within the intumesced materials.25
2.3 XRD structural studies on 1, 2c and 2d
The structure of 1 as a triborate salt of a dication was confirmed by single-crystal XRD studies. A drawing of 1 showing the ions present and the atomic numbering scheme is shown in Fig. 5. The structure contains two identical [B3O3(OH)4]− anions, as illustrated in Fig. 1(i), partnered by one [H2N(CH2CH2)2NH2]2+ cation which adopts the familiar chair conformation.26 The [B3O3(OH)4]− anion appears in significant concentrations in aqueous solution at pH 5–12 but rarely is crystalized from such solutions by non-metal cations which generally favour partnering pentaborate anions. As noted above, the solutions had excess base present, and the pH was higher than normal.5,15 Structurally characterized triborate(1-) salts are uncommon and are limited to two alkali metal salts Na[B3O3(OH)4] (ref. 27) and K[B3O3(OH)4]·H2O,28 and a NMC salt [HOCH2C(Me)2NH3][B3O3(OH)4].11 Interatomic distances and angles for the triborate anion of 1 are given in the caption to Fig. 5. The B3O3 ring in 1 deviates slightly from planarity and adopts an envelope conformation with 4-coordinate B1 0.034 Å from the plane containing B2B3O1O3. O2 is 0.008 Å below the same plane but this distance is not significant. Similar ring conformations are found for Na[B3O3(OH)4] (ref. 27) and K[B3O3(OH)4]·H2O.28 The anion in [HOCH2C(Me)2NH3][B3O3(OH)4] (ref. 11) adopts a half-chair conformation with the 4-coordinate B 0.206 Å out of the plane containing the 3 ring O atoms. The bond lengths and bond angles for 1 are not significantly different from those found for [HOCH2C(Me)2NH3][B3O3(OH)4] (ref. 11) with O–Btetra significantly longer than O–Btrig and vary systematically about the ring, as has been observed or related organotriboroxine adducts29 and organotriboroxinates,30 with B2–O1 and B3–O3 being significantly shorter.![ORTEP drawing of ions present in [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1) showing atomic numbering scheme. Selected bond distances (Å) and angles (°) for [B3O3(OH)4]− anion: B1–O4, 1.453(2); B1–O5 1.456(2); B1–O3 1.486(2); B1–O1 1.497(2); B2–O1 1.351(2); B2–O6 1.359(2); B2–O2 1.400(2); B3–O3 1.337(2); B3–O7 1.378(2); B3–O2 1.396(2). O1–B1–O3 110.16(13); B1–O1–B2 124.38(13); O1–B2–O2 120.59(15); B2–O2–B3 118.72(13); O2–B3–O3 122.39(15); B3–O3–B1 123.69(13).](/image/article/2013/RA/c3ra42387e/c3ra42387e-f5.gif) | ||
| Fig. 5 ORTEP drawing of ions present in [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1) showing atomic numbering scheme. Selected bond distances (Å) and angles (°) for [B3O3(OH)4]− anion: B1–O4, 1.453(2); B1–O5 1.456(2); B1–O3 1.486(2); B1–O1 1.497(2); B2–O1 1.351(2); B2–O6 1.359(2); B2–O2 1.400(2); B3–O3 1.337(2); B3–O7 1.378(2); B3–O2 1.396(2). O1–B1–O3 110.16(13); B1–O1–B2 124.38(13); O1–B2–O2 120.59(15); B2–O2–B3 118.72(13); O2–B3–O3 122.39(15); B3–O3–B1 123.69(13). | ||
Compound 1 has a giant H-bonded network involving both anion-anion and cation-anion interactions and where all four NH and all four OH groups participate as donors. For convenience, the O atom acceptor sites in 1 may be labelled, by analogy with pentaborate chemistry and distance from the 4 co-ordinate B1, α-, β- or γ-sites.2 The OH sites on B1 can be labelled δ. The triborate anions form infinite zig-zag chains through alternating γ and α R22(8) rings (Etter31). The D⋯A distances/angle at H for the α and γ interactions are 2.7453(16) Å/176.4° and 2.7179(16) Å/167.0°, respectively. These chains are then stacked into layers by H-bond interactions from the two OH groups on B1 to β-sites as C(6) interactions with D⋯A distances/angle at H of 2.8312(17) Å/162.8° and 2.8258(16) Å/169.5°. The layers are separated by layers of [H2N(CH2CH2)2NH2]2+ cations which are H-bonded to δ acceptor sites of the triborate anion via unusual R33(10) rings. The D⋯A distances/angle at H for the cation–anion interactions are 2.7182(18) Å/165.0° and 2.6786(18) Å/163.3°.
The structures of 2c and 2d were confirmed by single crystal XRD studies. Drawings of 2c and 2d are shown in Fig. 6 and 7, respectively. Both compounds contain isolated [B5O6(OH)4]− anions, as illustrated in Fig. 1(ii). B–O bond lengths and bond angles at B and O are not significantly different from those observed in other pentaborate structures containing non-metal cations, and related systems.1–4,14,19,32
![ORTEP drawing of ions present in [MeHN(CH2CH2)2NMe][B5O6(OH)4] (2c) showing atomic numbering scheme.](/image/article/2013/RA/c3ra42387e/c3ra42387e-f6.gif) | ||
| Fig. 6 ORTEP drawing of ions present in [MeHN(CH2CH2)2NMe][B5O6(OH)4] (2c) showing atomic numbering scheme. | ||
![ORTEP drawing of ions present in [Me2N(CH2CH2)2NMe2][B5O6(OH)4]2 (2d) showing atomic numbering scheme.](/image/article/2013/RA/c3ra42387e/c3ra42387e-f7.gif) | ||
| Fig. 7 ORTEP drawing of ions present in [Me2N(CH2CH2)2NMe2][B5O6(OH)4]2 (2d) showing atomic numbering scheme. | ||
The anions in both compounds form remarkably similar (despite the stoichiometry) extended H-bonded lattices, and the H-bond interactions for both 2c and 2d can be described as α,α,α,β. The α,α,α,β configuration is commonly found in ‘brickwall’ or ‘herringbone’ structures1,14 but the extended lattice found in 2c and 2d is not one of these types. Each pentaborate anion forms 3 reciprocal-pair R22(8) α-interactions with neighboring pentaborates, and 1 reciprocal-pair R22(12) β-interaction (Etter31). The R22(12) has not been previously observed in pentaborate systems and is responsible for the unusual extended lattices in 2c and 2d. The D⋯A distances/angle at H for these β interactions in 2c and 2d are 2.8508(10) Å/165.0° and 2.8182(19) Å/178.2°, respectively. Compound 2c has an additional H-bond contact between the cation and the α-position of a pentaborate (N12–H12⋯O4) with a N⋯O distance of 2.7548(11) Å and NHO angle of 169.2°.
3. Experimental
3.1 General
Reagents were purchased from Aldrich Chemical company and used without purification. Multi-element NMR spectra were recorded at room temperature (298 K) on a Bruker Avance 500 (11.7 T) spectrometer operating at 500 MHz for 1H, 125 MHz for 13C–{1H}, and 160 MHz for 11B–{1H} and referenced to SiMe4 or BF3·OEt2 using D2O as solvent. Chemical shifts (δ) are reported in ppm. Elemental analyses were performed at OEA Laboratories Ltd (Callington, UK). Powder XRD data were obtained on a Philips 1050/37 X-ray diffractometer, equipped with an iron filter, using Cu-Kα radiation (λ = 0.154056 nm) with a continuous scan between the range 2θ = 5–75°. Philips X'Pert System software was used to collect and manipulate the data. TGA and DSC analyses were obtained using a Netzch TG 209 instrument. BET multipoint analyses were performed on a Micromeritics Gemini III 2375 instrument.3.2 Synthesis of [H2N(CH2CH2)2NH2][B3O3(OH)4]2 (1)
B(OH)3 (4.31 g, 69.67 mmol) was dissolved in 50:50 H2O/MeOH (100 ml) with gentle warming. To this solution was added piperazine (2.00 g, 23.25 mmol) with stirring, and the solution was gently warmed (50 °C) for 1 h. The majority of the solvent (H2O/MeOH) was removed under reduced pressure (rotary evaporator), and the remaining solid, obtained by filtration, was placed in an oven to dry for 4 h at 100 °C. The product 1 was obtained as a white crystalline solid was obtained 4.5 g, (97%). Crystals were analytically pure and were suitable for X-ray diffraction studies. Elem. anal. (C4H20B6N2O14). Calc: C 12.5, H 5.2, N 7.3%; Found: C 12.7, H 5.3, N 7.1%. Mpt > 260 °C (dec). NMR: δH/ppm 2.96 (8H, s), 4.7 (12H). δC/ppm 42.4 (4CH2). δB/ppm 1.1 (5%), 13.2 (20%) 18.8 (75%) νmax/cm−1 3511(s), 3265(s), 1509(s), 1414(m), 1303(s), 1193(m), 1146(m), 985(s), 862(s), 696(m), 596(m). XRD: d-spacing/Å (% rel. int.): 3.49 (100), 5.45 (81), 2.63 (78), 4.22 (71), 5.51 (53), 3.70 (37).3.3 Synthesis of [NMC][B5O6(OH)4)] (2a–2d)
A similar procedure to that detailed in section 3.2 was used for 2a–c but with a B(OH)3 ratio of 5:1.NMR: δH/ppm 3.60 (12H, s), 3.88 (8H, s). 4.70 (4H, s). δC/ppm 30.2 (4CH3), 55.5 (8CH2).δB/ppm 1.2 (15%), 13.5 (25%), 18.9 (60%). νmax/cm−1 3386(s), 1409(s), 1321(s), 1201(m), 1142(m), 1040(s), 930(s), 868(s), 775(m), 475(m). XRD: d-spacing/Å (% rel. int.): 5.07 (100), 5.67 (88), 3.66 (83), 4.08 (23), 3.41 (22), 6.09 (19).
3.4 Thermolysis experiments of 1
Samples of 1 were individually placed within open topped ceramic bowls and positioned within a furnace (air atmosphere). The furnace temperature was set to increase from room temperature to either 250 °C (1a), 500 °C (1b) or 600 °C (1c) at 10 °C per minute. After the samples had reached the required temperature they were allowed to cool to room temperature, isolated and powdered in a mortar and pestle.Sample 1a was pale yellow. Samples 1b and 1c were black and had increased their volume ∼10 fold. 1a: 0.70 g obtained from 1.02 g 74% residue; Elem anal. (C4H12B6N2O10). Calc: C 15.4, H 3.9, N 8.9%; Found: C 15.1, H 4.0, N 8.6%. IR: νmax/cm−1: 3488(s), 3255(s), 1637(m), 1506(m), 1383(s), 1140(m), 1063(m), 864 (s), 809(m), 701(m). Density: 0.690 g cm−3. BET: surface area 0.21 m2 g−1. 1b: (0.70 g from 1.03 g, 68% residue). Elem anal. Found: C 5.3, H 2.6, N 1.9%. IR: νmax/cm−1: 3692(s), 3209(s), 1727(w), 1627(w), 1437(s), 1195(m), 883(w), 721(m), 644 (m), 547(w). Density: 0.096 g cm−3. BET: surface area 1.90 m2 g−1. 1c: (0.61 g from 1.02 g, 59% residue). Elem anal. Found: C 2.4, H 4.7, N 0.8%. IR: νmax/cm−1: 3600(m), 3220(s), 1449(s), 1196(s), 883(w), 795(m), 720(m), 644(m), 547(m). Density: 0.214 g cm−3. BET: surface area 1.85 m2 g−1. In a similar experiment 1 (0.4999 g) was placed within open topped Ni crucible and positioned within the furnace (air atmosphere). The furnace temperature was set to increase from room temperature to 1000 °C at 10 °C per minute, and then held at 1000 °C for 3 h, and then allowed to cool to room temperature, yielding a glassy black solid (1d), which was powdered in a mortar and pestle and weighed (0.2759 g, 44.2% weight loss). 1d: Elem anal. Found: C 0.1, H 0.4, N 0.1%. IR: νmax/cm−1: 3200(s), 1445(s), 1194(s), 795(m), 720(m), 550(m). BET: surface area 0.16 m2 g−1.
3.5 X-ray crystallography
The crystallographic data collection of compounds 1, 2c and 2d were performed using a Nonius Kappa CCD diffractometer with Mo-Kα radiation (λ = 0.71073 Å) controlled by the Collect33 software package and an Oxford Cryosystem N2 open flow cryostat at 120(2) K. The data were processed using Denzo34 and absorption corrections were applied using SORTAV.35 Crystallographic data are in Table 1. The structures were solved by direct methods and refined by full-matrix least-square procedures on F2 using SHELXS-97 and SHELXL-97 respectively.36 All non-hydrogen atoms were fixed using a standard riding model.| Crystal | 1 | 2c | 2d |
|---|---|---|---|
| Empirical formula | C4H20B6N2O14 | C6H19B5N2O10 | C8H28B10N2O20 |
| Formula wt (g mol−1) | 385.08 | 333.28 | 580.42 |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | Triclinic, P | Monoclinic, P21/c |
| a (Å) | 5.7619(2) | 8.8129(2) | 8.1624(6) |
| b (Å) | 6.3796(2) | 8.8960(2) | 11.3609(8) |
| c (Å) | 11.6206(5) | 10.3618(2) | 13.7429(9) |
| α (°) | 92.684(2) | 74.043(1) | 90 |
| β (°) | 98.060(2) | 78.854(1) | 94.416(4) |
| γ (°) | 114.750(2) | 88.962(1) | 90 |
| Vol (Å3) | 381.43(2) | 765.80(3) | 1270.63(15) |
| Z, Calc density (Mg m−3) | 1, 1.676 | 2, 1.445 | 2, 1.517 |
| μ (mm−1) | 0.155 | 0.125 | 0.136 |
| F(000) | 200 | 348 | 600 |
| Crystal | Colourless cut block | Colourless cut block | Colourless cut block |
| Crystal dimensions (mm3) | 0.08 x 0.05 x 0.03 | 0.28 x 0.23 x 0.18 | 0.42 x 0.16 x 0.10 |
| θ range (°) | 3.54–27.48 | 3.41–27.48 | 2.97–27.48 |
| No. of reflections collected | 7312 | 16010 | 14949 |
| Rint | 0.0440 | 0.0281 | 0.0486 |
| No. of data/restraints/parameters | 1746/0/122 | 3493/0/214 | 2909/0/187 |
| Final R indices [F2 > 2σ(F2)]: R1, wR2 | 0.0416, 0.0870 | 0.0322, 0.0817 | 0.0481, 0.1194 |
| R indices (all data): R1, wR2 | 0.0525, 0.0941 | 0.0358, 0.0842 | 0.0716, 0.1328 |
| Largest diff. peak and hole (e Å−3) | 0.296, −0.315 | 0.234, −0.243 | 0.442, −0.298 |
Crystallographic data for 1, 2c, 2d have been deposited with the Cambridge Crystallographic Data Centre with CCDC 930889–930891. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ (fax: +44 1223 336033) or email: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk.
3.6 Conclusion
Salts containing cations derived from N-substituted piperazines, partnered with either triborate(1−) or pentaborate(1−) anions have been prepared. The triborate salt 1 possesses an alternating cationic/anionic layered solid-state structure and contains extensively H-bonded triborate sheets. Compound 1 is thermally decomposed in air via a multistage process to glassy B2O3. Materials thermally derived from 1 were amorphous and non-porous. Similar thermal behaviour was observed for the pentaborate salts (2a–2d) which possess three dimensional H-bonded lattices.Acknowledgements
We thank the EPSRC for use of the X-ray crystallographic service (Southampton),37 and Ryan Wyn-Thomas who helped with some thermolysis experiments.Notes and references
- (a) C. C. Freyhardt, M. Wiebcke, J. Felsche and G. Englehardt, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 161–175 CrossRef CAS; (b) M. Wiebcke, C. C. Freyhardt, J. Felsche and G. Englehardt, Z. fur. Naturforsch B, 1993, 48, 978–985 CAS.
- M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan and D. M. Schubert, Cryst. Growth Des., 2006, 6, 538–545 CAS.
- Z.-H. Liu, J.-J. Zhang and W.-J. Zhang, Inorg. Chim. Acta, 2006, 359, 519–524 CrossRef CAS.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse, D. A. Knox and J. L. Timmis, Dalton Trans., 2010, 39, 3944–3951 RSC.
- G. J. de A. A. Soler-Illa, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef.
- (a) S. Yang, G. Li, S. Tian, F. Liao and J. Lin, Cryst. Growth Des., 2007, 7, 1246–1250 CrossRef CAS; (b) H.-X. Liu, Y.-X. Liang and X. Jiang, J. Solid State Chem., 2008, 181, 3243–3247 CrossRef CAS; (c) P. Becker, P. Held and L. Bohaty, Cryst. Res. Technol., 2000, 35, 1251–1262 CrossRef CAS.
- (a) W. R. Cook Jr. and H. Jaffe, Acta Crystallogr., 1957, 10, 705–707 CrossRef; (b) M. Touboul and E. Betourne, Solid State Ionics, 1993, 63, 340–345 CrossRef; (c) A. Garja, Phys. Status Solidi B, 1968, 27, K93 CrossRef.
- (a) V. P. Dotsenko, N. P. Efryshina and I. V. Berezovskaya, Mater. Lett., 1996, 28, 517–520 CrossRef CAS; (b) A. Akella and D. A. Keszler, Mater. Res. Bull., 1995, 30, 105–111 CrossRef CAS.
- (a) C. G. Salentine, Inorg. Chem., 1983, 22, 3920–3924 CrossRef CAS; (b) J. L. Anderson, E. M. Eyring and M. P. Whittaker, J. Phys. Chem., 1964, 68, 1128–1132 CrossRef CAS.
- R. C. Petersen, M. Finkelstein and S. D. Ross, J. Am. Chem. Soc., 1959, 81, 3264–3267 CrossRef CAS.
- D. M. Schubert, M. Z. Visi and C. B. Knobler, Inorg. Chem., 2008, 47, 2017–2023 CrossRef CAS.
- (a) T. J. R. Weakley, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 377–379 CrossRef; (b) G.-M. Wang, Y.-Q. Sun and G.-Y. Yang, J. Solid State Chem., 2004, 177, 4648–4654 CrossRef CAS.
- (a) C.-Y. Pan, G.-M. Wang, S.-T. Zheng and G.-Y. Yang, Z. Anorg. Allg. Chem., 2007, 633, 336–340 CrossRef CAS; (b) D. M. Schubert, M. Z. Visi, S. Khan and C. B. Knobler, Inorg. Chem., 2008, 47, 4740–4745 CrossRef CAS.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse, J. L. Timmis and K. S. Varma, Dalton Trans., 2012, 41, 4396–4403 RSC.
- D. M. Schubert, R. A. Smith and M. Z. Visi, Glass Technol., 2003, 44, 63–70 CAS.
- D. M. Schubert, M. Z. Visi and C. B. Knobler, Inorg. Chem., 2000, 39, 2250–2251 CrossRef CAS.
- Z.-H. Liu, L.-Q. Li and W.-J. Zhang, Inorg. Chem., 2006, 45, 1430–1432 CrossRef CAS.
- (a) M. Li, J.-Z. Chang, Z.-L. Wang and H.-Z. Chu, J. Solid State Chem., 2006, 179, 3265–3269 CrossRef CAS; (b) S. Yang, G. Li, S. Tian, F. Liao, M. Xiong and J. Lin, J. Solid State Chem., 2007, 180, 2225–2232 CrossRef CAS; (c) G.-M. Wang, C.-Y. Pan, S.-T. Zheng and G.-Y. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1101–o1103 CAS; (d) G.-M. Wang, C.-Y. Pan, S.-T. Zheng and G.-Y. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1104–o1105 CAS.
- M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse and K. S. Varma, J. Organomet. Chem., 2007, 692, 2832–2838 CrossRef CAS.
- M. A. Beckett, P. N. Horton, S. J. Coles and D. W. Martin, Inorg. Chem., 2011, 50, 12215–12228 CrossRef CAS.
- (a) M. Wojtas, A. Gagor, O. Czupinski, W. Medycki and R. Jakubas, J. Solid State Chem., 2012, 187, 35–44 CrossRef CAS; (b) G. J. Perpetuo and J. Janczak, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, E61, o2531–o2533 Search PubMed.
- J. Li, S. Xia and S. Goa, Spectrochemica Acta, 1995, 51A, 519 Search PubMed , 532.
- M. A. Beckett, P. N. Horton, S. J. Coles, D. A. Kose and A.-M. Kreuziger, Polyhedron, 2012, 38, 157–161 CrossRef CAS.
- A. V. Paul and S. Natarajan, Cryst. Growth Des., 2010, 10, 765–774 CAS.
- U. Schubert and N. Husing, Synthesis of Inorganic Materials, Wiley-VCH, 2nd edn, 2007, Chapter 6, pp. 305–352 Search PubMed.
- (a) S. G. Hou, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, o3292 Search PubMed; (b) C. Peng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, o2214 Search PubMed; (c) S. Murugavel, R. Selvakumar, S. Godindarajan, P. S. Kannan and A. SubbiahPandi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, o1004 Search PubMed.
- A. Dal Negro, J. M. Martin Pozaz and L. Ungaretti, Am. Mineral, 1975, 60, 897–883 Search PubMed.
- C. G. Salentine, Inorg. Chem., 1987, 26, 128–132 CrossRef CAS.
- (a) M. A. Beckett, G. C. Strickland, K. S. Varma, D. E. Hibbs, K. M. A. Malik and M. B. Hursthouse, J. Organomet. Chem., 1997, 535, 33–41 CrossRef CAS; (b) M. A. Beckett, G. C. Strickland, K. S. Varma, D. E. Hibbs, M. K. A. Malik and M. B. Hursthouse, Main Group Chem., 1998, 2, 251–258 CrossRef CAS.
- (a) M. A. Beckett, S. J. Coles, M. E. Light, L. Fischer, B. M. Stiefvater-Thomas and K. S. Varma, Polyhedron, 2006, 25, 1011–1016 CrossRef CAS; (b) M. A. Beckett, E. L. Bennett, P. N. Horton and M. B. Hursthouse, J. Organomet. Chem., 2010, 695, 1080–1083 CrossRef CAS; (c) M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse and K. S. Varma, Inorg. Chem., 2007, 46, 3801–3803 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- P. N. Horton, M. B. Hursthouse, M. A. Beckett and M. P. Rugen-Hankey, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, E60, o2204–o2206 Search PubMed.
- R. Hooft, Collect, Data collection software, Nonius BV, Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- (a) R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A51, 33–37 Search PubMed; (b) R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421–426 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra42387e |
| This journal is © The Royal Society of Chemistry 2013 |