DOI:
10.1039/C3RA42123F
(Review Article)
RSC Adv., 2013,
3, 17602-17632
The role of biocatalysis in the asymmetric synthesis of alkaloids
Received 29th April 2013, Accepted 28th June 2013
First published on 4th July 2013
Abstract
Alkaloids are not only one of the most intensively studied classes of natural products, their wide spectrum of pharmacological activities also makes them indispensable drug ingredients in both traditional and modern medicine. Among the methods for their production, biotechnological approaches are gaining importance, and biocatalysis has emerged as an essential tool in this context. A number of chemo-enzymatic strategies for alkaloid synthesis have been developed over the years, in which the biotransformations nowadays take an increasingly ‘central’ role. This review summarises different applications of biocatalysis in the asymmetric synthesis of alkaloids and discusses how recent developments and novel enzymes render innovative and efficient chemo-enzymatic production routes possible.
 Joerg H. Schrittwieser | Joerg H. Schrittwieser, born 1984, studied Chemistry at the University of Graz. In 2007, he joined the research group of Prof. Wolfgang Kroutil for working on his diploma thesis dealing with biocatalytic cascades involving alcohol dehydrogenases. After participating in two short-term research projects at the Universidad Complutense in Madrid and the Research Centre Applied Biocatalysis in Graz, he returned to Prof. Kroutil's labs where he completed his PhD, which dealt with the chemo-enzymatic synthesis of benzylisoquinoline and berbine alkaloids, in 2011. Since March 2012, he is a post-doctoral researcher with Dr Frank Hollmann at Delft University of Technology, via an Erwin-Schrödinger Fellowship from the Austrian Science Fund. His research interests are the development and optimisation of multi-enzymatic and chemo-enzymatic cascade systems, the chemo-enzymatic synthesis of natural products, as well as the utilisation of flavoenzymes for “green” oxidation chemistry. |
 Verena Resch | Verena Resch completed her MSc in biochemistry and molecular biology in 2008 at the University of Graz under the supervision of Prof. Wolfgang Kroutil establishing multi-enzyme cascades. Staying in the same group she received her PhD in organic chemistry in 2011 working on the use of alkaloid pathway enzymes in organic synthesis. After a short project as a contract researcher at the ACIB (Austrian Centre of Industrial Biotechnology) she started as a post-doctoral fellow with an Erwin-Schrödinger Fellowship from the Austrian Science Fund at the University of Technology in Delft in the group of Prof. Ulf Hanefeld. Her main research interests are alkaloid pathway enzymes, (chemo)-enzymatic cascade reactions, and protein expression and characterisation. |
1. Introduction
The alkaloids are a structurally diverse group of nitrogen-containing secondary metabolites with a relatively wide distribution in nature and a broad range of biological activities.1 While the original definition of the term ‘alkaloid’, introduced by the German pharmacist Carl F. W. Meissner in 1819,2 covers only plant-derived substances with alkaline character, modern conceptions are usually much broader. For instance, S. William Pelletier proposed a very simple definition in 1983, saying that “An alkaloid is a cyclic organic compound containing nitrogen in a negative oxidation state which is of limited distribution among living organisms” .1d,3 The total number of known structures that can be categorised as alkaloids according to these general criteria exceeds 20,000,4 and around 100 new compounds are still discovered annually.1b The higher plants are a major source of alkaloids, as approximately 25% of all plant species contain these constituents,1b but their occurrence in animals, insects, microorganisms, and lower plants is also well documented.1dThe natural functions of plant alkaloids are thought to be mainly associated with the interactions of the plant with other organisms–for instance, protection against herbivores or pathogens, or attraction of pollinating insects, seed dispensing animals or root nodule bacteria.1d,5 Hence, it is not surprising that many alkaloids exert potent pharmacological activities, a fact that has been recognised by humans and exploited for both therapeutic and recreational purposes for thousands of years.1c,6 Also in modern medicine, many alkaloids and their derivatives have found application as drugs and even more are currently under investigation in clinical trials (for a selection of therapeutically important alkaloids, see Table 1). Estimated global sales of over 4 billion US$ (2002) for alkaloids applied in medicine illustrate both the interest in this substance class and the necessity for efficient production routes.7 However, not all alkaloids of public demand can be isolated from their natural source in sufficient quantities, and even if isolation is possible on large scale, it is often a very expensive and time-consuming procedure.7 Especially for minor constituents the purification effort to arrive at a chemically well-defined product can be enormous, and isolation yields are typically low.
Table 1 Examples of alkaloids with medicinal applicationa
| Alkaloid (active ingredient) | Natural source1d | Therapeutic use |
|---|
| Apart from galantamine, all listed substances are part of the World Health Organization's Model List of Essential Medicines (17th ed., for adults).25 |
|---|
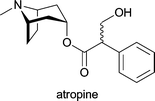 | Atropa belladonna; industrially isolated from Duboisia species, Hyoscyamus muticus and Hyoscyamus niger | antispasmodic; treatment of organo-phosphate poisoning; against myasthenia gravis; against arrhythmias; local treatment for muscular rheumatism, sciatica and neuralgia; in ophthalmology as mydriatic and cycloplegic drug1d |
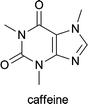 | Caffea arabica, Paullinia cupana, Cola nitita and Cola acuminata | added to analgesics to increase their activity; used in the treatment of neonatal apnoea and atopic dermatitis1d |
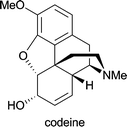 | Papaver somniferum | as antitussive in treatment of cough; as analgesic and mild sedative1d |
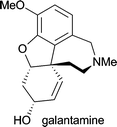 | Galanthus caucasicus, Galanthus woronowii and Galanthus nivalis | in the treatment of Alzheimer's disease24 |
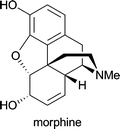 | Papaver somniferum | pain control; treatment of diarrhoea1d |
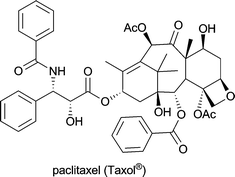 | Taxus brevifolia; produced industrially by Taxus plant cell fermentation | in treatment of mamma and ovary carcinoma and several other malignancies1d |
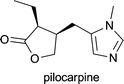 | Pilocarpus jaborandi, Pilocarpus pennatifolius, Pilocarpus racemosus and Pilocarpus microphyllus | in treatment of open-angle glaucoma1d |
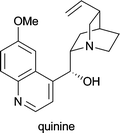 | Cinchona succirubra | in treatment of malaria, babesiosis and myotonic disorders1d |
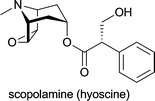 | Duboisia myoporoides, Duboisia leichhardtii | against acute motion sickness; as premedicant before induction of general anaesthesia1d |
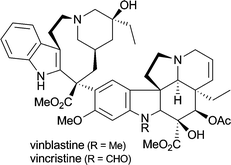 | Catharanthus roseus | in treatment of Hodgkin's disease and other lymphoma, testicular cancer and a variety of solid neoplasms1d,5a |
On the other hand, the intricate molecular architecture of many alkaloids–with diverse functional groups and often multiple stereogenic centres–also makes them challenging targets for total synthesis. In view of these difficulties it is just the more impressive that many complex alkaloids have indeed been synthesised in the lab; however, long sequences involving extensive protective group chemistry are often required, resulting in very limited overall yields. Consequently, the transfer of these synthetic routes to production scale usually represents a major problem. An excellent example for this discrepancy–discussed in a recent account by Walji & MacMillan8–is the case of paclitaxel (Taxol®): Six ingenious and highly acclaimed total syntheses of this complex alkaloid (for the structure, see Table 1) have been developed to date, the most efficient of which gives the target compound in 0.4% yield over 37 steps.9 Unfortunately, large-scale production of paclitaxel by this route is not practical or economically feasible, and neither is its isolation from natural sources. Today, the growing demand in this natural anti-tumour agent is met by plant cell fermentation,10 and the environmental benefits of this production method have been recognised with the 2004 Presidential Green Chemistry Award.11
The paclitaxel example already illustrates that biological methods can represent favourable options for alkaloid production. Indeed, biotechnology in its broadest sense offers several strategies that may help to overcome some of the above-mentioned supply limitations. For instance, genetic modification of alkaloid-containing plants can be used to increase production levels and hence facilitate isolation.12 Plant cell fermentation can be used to achieve production of alkaloids by the native source organism under controlled and scalable conditions. Entire secondary metabolic pathways can be engineered into suitable microbial hosts, which thus become able to biosynthesise the corresponding natural products.13 This approach has recently been applied to the fermentative production of the benzylisoquinoline alkaloid (S)-reticuline from simple carbon sources such as glycerol or glucose, using an engineered E. coli strain.14 In a related study, (S)-reticuline has been obtained by fermentation from dopamine, and has been further transformed into aporphine or protoberberine alkaloids using transgenic Saccharomyces cerevisiae.15 The implementation of an entire biosynthetic pathway for the production of benzylisoquinolines into Saccharomyces cerevisiae has also been reported.13,16
In more chemistry-oriented settings, biocatalysis can be employed for the selective modification of alkaloids to access non-natural or less abundant derivatives. A classical example is the use of the enzymes morphine dehydrogenase and morphinone reductase from Pseudomonas putida M10 for the interconversion of morphinan alkaloids.17 More recent examples include the laccase-catalysed coupling of catharanthine and vindoline to yield anhydrovinblastine,18 the laccase-catalysed hydroxylation of ergot alkaloids,19 or the coupling of the alkaloid sinomenine with electron-rich arenes mediated by the fungi Antrodiella semisupina20 and Coriolus unicolor.21 Finally, biocatalytic methods can be employed in the asymmetric total synthesis of alkaloids, thereby combining the flexibility of chemical routes with the high degree of chemo- and enantioselectivity offered by enzymes. This interdisciplinary approach seems to be particularly fruitful, as a considerable number (approx. 150) of chemo-enzymatic total syntheses of alkaloids have been reported over the last 25 years, which in many cases prove more efficient than either a purely chemical synthesis or isolation of the target compound from natural sources.
Three main strategies for chemo-enzymatic alkaloid synthesis can be distinguished: (1) the biocatalytic preparation of chiral building blocks, which are chemically transformed into the target compounds, (2) the biocatalytic kinetic resolution, desymmetrisation or deracemisation of alkaloids that have been synthesised by chemical means, and (3) the chemo-enzymatic synthesis of alkaloids via biocatalytic C–N and/or C–C bond formation in the asymmetric key step. The first approach is definitely the most versatile, and also by far the most explored, but in recent years biocatalytic deracemisation and asymmetric biocatalytic C–C bond formation have gained momentum. This review provides an overview of all three above-mentioned strategies and discusses recent developments that are likely to change the role of biocatalysis in future alkaloid synthesis.
2. Biocatalytic asymmetric synthesis of chiral building blocks
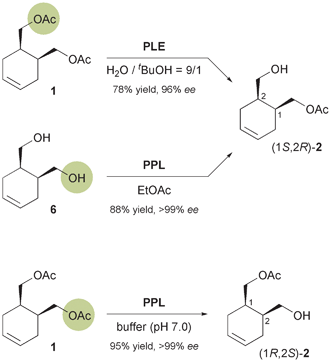
The chiral building block (also called ‘chiral synthon’ or ‘chiron’) approach to asymmetric synthesis involves the recognition of structural elements in the target molecule that can be traced back retrosynthetically to readily available chiral molecules.22 Classically, the latter are derived from the ‘chiral pool’ of amino acids, carbohydrates, terpenes, etc., while in chemo-enzymatic approaches the chiral building blocks are compounds that can be obtained in high enantiomeric purity by a biocatalytic reaction.23 In the context of alkaloid chemistry, lipases and esterases as well as toluene dioxygenase are most frequently used for the generation of chiral building blocks, and a large number of structurally diverse alkaloids have been synthesised using these enzymes.2.1 Lipases and esterases
Their excellent stereoselectivity and broad substrate scope, their ability to work in organic solvents, and also their broad commercial availability have made lipases and esterases the most widely used biocatalysts in organic synthesis. Therefore, it is not surprising that also the majority of chemo-enzymatic syntheses of alkaloids rely on these enzymes. When applied in a kinetic resolution, they can provide access to both enantiomers of a chiral building block in 50% maximum yield. However, desymmetrisation of meso-compounds–which does not suffer from this limitation–is common as well. This strategy can also afford both enantiomers of the product, provided that stereocomplementary enzymes are available, or that the desymmetrisation can be run in both hydrolytic and acylative direction (cf.Scheme 2). Both reaction types have been used to prepare a considerable number of chiral building blocks, most of which can be categorised structurally into the groups of cyclic alcohols, piperidine derivatives, and nitrones.2.1.1 Cyclic alcohol building blocks. The earliest report on the application of a biocatalytically obtained building block in the asymmetric total synthesis of an alkaloid is a brief communication by Renata Riva and co-workers published in 1987, which describes the preparation of (−)-alloyohimbane (5) from the hydroxy ester (1S,2R)-2 (Scheme 1).26 This building block was obtained in 96% ee by hydrolytic desymmetrisation27 of the corresponding meso-diacetate 1 using pig liver esterase (PLE) as biocatalyst. The hydroxy ester was then converted into the lactone (S,S)-3, which was coupled with tryptamine to give amide intermediate 4, from which 5 was prepared following a literature procedure.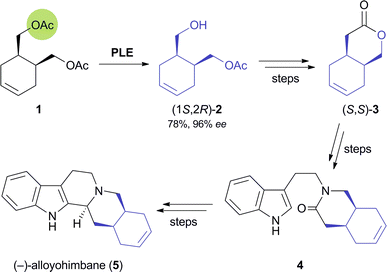 |
| | Scheme 1 Chemo-enzymatic synthesis of (−)-alloyohimbane (5).28 | |
 |
| | Scheme 2 Options for the enantioselective preparation of building block 2 using hydrolases. | |
A later study found it difficult to reproduce the excellent optical purity of (1S,2R)-2 in the PLE-catalysed hydrolysis process and moved to stereoselective monoacetylation of the corresponding meso-diol 6 (Scheme 2), employing porcine pancreatic lipase (PPL) in anhydrous ethyl acetate, to access the same compound in 88% yield and >99% ee on gram scale.29 The building block was again converted into (S,S)-3,30 which via 10 additional steps was further elaborated into the Strychnos alkaloid (−)-akagerine (7, Fig. 1), obtained in 19% overall yield from 2. Very recently, (1S,2R)-2 has been ‘rediscovered’ and used in the asymmetric total synthesis of the complex polycyclic indole alkaloid (+)-scholarisine A (8, Fig. 1), first isolated from the leaves of Alstonia scholaris in 2008. The target compound was obtained in 0.4% overall yield via 20 synthetic steps, which include a nitrile hydrogenation/epoxide aminolysis cascade, a Fischer-type indole synthesis, and an oxidative lactonisation.31
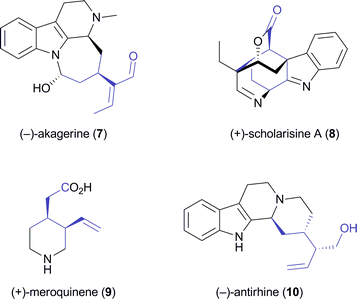 |
| | Fig. 1 Alkaloids synthesised from the chiral building block 2. | |
The opposite enantiomer of 2, accessible via stereoselective monohydrolysis of 1 with PPL (Scheme 2) has also found application in the asymmetric synthesis of alkaloids: It has been used by Danieli and co-workers in the preparation of (+)-meroquinene (9), a degradation product of cinchonine and key intermediate in the synthesis of Cinchona alkaloids,32 and of the indole alkaloid (−)-antirhine (10, Fig. 1).33
Lesma and co-workers, on the other hand, have extended the concept of hydrolytic desymmetrisation of meso-diacetates to the cycloheptene derivative 11, obtaining the monoacetate (1S,6R)-12 (Scheme 3) in 95% yield and >97% ee using lipase PS.34,35 From this building block, several 4-hydroxypiperidine derivatives such as cis-4-hydroxy-2-pipecolic acid, a hydroxylated quinolizidine 13, and the piperidine alkaloid (−)-halosaline (14, Scheme 3) were prepared,34,36 the latter in a very elegant sequence involving ruthenium-catalysed ring-rearrangement metathesis as a key step.
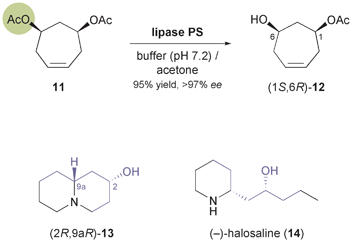 |
| | Scheme 3 Desymmetrisation of meso-diacetate 11 using lipase PS affords compound 12, a building block used in the synthesis of alkaloids 13 and 14. | |
Another common class of chiral building blocks in alkaloid synthesis are cyclic allylic alcohols with substituents on the double bond, such as 3-ethylcyclohexenol 15 (Scheme 4). The kinetic resolution of this and similar compounds with various lipases has been studied by Palmisano and co-workers,37 who also demonstrated the conversion of (S)-15, obtained from the racemate in 43% yield and 99.5% ee using Burkholderia cepacia lipase35 in neat vinyl acetate, into alkaloids of the eburnane type (e.g.18).38 A key step in their synthesis is a transfer of chirality from C1 of the cyclohexenol structure to C3 by means of a decarboxylative Claisen rearrangement, thus setting up a quaternary carbon chiral centre in excellent enantiomeric purity (ee = 96%). The authors followed a similar strategy in the asymmetric synthesis of (+)-decarbomethoxy-15,20;16,17-tetrahydrosecodine (22), where a Claisen rearrangement of the ester (S)-20 was used to establish a tertiary carbon chiral centre (Scheme 4).39 Immobilised porcine pancreatic lipase (PPL) in neat vinyl acetate was used for the kinetic resolution of rac-19 (the precursor to 20), and the bromine atom in the substrate served the sole purpose of enabling a sufficiently high enantioselectivity (E = 133) in this reaction by increasing the steric demand of one side of the substrate. A ‘transfer of chirality’ concept was also applied by Yamane & Ogasawara in their chemo-enzymatic syntheses of spirocyclic piperidine alkaloids:40 Kinetic resolution of carbethoxycyclohexenol 23 (Scheme 4) with lipase PS35 gave the corresponding (R)-acetate 24 in 40% yield and 99% ee, along with 53% of the remaining alcohol in 95% ee. Derivatisation of the alcohol (R)-23 to an α-bromoacetal followed by a highly diastereoselective radical cyclisation afforded the building block (1R,2S)-26, which was further elaborated into the spirocyclic alkaloids (+)-nitramine (27) and (+)-isonitramine (28). Similarly, its enantiomer (1S,2R)-26 was transformed into (−)-sibirine.40
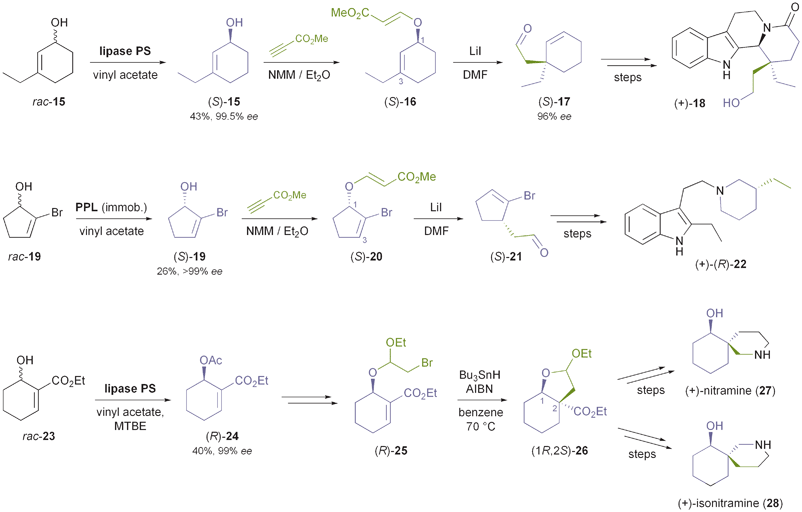 |
| | Scheme 4 Use of enzymatically derived cycloalkenol building blocks in the synthesis of indole alkaloids and spirocyclic piperidine alkaloids. | |
Ogasawara and co-workers have developed a chemo-enzymatic route towards several cyclopentanoid chiral building blocks, using cyclopentadiene as starting material.41 The asymmetric key step is the kinetic resolution of rac-cis-29 employing lipase PS35 in methyl tert-butyl ether (MTBE) at room temperature (Scheme 5). This transformation proceeds with excellent enantioselectivity, affording both the (−)-(1S,4R)-acetate 30 and the (+)-(1R,4S)-alcohol 29 in optically pure form (ee > 99%) and in high isolated yields (43% and 50%, respectively). Compound 29 has served as starting material in the (formal) total synthesis of various indole alkaloids, including (−)-physostigmine (31) and (+)-aspidospermidine (32), as well as the benzomorphan alkaloid (−)-aphanorphine (33, Scheme 5).42 In addition, it has been elaborated into the cyclohexenone derivative 34, which is an intermediate en route to Sceletium alkaloids.43
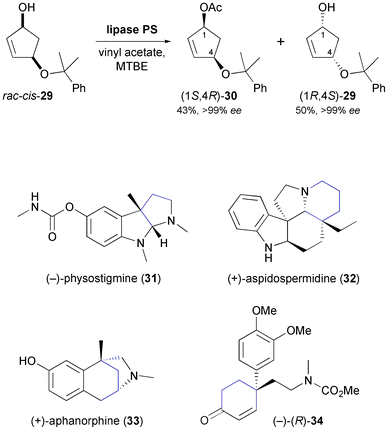 |
| | Scheme 5 Lipase-catalysed kinetic resolution of building block 29, and examples of alkaloids prepared from this building block. | |
While the chiral building blocks discussed so far have been used in multiple syntheses of alkaloids, lipase catalysis has also been employed to access some ‘purpose-made’ chiral intermediates for single synthetic endeavours. A notable example is Fukuyama's total synthesis of (−)-morphine (43, Scheme 6), which uses the alcohol 36, obtained in 99% ee by hydrolytic kinetic resolution of the corresponding acetate 35 using lipase AK.35,44 All five stereocentres of the target molecule were established from 36 with excellent diastereocontrol, and (−)-morphine was obtained in 5% overall yield over 17 linear steps.
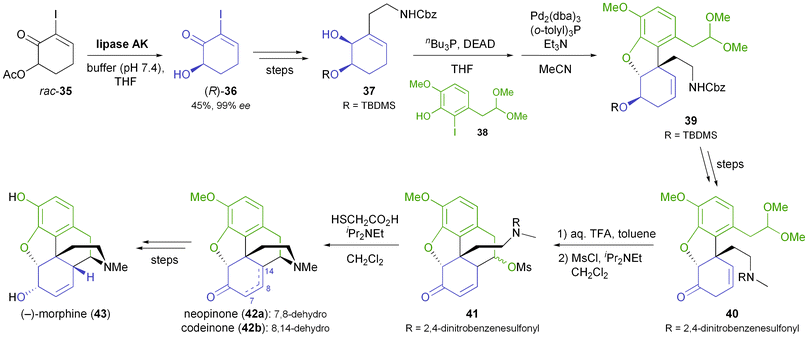 |
| | Scheme 6 Fukuyama's chemo-enzymatic total synthesis of (−)-morphine (43). | |
2.1.2 Piperidine building blocks. The first nitrogen-containing chiral building block derived from an enzymatic reaction was introduced to the asymmetric synthesis of alkaloids in 1992: Momose and co-workers have investigated the stereoselective transesterification of N-protected meso-9-azabicyclo[3.3.1]nonanediol 44 (Scheme 7), as well as the stereoselective hydrolysis of the corresponding diacetate, catalysed by various lipases.45 In both cases, a commercial lipase from Humicola lanuginosa35 gave the best results, providing access to both enantiomers of the monoacetate 45 in high yield and good enantiomeric excess (ee = 90% for R, 80% for S). The monoacetate was oxidised to the corresponding ketone 46, which could be obtained in optically pure form by a single recrystallisation from diisopropyl ether. This ketone, in turn, was transformed into the trisubstituted piperidine building block 47via enol ether formation and ozonolytic cleavage of the bicyclic core structure (Scheme 7). Compound 47 proved useful for further elaboration into different piperidine alkaloids, such as (+)-dihydropinidine (48) and (−)-cassine (49),45,46 as well as indolizidines and quinolizidines sequestered by the poison-dart frogs of the Dendrobatidae family, e.g. (−)-indolizidine 235B′ (50, Scheme 7).47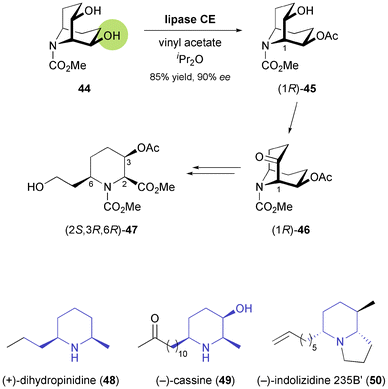 |
| | Scheme 7 Lipase-catalysed desymmetrisation of diol 44, its conversion into the trisubstituted piperidine derivative 47, and examples of alkaloids prepared from this building block. | |
The obvious advantages of nitrogen-containing chiral building blocks in the asymmetric synthesis of alkaloids led several other research groups to investigate biocatalytic methods for their preparation, and piperidine derivatives have attracted particular interest. Chênevert and Dickman have investigated the hydrolytic desymmetrisation of meso-diacetates 51 (Scheme 8) with various lipases and have identified the enzyme from Aspergillus niger as the most selective biocatalyst.48 The (2R,6S)-monoacetates 52 were isolated in good yields (76–92%) and excellent optical purity (ee > 98%), but the reaction took several days (72–108 h) to complete. Still, the authors could demonstrate the synthetic utility of the method by converting the monoacetates into (+)-dihydropinidine (48, Scheme 7) and the dendrobate frog alkaloids (+)-hydroxypiperidine 241D (54) and (−)-indolizidine 167B (55, Scheme 8).48b,49 Later studies avoided the time-consuming hydrolysis protocol and focused on the preparation of the opposite enantiomer of 52 by lipase-catalysed stereoselective monoacetylation. The lipases from Candida antarctica and Candida cylindracea (the latter employed in the ionic liquid BMIM-PF6 as reaction medium) were found to be suitable enzymes for this transformation, affording (2S,6R)-38 in 80% yield (ee = 95%) and 90% yield (ee = 98%), respectively.50 In the former case, the piperidine intermediate was further transformed into the non-natural enantiomer of the Dendrobatidae alkaloid indolizidine 209D [(+)-56],50a while in the latter it was elaborated into polyhydroxylated indolizidines and quinolizidines, e.g. (−)-57 (Scheme 8), via a sequence featuring ring-closing metathesis and OsO4-catalysed dihydroxylation as key steps.50b
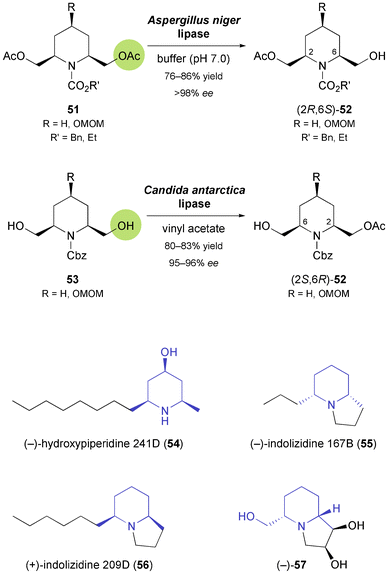 |
| | Scheme 8 Biocatalytic preparation of both enantiomers of the 2,6-disubstituted piperidine building block 52, and examples of alkaloids prepared from this building block. | |
3,5-cis-Disubstituted piperidines also form a common structural motif in natural products, for example in indole alkaloids of the ibogan and tacaman type, or in (−)-sparteine, which has found broad application as a chiral ligand. Lesma and co-workers first tried to establish an asymmetric entry to 3,5-disubstituted piperidine building blocks via stereoselective enzymatic hydrolysis of meso-diesters 58 (Scheme 9), but both yield and optical purity of the obtained monoesters 59 were only moderate.51 Stereoselective monoacetylation of the analogous piperidine-3,5-dimethanols 60 (Scheme 9) catalysed by the lipase from Pseudomonas fluorescens turned out to be more efficient, providing the (3S,5R)-enantiomer of monoacetates 61 in good yield (74–78%) and excellent optical purity (ee > 98%).52 The opposite enantiomer could conveniently be accessed by hydrolytic desymmetrisation of the diacetates 62 using the same enzyme. The chiral building blocks thus obtained served as a basis for the preparation of several compounds featuring cis-fused piperidine rings as core structure, e.g. the 3,7-diazabicyclo[3.3.1]nonane derivative 63,53 the Leguminosae alkaloid (−)-cytisine (64, Scheme 9),54 and a truncated (+)-sparteine analogue.55 Furthermore, the conversion of derivatives of 61 into the ibogan type indole alkaloid (+)-dihydrocleavamine (65), featuring an unusual tetracyclic framework that contains a 9-membered ring, has been reported.56
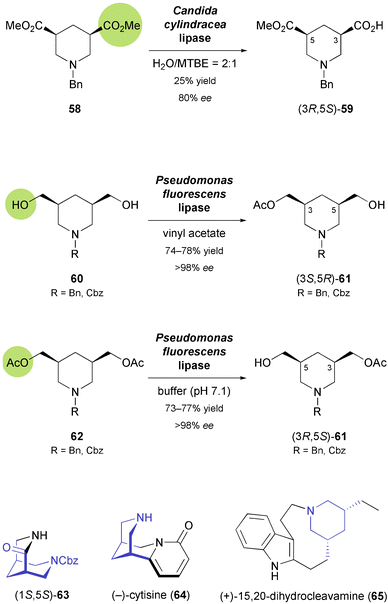 |
| | Scheme 9 Biocatalytic preparation of the 3,5-disubstituted piperidine building blocks 59 and 61, and examples of alkaloids prepared from 61. | |
In comparison to the di- or polysubstituted piperidines described above, monosubstituted derivatives have attracted less attention, although several monosubstituted piperidine alkaloids are known. Passarella, Riva, and co-workers have investigated N-Boc-piperidine-2-ethanol 66 as chiral building block, but this compound proved a challenging substrate for lipase-catalysed kinetic resolution due to its conformational flexibility and the distance of the alcohol moiety from the stereogenic centre.57 The enantioselectivities observed were generally low (E ≤ 8), but the identification of two enantiocomplementary enzymes–(S)-selective porcine pancreatic lipase (PPL), and (R)-selective lipase PS (from Burkholderia cepacia)35–allowed the preparation of both enantiomers of acetate 67 in decent optical purity (ee = 90% for R, 95% for S) via a complex, but scalable triple kinetic resolution protocol (Scheme 10). The optically enriched building blocks were transformed into both enantiomers of sedamine (69) and allosedamine (70, Fig. 2) in three additional steps. In a similar fashion, (+)-dumetorine (71),58 (−)-coniine (72),59 and (−)-epi-dihydropinidine (73) were prepared, whereby in the latter case the N-Boc group served as a lithiation director in the introduction of the 6-methyl substituent.58 Furthermore, the lupinine alkaloid (+)-aloperine (74) was synthesised from (R)-67 in a 12-step sequence that featured the use of a laccase–TEMPO system for the oxidation of primary alcohol moieties, and of a Diels–Alder reaction for setting up the correct stereochemistry at three chiral centres.60
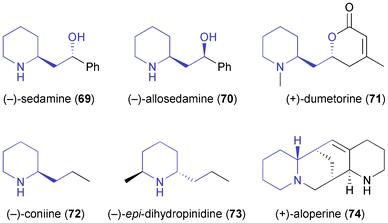 |
| | Fig. 2 Examples of alkaloids prepared from building block 67. | |
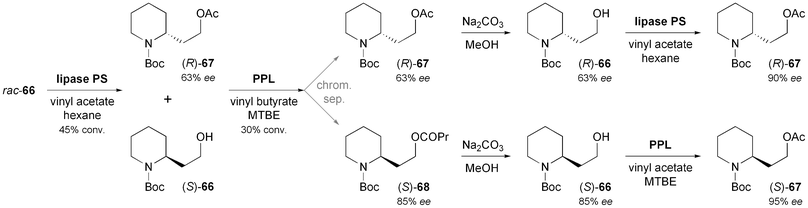 |
| | Scheme 10 Preparation of both enantiomers of the 2-substituted piperidine building block 67via a triple kinetic resolution protocol employing enantiocomplementary lipases. | |
2.1.3 Nitrone building blocks. Optically active nitrones represent chiral building blocks that are particularly interesting for the synthesis of nitrogen heterocycles. The nitrone moiety can react with olefins in inter- or intramolecular (3 + 2)-cycloadditions, which often proceed with excellent regio- and diastereoselectivity, and furthermore this functional group can provide a nitrogen atom present in the target molecule. These features were exploited by Holmes and co-workers,61 who prepared nitrone 76 from the chiral alcohol (R)-75 (Scheme 11), which was obtained in 47% yield and 92% ee by kinetic resolution of the racemate using lipase PS35 under previously reported conditions.62 Intramolecular cyclisation of 76 afforded a separable mixture of isomers, of which the major one (77, 32%) was converted into the indolizidine (−)-79,61a and further into the dendrobate frog alkaloid (+)-allopumiliotoxin 323B′ (80).61b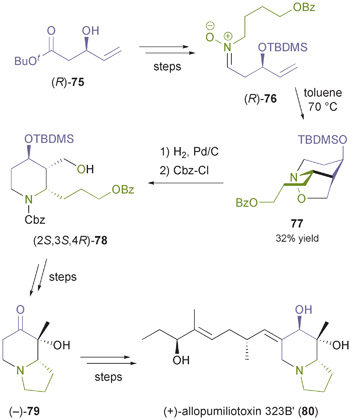 |
| | Scheme 11 Synthesis of (+)-allopumiliotoxin 323B′ (80) from the lipase-derived building block (R)-75. | |
Hatakeyama and co-workers have devised a synthetic sequence towards the antimalarial Hydrangea alkaloids (+)-febrifugine (86) and (+)-isofebrifugine (87), in which the nitrone 84 is a key intermediate (Scheme 12).63 Kinetic resolution of alcohol 81 using immobilised Candida antarctica lipase B (CAL-B; Novozym 435)35 provided the corresponding (S)-acetate 82 in 43% yield and 91% ee (enantioselectivity E = 50). The key step of the synthesis is a cascade nitrone formation–cycloaddition reaction, which joins three molecules and sets up two rings and two chiral centres in one operation, albeit in only moderate diastereoselectivity. The mixture of isomers obtained was transformed into the target alkaloids 86 and 87 in six additional steps. An even more elegant one-pot procedure forms the cornerstone of Kita's asymmetric synthesis of (−)-rosmarinecine (92): the racemic hydroxynitrone 88 was stereoselectively acylated with the maleate ester 89 using immobilised CAL-B (Chirazyme L-2),35 and the acylation product underwent a spontaneous intramolecular (3 + 2)-cycloaddition to afford compound 91, featuring 4 contiguous stereocentres, as a single diastereomer in 58% yield and 92% ee (Scheme 12). Recrystallisation and minor functional group interconversions completed the synthesis of optically pure 92, which was obtained in 45% overall yield from rac-88.64 An improved procedure for the preparation of rac-88 published some years later even rendered the use of protective groups unnecessary.65
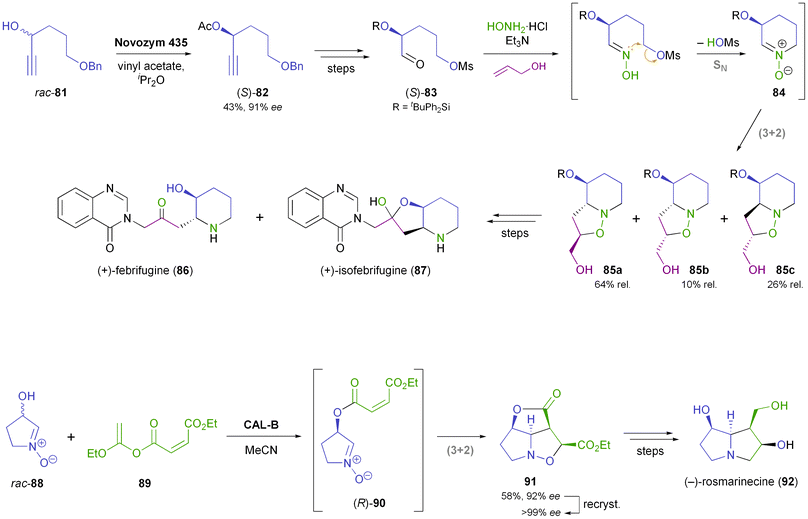 |
| | Scheme 12 Cascade reactions involving nitrone–olefin (3 + 2)-cycloadditions in the chemo-enzymatic syntheses of (+)-febrifugine (86), (+)-isofebrifugine (87), and (−)-rosmarinecine (92). | |
2.2 Toluene Dioxygenase
Arene cis-dihydrodiols are versatile chiral building blocks offering ample possibilities for further functionalisation (Scheme 13).66 While the asymmetric preparation of these labile compounds represents an enormous challenge for chemical methods, they are formed with excellent selectivity from the corresponding arenes by the action of bacterial dioxygenases. Toluene dioxygenase (TDO) from Pseudomonas putida is the best-known of these enzymes, and the cis-dihydroxylation of various simple arenes by Pseudomonas has been reported by David Gibson and co-workers as early as 1968.67 It took almost two decades until the preparative value of this biotransformation was recognised, but since the late 1980s, biocatalytically prepared arene cis-dihydrodiols have been used in the asymmetric synthesis of numerous natural products, including many alkaloids.66 Early studies relied on Pseudomonas putida 39/D, a mutant strain developed by Gibson's group, for fermentative production of the desired metabolites, but later the responsible toluene dioxygenase has been cloned and heterologously expressed in E. coli, which soon became the preferred biocatalyst. Today, several optically pure arene cis-dihydrodiols (obtained by TDO catalysis) are commercially available.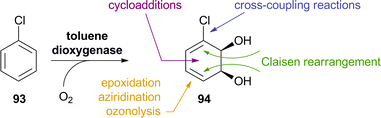 |
| | Scheme 13 Preparation of aromatic cis-dihydrodiols, e.g.94, by biocatalytic dioxygenation of arenes, and some general methods for their further functionalisation. | |
2.2.1 TDO in the synthesis of pyrrolizidine and iminocyclitol alkaloids. The first asymmetric synthesis of an alkaloid from an arene cis-dihydrodiol was reported by Hudlicky et al. in 1990:68 The authors prepared both enantiomers of acetonide-protected trihydroxyheliotridane 98, in nine steps for the (+)-isomer and twelve for its (−)-counterpart, from chlorobenzene 93. Thereby, they made elegant use of ozonolysis and selective redox transformations to obtain either enantiomer of the key intermediate 96 from the same cis-dihydrodiol (Scheme 14).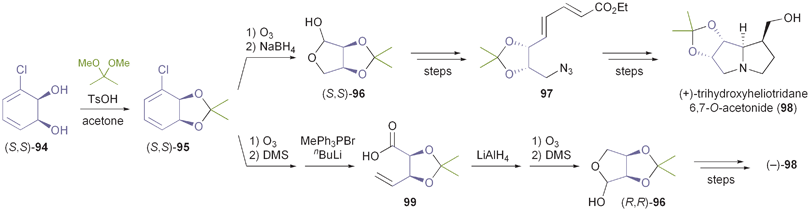 |
| | Scheme 14 Stereodivergent synthesis of trihydroxyheliotridane acetonide (98) from the TDO-derived diol 94. | |
A related ozonolysis approach (in this case cleaving only one double bond) has been followed in the synthesis of the iminocyclitols (+)-kifunensine (100), (+)-mannojirimycin (101), (+)-1-deoxygalactonojirimycin (102), and (−)-1-deoxymanno-jirimycin (103, Fig. 3), which are glycosidase inhibitors with potential anti-viral properties.69
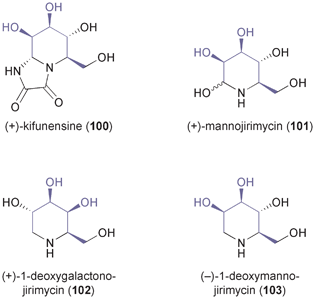 |
| | Fig. 3 Examples of iminocyclitol alkaloids prepared from TDO-derived arene cis-dihydrodiols. | |
2.2.2 TDO in the synthesis of Amaryllidaceae isocarbostyril alkaloids. Over the last 20 years, Hudlicky's research group has made extensive use of arene cis-dihydrodiols as building blocks in natural product synthesis, most notably in the preparation of the cytotoxic Amaryllidaceae alkaloids pancratistatin (104), narciclasine (105), lycoricidine (106), and their analogues (Fig. 4), which exhibit potent in vitro and in vivo anti-cancer activity.66,70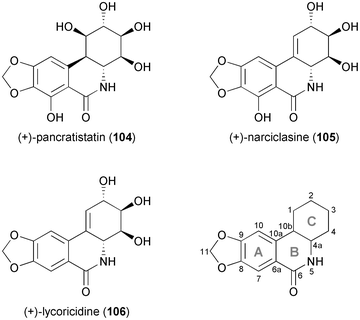 |
| | Fig. 4 Structures of pancratistatin (104), narciclasine (105) and lycoricidine (106), and general ring denotation and atom numbering conventions for isocarbostyril alkaloids. | |
In all these synthetic studies, the biocatalytically prepared cis-diol served as precursor for the C-ring of the target compounds, while different strategies were explored for the construction of ring B: (i) In the case of (+)-lycoricidine (106), the first Amaryllidaceae alkaloid obtained by this chemo-enzymatic approach, the piperonyl unit 108 was attached to the diol building block 107via an acyl-nitroso–diene [4 + 2] cycloaddition (nitroso-Diels–Alder reaction).71 Subsequent closure of the B-ring by a Heck reaction proved problematic, as desilylation and transacetylation occurred as side reactions, resulting in a yield of only 27% (Scheme 15a). (ii) Later studies focused on nucleophilic opening of aziridines derived from the arene cis-diol as a way to connect rings A and C, and this approach was first used in the asymmetric synthesis of (+)-pancratistatin (104):72 The tosylaziridine 112, obtained in 4 steps from bromobenzene, was coupled with the cyanocuprate 113, whose dimethylamide moiety was meant to serve as direct precursor of the amide functionality in the target compound. However, the intended intramolecular transamidation on intermediate 114 could not be achieved, making various additional functional group interconversions necessary (Scheme 15b). (iii) This problem was addressed in the synthesis of 7-deoxypancratistatin (122, Scheme 15c), where the tosylaziridine was replaced by the corresponding carbomethoxy analogue 117, and cyanocuprate 118–lacking the amide moiety of 113–was used as electrophile.73 Ring-closure was achieved in fair yield (61%) by a Bischler–Napieralski-type cyclisation. (iv) The same method for closing ring B was employed in the synthesis of narciclasine (105, Scheme 15d), while the strategy for coupling the diol and piperonyl units was again changed:74 The presence of an additional bromine atom in the acetonide 123 made Suzuki coupling possible, avoiding the need for the rather unstable cyanocuprate. For the construction of 123, the authors returned to a nitroso-Diels–Alder approach, providing an elegant means of differentiating between the two vinylic bromide moieties of the TDO-derived cis-diol. (v) In the synthesis of 10b-epi-7-deoxypancratistatin the B ring was constructed via an aza-Payne rearrangement on aziridine 127 and Lewis-acid catalysed intramolecular epoxide ring-opening on the resulting tosylamide 130 (Scheme 15e).74b,75 Several derivatives of the natural Amaryllidaceae alkaloids have been prepared following variations of these synthetic strategies, including a carboline analogue,76 derivatives featuring various functional groups at C1,77 and others.74b,78
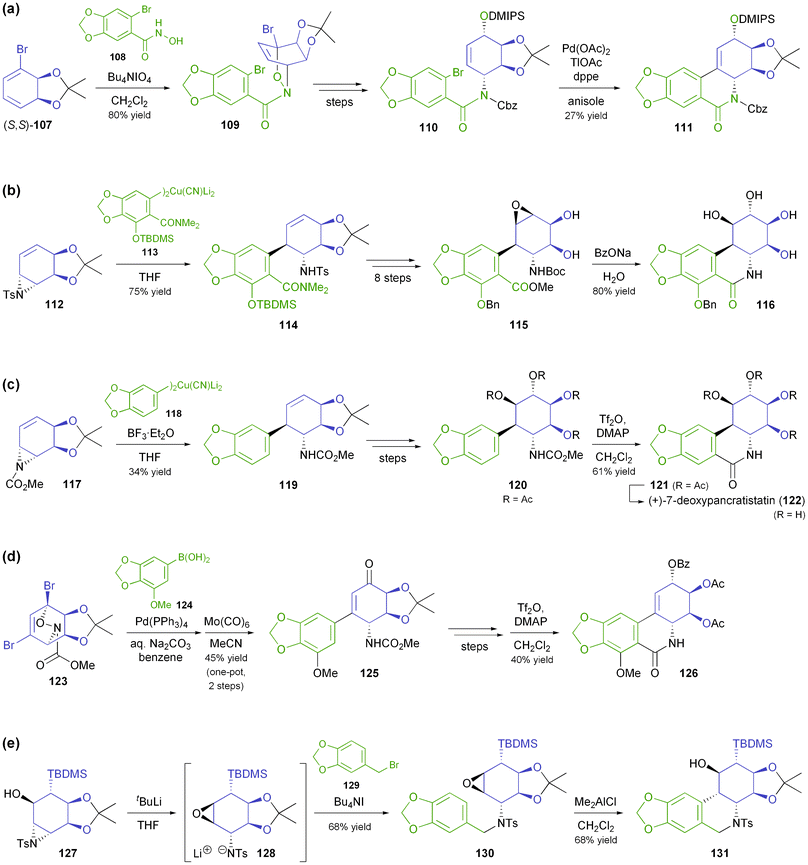 |
| | Scheme 15 Different strategies for construction of the B ring of isocarbostyril alkaloids: (a) nitroso-Diels–Alder reaction and Heck reaction, (b) aziridine ring-opening and intramolecular ester aminolysis, (c) aziridine ring-opening and Bischler–Napieralski cyclisation, (d) Suzuki cross-coupling and Bischler–Napieralski cyclisation, and (e) aza-Payne rearrangement/alkylation and intramolecular epoxide ring-opening. | |
One of the challenges when using arene cis-dihydrodiols as building blocks in asymmetric synthesis arises from the fact that the highly stereoselective TDO-catalysed dioxygenation only provides access to one enantiomer of the desired metabolite. As a consequence, enantiodivergent syntheses are much more difficult to realise by this approach than by hydrolase catalysis, where stereocomplementary enzymes are often available and reactions can be run in two ‘modes’ (acylation vs. hydrolysis). In the particular case of the Amaryllidaceae alkaloids, two strategies to overcome this problem, and thereby make the non-natural enantiomers available, have been developed. Hudlicky's research group has carried out the TDO-catalysed oxidation of para-dihalobenzenes 132a and 132b79 followed by reductive removal of the iodine atom, to obtain the (R,R)-diols 134a and 134b in 20% and 88% enantiomeric excess, respectively (Scheme 16).74b,80 In the first case, the optically enriched intermediate was converted into the conduramine A derivative 135, which was subjected to lipase-catalysed kinetic resolution, affording the alcohol 136 in 98% ee (Scheme 16). From this building block, (−)-7-deoxypancratistatin (ent-121) was synthesised in eight steps following the approach developed for the preparation of the natural alkaloid. The fluorodiol 134b, on the other hand, was not easily converted into 136, but instead gave the corresponding ketone, which can also serve as building block for alkaloid synthesis.
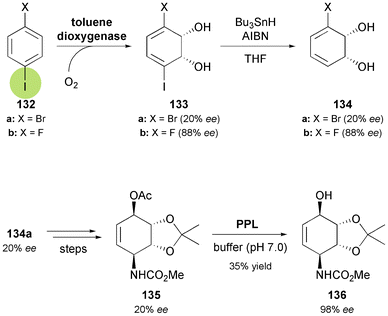 |
| | Scheme 16 Chemo-enzymatic preparation of (R,R)-3-halocyclohexadiene-1,2-diols 134via TDO-catalysed oxygenation of para-dihalobenzenes 132, and lipase-catalysed kinetic resolution of conduramine A derivative 135. | |
A completely different approach towards the ent-series of Amaryllidaceae alkaloids has been presented by Banwell and co-workers, who prepared non-natural (−)-lycoricidine (ent-106) from bromodiol (S,S)-137, the same compound used by Hudlicky for the preparation of the (+)-enantiomer.81 For the ent-series, the cis-vicinal diol moiety of the target compound can obviously not derive directly from the hydroxyl groups in 137. Instead, a diol of correct absolute configuration was established by an OsO4-catalysed dihydroxylation reaction on derivative 138. Further key transformations of the 10-step synthesis are an Overman rearrangement providing the amine moiety in 141, and a Suzuki coupling used to establish the B ring (Scheme 17). The syntheses of 3-epi-ent-lycoricidine, 4-deoxy-3-epi-ent-lycoricidine, and (−)-narciclasine (ent-104) following similar lines have also been reported.81,82
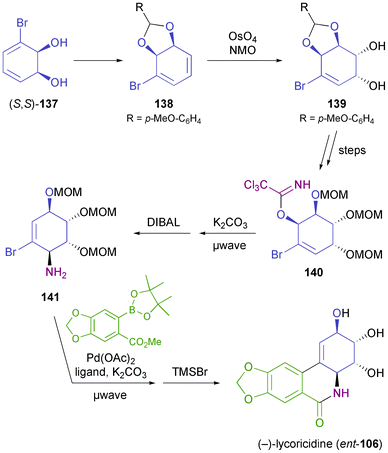 |
| | Scheme 17 Chemo-enzymatic synthesis of (−)-lycoricidine (ent-106) from TDO-derived diol (S,S)-137. | |
2.2.3 TDO in the synthesis of morphinan alkaloids. Considerable effort has also been devoted to the synthesis of morphinan alkaloids (Fig. 5) from arene cis-dihydrodiols.70a,83 Hudlicky's research group has first established a route to the complete morphinan core structure from two building blocks obtained with the help of biocatalytic dioxygenation:84 The oxidation of 143 by TDO afforded the diol 144, as the regio- and stereochemistry of the bioconversion is directed by the larger bromoethyl substituent. The diol was subsequently converted into compound 145. Furthermore, the incubation of bromobenzene 146 with an E. coli strain expressing both TDO and catechol dehydrogenase (CDH) yielded bromocatechol 147, which was transformed into the arene building block 148. A radical cyclisation strategy was then followed to arrive at a key intermediate 151, and finally the morphinan skeleton was established (in non-natural absolute configuration) by C10–C11 ring closure (Scheme 18a). Studies aimed at preparing the natural enantiomer from building block 153 using a related cascade radical cyclisation of intermediate 154 as key step met with limited success, as the cyclisation proceeded with low diastereoselectivity and afforded the epi-C14 isomer 155 as the main product (Scheme 18b).84b An alternative route based on an intramolecular Diels–Alder reaction was also explored but did not give satisfactory results either.85 An entry to the natural series of morphinan alkaloids was finally established by a Heck cyclisation approach (Scheme 18c), using the chemo-enzymatically prepared building blocks 148 and 153.86 A Heck-based enantiodivergent synthesis of both enantiomers of codeine (142), which uses mercury(II)-catalysed hydroamination for closing ring D (Scheme 18d), has also been reported recently.87 A further variation of this route has been employed in the synthesis of ent-neopinone (ent-42a), in which the D ring is closed by a 1,6-conjugate addition of the amine to C9.88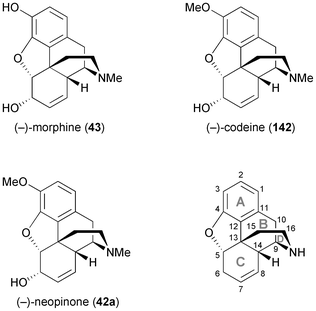 |
| | Fig. 5 Structures of morphine (43), codeine (142) and neopinone (42a), and general ring denotation and atom numbering conventions for morphinan alkaloids. | |
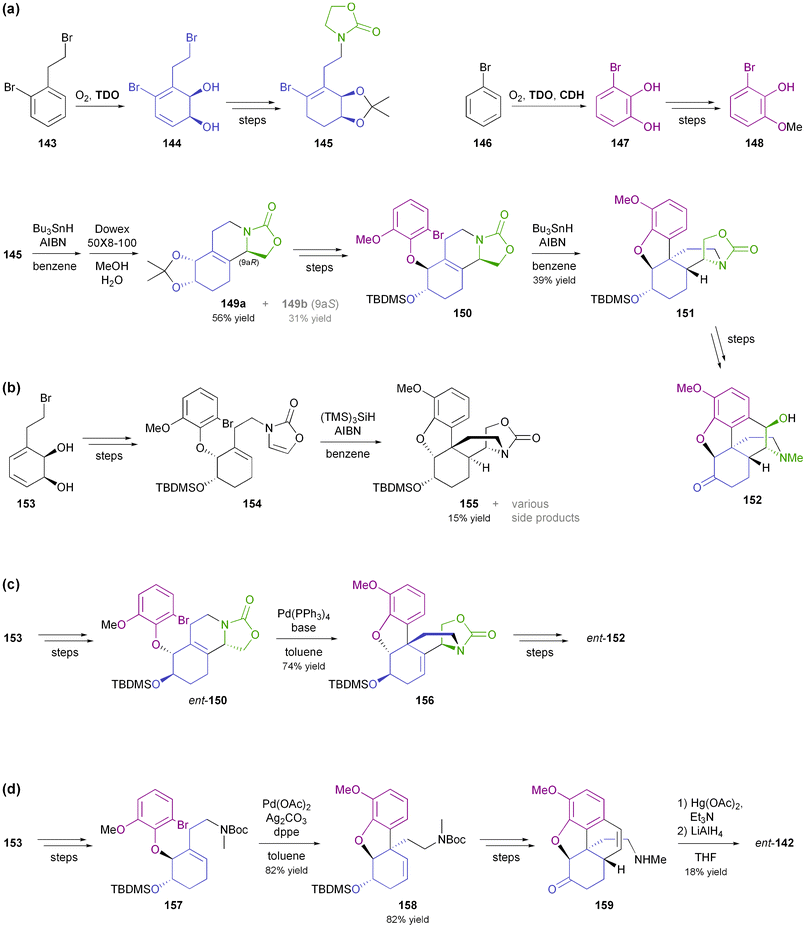 |
| | Scheme 18 Chemo-enzymatic synthesis of morphinan alkaloids: (a) preparation of building blocks 145 and 148 using biocatalytic dioxygenation, and synthesis of morphinan 152 from these building blocks using radical cyclisations, (b) radical cyclisation cascade affording compound 155, (c) synthesis of ent-152 from TDO-derived diol 153 by a Heck-cyclisation approach, and (d) synthesis of (+)-neopinone (ent-142) from TDO-derived diol 153via Heck-cyclisation and hydroamination as key steps. | |
2.2.4 Recent application of TDO in the synthesis of various plant alkaloids. In recent years, Banwell and co-workers have considerably broadened the application of arene cis-dihydrodiols in the synthesis of alkaloids.89 For instance, the authors have established a general route towards the montanine class of Amaryllidaceae alkaloids, which uses (S,S)-94 (Scheme 14) as starting material and employs a radical cyclisation as well as a Pictet–Spengler reaction for construction of the pentacyclic target compounds. Thus, the closely related alkaloids (+)-brunsvigine (160) and (+)-nangustine (non-natural enantiomer in both cases), and the structure 161 preliminarily assigned to (+)-montabuphine (Fig. 6) have been prepared.90 The last synthesis demonstrated that montabuphine is not identical to structure 161. Further synthetic studies on Amaryllidaceae alkaloids conducted by Banwell's group have resulted in the asymmetric preparation of (+)-amabiline (162) and the lycorine derivative 163 (Fig. 6) in 15 and 10 steps, respectively, from the TDO-derived building block (S,S)-137 (Scheme 21).91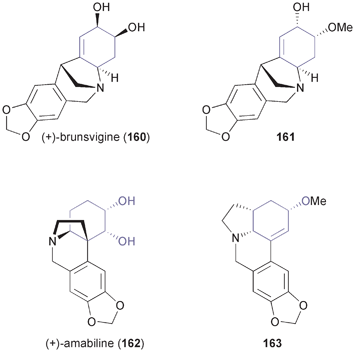 |
| | Fig. 6 Examples of Amaryllidaceae alkaloids synthesised from TDO-derived building blocks. | |
Banwell's group has also developed a chemo-enzymatic synthetic approach towards the lycorenine-type alkaloids (+)-clividine (169) and (−)-narseronine (172).92 Both syntheses employ Suzuki coupling as a key step and close ring D through the formation of the N–C11c bond, in the former case via a nitrogen-centred radical, in the latter by a conjugate addition/esterification cascade (Scheme 19). The structure assigned to the alkaloid nobilisitine A has been obtained in an analogous manner, and also in this case the spectral data indicated that the structural assignment was incorrect.93
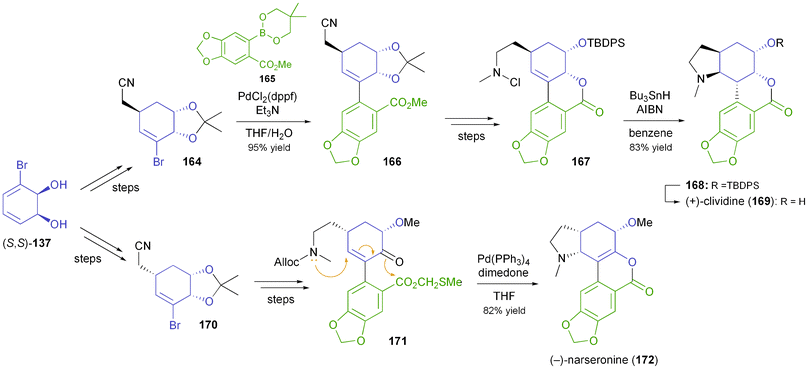 |
| | Scheme 19 Synthesis of lycorenine-type alkaloids (+)-clividine (169) and (−)-narseronine (172) from TDO-derived diol 137. | |
2.3 Baker's yeast
While chiral alcohols are frequently used as building blocks in the synthesis of alkaloids, biocatalytic asymmetric reduction of the corresponding ketones has only rarely been explored as a way to access them.94 A notable exception is the highly functionalised piperidinol 174 (Scheme 20) described by Momose and co-workers:95 Although this compound can be obtained by kinetic resolution of the racemate using lipase PS35, the authors found it more efficient to subject the corresponding ketone 173 to reduction by baker's yeast. This biotransformation afforded (2R,3S)-174 in 98% ee and 88% yield, as the chiral centre at C2 was dynamically resolved in the process. Several 2,6-disubstituted 3-piperidinols of all four possible diastereomeric forms have been prepared from 174 and used in the asymmetric synthesis of various alkaloids.95,96 For instance, the preparation of (−)-iso-6-cassine (cf.Scheme 7), clavepictines A [(−)-175a] and B [(+)-175b], and (−)-lepadin B (176) has been reported (Scheme 20).97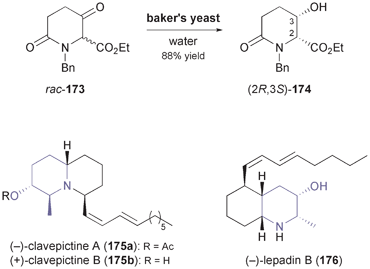 |
| | Scheme 20 Asymmetric reduction/dynamic kinetic resolution of rac-173 by bakers yeast affording building block 174, and examples of alkaloids prepared from this building block. | |
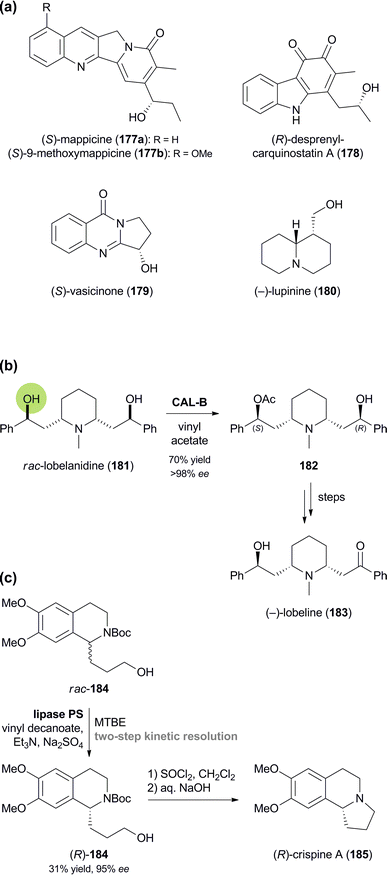 |
| | Scheme 21 Lipase-catalysed kinetic resolution and desymmetrisation of alkaloids: (a) examples of alkaloids obtained by kinetic resolution, (b) desymmetrisation of lobelanidine (181) and conversion into (−)-lobeline (183), and (c) kinetic resolution of alcohol 184 and conversion into (R)-crispine A (185). | |
3. Biocatalytic kinetic resolution, desymmetrisation, and deracemisation of alkaloids
The enormous flexibility of the ‘chiral building block’ approach has enabled the chemo-enzymatic synthesis of alkaloids of remarkable structural complexity and diversity, often even in both enantiomeric forms. However, as chirality is introduced early in the synthetic sequence, an enantiodivergent synthesis requires several identical steps to be carried out independently for each enantiomer. In some cases it would clearly be more efficient to prepare the racemic alkaloid (or a late synthetic intermediate) and subject it to kinetic resolution. On the other hand, even if only one enantiomer of an alkaloid is to be synthesised, preparation of the racemate followed by deracemisation might be an attractive option. Biocatalysis offers possibilities for both these approaches; however, the sheer size and functional complexity of alkaloids make them challenging substrates even for enzymes, and hence examples are limited in number.3.1 Lipase-catalysed kinetic resolution and desymmetrisation
Amongst the largest alkaloids that have been kinetically resolved are mappicine (177a) and 9-methoxymappicine (177b, Scheme 21a), two minor constituents of Nothapodytes foetida, which can be obtained in racemic form from the more abundant camptothecin. Kinetic resolution of the side-chain alcohol in 177a and 177b has been achieved by enantioselective hydrolysis of the corresponding acetates with baker's yeast or lipase PS35 (with enantioselectivities E ranging from 31 to >200), and by enantioselective acetylation with Candida cylindracea lipase (177a: E > 200, 177b: E = 146).98 Similarly, a side-chain secondary alcohol has been resolved in the chemo-enzymatic preparation of the quinoid indole alkaloid (R)-(−)-desprenylcarquinostatin A (178, Scheme 21a):99 In this case, a late synthetic intermediate of 178 was subjected to kinetic resolution by commercial lipase QLM,35 which proceeded with excellent enantioselectivity (E > 200) to afford the (R)-acetate and the (S)-alcohol in 98% and 97% ee, respectively. The synthesis was completed via oxidation to the quinone and, for the natural (R)-enantiomer, hydrolysis of the acetate. Lipase catalysis has also been employed in the kinetic resolution of vasicinone (179; lipase PS35, E > 200), sedamine (69, Fig. 2; porcine pancreatic lipase, E = 35), as well as lupinine (180; lipase AK,35E = 24),100 and in the desymmetrisation of lobelanidine (181) with CAL-B, which afforded the (S)-monoacetate 182 in 70% yield and >98% ee (Scheme 21b).101 The latter compound was transformed into (−)-lobeline (183), a natural product with anti-addictive properties, by oxidation and ester hydrolysis.A remarkable case of remote stereocentre discrimination in lipase-catalysed kinetic resolution has been reported by Fülöp and co-workers:102 The authors prepared both enantiomers of the tetrahydroisoquinoline alkaloid crispine A (185) via the open-chain analogue 184, featuring a primary alcohol in δ-position of the chiral centre (Scheme 21c). Decanoylation of rac-184 was found to be catalysed by lipase PS35 with surprisingly high enantioselectivity (E = 68), and by employing a two-step kinetic resolution protocol both the (S)-ester and the (R)-alcohol could be obtained in good optical purity (ee = 94% and 95%, respectively). Hydrolytic kinetic resolution of the racemic decanoate was also possible, but proceeded with somewhat lower enantioselectivity (E = 52). Conversion of the enantiomerically enriched alcohol 184 into crispine A was accomplished in a single step by treatment with thionyl chloride.
3.2 Monoamine oxidase in chemo-enzymatic deracemisation systems
Monoamine oxidases (MAOs) are flavin-dependent enzymes catalysing the aerobic oxidation of amines to the corresponding imines or iminium ions. The one-pot combination of this reaction with chemical reduction of the imine leads–over several cycles of enantioselective oxidation and non-stereoselective reduction–to accumulation of the slower reacting amine enantiomer (Scheme 22). Over the last ten years, Nicholas Turner's research group has developed several highly enantioselective variants of MAO from Aspergillus niger (MAO-N), which have been engineered for the deracemisation of primary, secondary, and tertiary amines.103 Recent variants featuring a comparably broad substrate scope are interesting biocatalysts in the context of alkaloid synthesis,104 as has first been demonstrated by stereoinversion of (S)-nicotine (188) to the non-natural (R)-enantiomer by MAO-N-5 (Scheme 22).105 The same enzyme variant has also been found capable of deracemising crispine A (185, Scheme 21c), although in this case the reaction proceeded rather slowly.106 Further optimisation of the biocatalyst through directed evolution has led to the identification of a variant termed MAO-N-9, which displays exceptionally high activity (>5.5 kU/mg) towards crispine A.107 Very recent studies have demonstrated the broad applicability of MAO-N in the asymmetric synthesis of alkaloids:108 Variant MAO-N-9 was applied in the chemo-enzymatic preparation of the indole alkaloids (R)-eleagnine (189) and (R)-harmicine (190), while MAO-N-5 was used in the deracemisation of the hemlock neurotoxin coniine (72). In addition, a variant with opposite enantioselectivity (MAO-N-11) has been developed and applied in the synthesis of (S)-1-phenyltetrahydroisoquinoline (191), a building block for the urinary antispasmodic drug solifenacin.108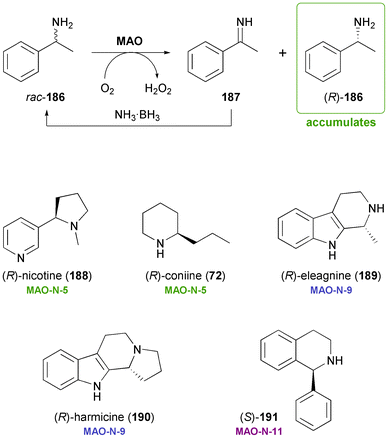 |
| | Scheme 22 Chemo-enzymatic deracemisation of amines via MAO-catalysed oxidation illustrated with 1-phenylethylamine 186, and examples of alkaloids that have been obtained employing engineered variants of MAO-N. | |
3.3 Oxidative kinetic resolution of benzylisoquinolines by berberine bridge enzyme
The berberine bridge enzyme (BBE, EC 1.21.3.3) is a bicovalent flavoenzyme found in various alkaloid-producing plants, most notably those of the Papaveraceae (poppy) and Fumariaceae (fumitory) families.109 It is a branch-point enzyme in benzylisoquinoline alkaloid biosynthesis and catalyses an extraordinary intramolecular oxidative carbon-carbon bond formation: At the expense of molecular oxygen as terminal electron acceptor, the natural substrate, (S)-reticuline (192), is oxidatively cyclised to give (S)-scoulerine (193) as sole product (Scheme 23)–a reaction that lacks any direct counterpart in synthetic chemistry. Studies on the mechanism110 and the structure110a,111 of BBE have provided deeper insight in the nature of this remarkable reaction, and the development of an efficient heterologous expression system (secretory expression in Pichia pastoris)112 enabled the production of the pure enzyme in amounts sufficient for detailed biocatalytic investigations.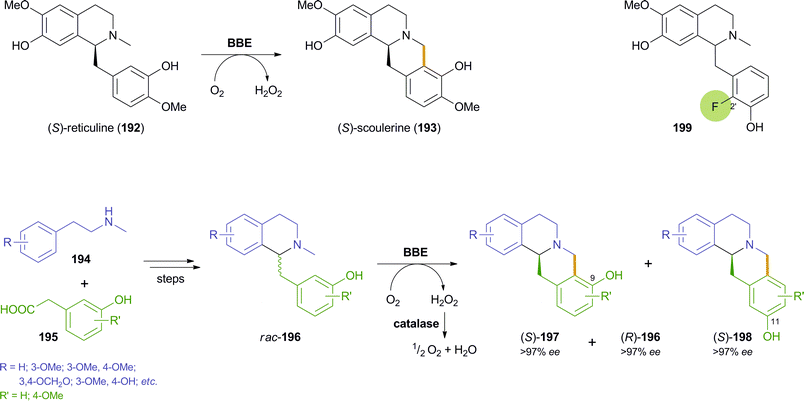 |
| | Scheme 23 Oxidative cyclisation of (S)-reticuline 192 catalysed by BBE, application of BBE in the chemo-enzymatic asymmetric synthesis of (S)-berbines 197 and (R)-benzylisoquinolines 196via kinetic resolution, and example of a substrate 199 that is cyclised to an 11-hydroxyberbine by BBE. | |
Optimisation studies aimed at finding suitable reaction conditions for the conversion of non-natural benzylisoquinoline derivatives showed that BBE is a fairly stable biocatalyst able to work in the presence of various organic solvents and at relatively high substrate concentrations.113 Most importantly, however, BBE has been shown to convert several non-natural racemic substrates rac-196–bearing varied substituents on both aromatic rings–with excellent enantioselectivity (E > 200), thus affording both the berbine-type cyclisation products (S)-197 and the remaining benzylisoquinolines (R)-196 in optically pure form (ee > 97%; Scheme 23).114 In addition, the reactions proceeded with high selectivity towards the 9-hydroxyberbines 197, while the regioisomeric products 198 were only formed in minor amounts (ratio 197/198 ≥ 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14). Preparative transformations on 0.5 gram scale demonstrated the applicability of the BBE-catalysed kinetic resolution in the chemo-enzymatic synthesis of various benzylisoquinoline and berbine alkaloids, including the sedative and muscle-relaxing agent (S)-scoulerine (193), which was obtained in 7.4% yield over 9 steps.114b
:14). Preparative transformations on 0.5 gram scale demonstrated the applicability of the BBE-catalysed kinetic resolution in the chemo-enzymatic synthesis of various benzylisoquinoline and berbine alkaloids, including the sedative and muscle-relaxing agent (S)-scoulerine (193), which was obtained in 7.4% yield over 9 steps.114b
The regiochemistry of the cyclisation was investigated in more detail in a follow-up study, which showed that the ratio of products 197 and 198 is influenced by the substitution pattern of the substrate, the pH of the reaction medium and the type and amount of co-solvents used. A complete switch in regioselectivity could be achieved by using substrates like compound 199, in which the usual site of C–C coupling, position 2′, is blocked by a fluorine atom. Preparative-scale biotransformations of 199 and two related derivatives gave good to excellent yields (32–50%) of the (S)-11-hydroxyberbines and the remaining (R)-benzylisoquinolines, and even though the regioselectivity of the reaction had been changed, the enantioselectivity was fully conserved (ee > 97% for all compounds).115
3.4 Kinetic resolution of berbines by whole cells
Another extraordinary example for a biocatalytic kinetic resolution of alkaloids has recently been reported by Ge et al.:116 The authors describe the kinetic resolution of tetrahydroberberrubine (rac-200) via kinetic glycosylation and enantioselective sulfation, both catalysed by whole cells of the fungus Gliocladium deliquescens NRRL 1086 (Scheme 24). Fermentation of 200 with the fungal culture in glucose-containing medium resulted in the formation of the diastereomeric β-glucosides (14S)- and (14R)-201 (ratio 15:1), which could be separated by chromatography.117 Following the time course of the fermentation revealed that the (14S)-product was formed much faster than the (14R)-isomer, and that upon long-term incubation (120 h) the latter was converted further into the sulfate ester (R)-202. This sulfation reaction proved more selective than the glycosylation, as (14S)-201 was not converted at all. Sulfate ester (R)-202 was hydrolysed using commercially available sulfatase type H1 to give (R)-200 in 43% overall yield and >99% ee. Likewise, the (14S)-glycoside 201 was subjected to acid-catalysed hydrolysis to afford (S)-200 in 48% yield, also maintaining an ee of >99%. Testing the method on five closely related berbine alkaloids showed that the glycosylation only takes place when the hydroxyl group of the substrate is located in position 9 of the berbine scaffold.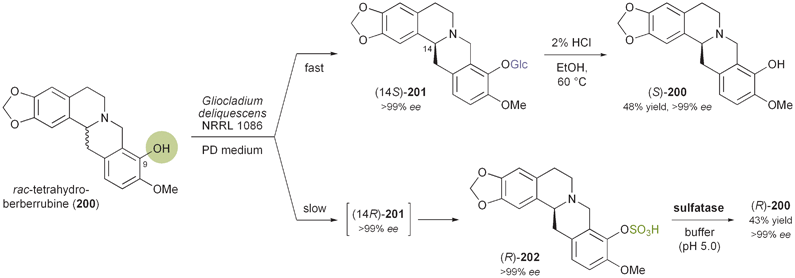 |
| | Scheme 24 Kinetic resolution of tetrahydroberberrubine (rac-200) via kinetic glycosylation and enantioselective sulfation catalysed by Gliocladium deliquescens, and subsequent hydrolysis. | |
4. Biocatalytic asymmetric key C–C and C–N bond formation
In the chemo-enzymatic syntheses discussed in the previous sections, the biocatalytic step appears either relatively early or at the very end of a longer sequence. However, enzymes can also take a more central role in alkaloid synthesis by catalysing key bond forming reactions in a highly stereoselective fashion. This strategic application of biocatalysis for chemically challenging reactions does not only open up novel routes towards the target compounds, it also often reduces the requirement for protective-group manipulations and thus leads to shorter and more practical syntheses.4.1 Aldolases
The synthesis of iminocyclitols (‘aza-sugars’) provides some excellent examples of the use of enzymes in key C–C bond forming reactions. Early contributions to the field have come from the research groups of Chi-Huey Wong and of Franz Effenberger, who employed D-fructose-1,6-diphosphate aldolase (FDPA) for highly stereoselective aldol reactions en route to (−)-D-1-deoxymannojirimycin (103, Fig. 3), (+)-D-1-deoxynojirimycin (206), and (+)-D-fagomine (207, Scheme 25).118 In these chemo-enzymatic syntheses, the aldolase catalyses the coupling of dihydroxyacetone phosphate (DHAP) with an aldehyde featuring an azide or Cbz-amine substituent. Upon catalytic hydrogenation of the dephosphorylated linear aldol product, a free amine is formed and the iminocylitol ring is closed by intramolecular reductive amination. The target compounds were obtained in overall yields ranging from 30% to 80%. Aza-analogues of N-acetylglucosamine and N-acetylmannosamine, e.g.208 (Scheme 25), as well as derivatives featuring a phosphonic acid moiety or coupled to guanosine diphosphate have been prepared in a similar manner.119 Epimeric products have been synthesised using DHAP-dependent aldolases with different stereoselectivity, e.g.L-fuculose-1-phosphate aldolase (FucA) or L-rhamnulose-1-phosphate aldolase (RhuA).120 In addition, the scope of accessible products has been extended considerably by incorporating an enzyme-catalysed isomerisation of the linear sugar analogues (using e.g. glucose isomerase or fuculose isomerase) into the synthetic sequence. Thus, unusual 7-membembered iminocyclitols such as 209 have been prepared.121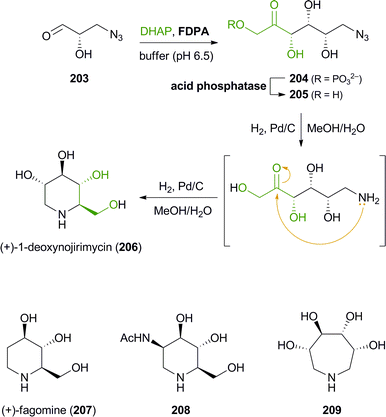 |
| | Scheme 25 Chemo-enzymatic synthesis of (+)-1-deoxynojirimycin (206) via aldolase catalysis and intramolecular reductive amination, and examples of further iminocyclitols synthesised in a similar manner. | |
In contrast to azidoaldehydes such as 203, Cbz-protected amino aldehydes are rather poor substrates for DHAP-dependent aldolases, a fact that has been attributed to their low aqueous solubility. Espelt et al. have demonstrated that the use of emulsion systems can alleviate this problem and can thus lead to higher yields of the desired products.122 A more serious practical limitation of the above-mentioned aldolases is the requirement for expensive and unstable DHAP as a donor substrate. Although it has been demonstrated that in situ formation of DHAP from cheap precursors is possible,123 it would clearly be advantageous if the involvement of DHAP could be avoided completely. This is indeed possible using D-fructose-6-phosphate aldolase (FSA) from E. coli, an enzyme that is structurally related to transaldolases and accepts dihydroxyacetone and other non-phosphorylated ketones as donor substrates.124 Using this biocatalyst, the efficiency of chemo-enzymatic iminocyclitol synthesis has been considerably improved over the FDPA-based processes (D-fagomine, for instance, has been obtained in 51–65% isolated yield from dihydroxyacetone within two synthetic steps) and novel aza-sugars derived from hydroxyacetone and 1-hydroxy-2-butanone have become accessible.125
Besides the preparation of iminocyclitols, aldolases have also found application in the asymmetric synthesis of polyhydroxylated pyrrolizidine alkaloids: Romero & Wong have employed FDPA in the syntheses of (−)-7-epi-alexine (214), (+)-3-epi-australine (216), and (+)-australine (217, Scheme 26a).126 DHAP served as aldol donor in the biotransformation, while the aldol acceptor was the complex aldehyde 211, generated in situ by oxidative cleavage of the corresponding vicinal diol 210 with NaIO4 in water. Divergence between australine and 3-epi-australine was achieved through choice of conditions in the final intramolecular reductive amination step: Catalytic hydrogenation afforded 216 as a single stereoisomer, while the use of sodium cyanoborohydride led to the formation of australine (217; albeit in an 8:1 mixture with 216). The synthesis of structurally related pyrrolizidines, which represent stereoisomers of the naturally occurring alkaloids hyacinthacine A1 and A2, has recently been reported by Clapés and co-workers.127 In this case, Cbz-protected prolinal 218 served as acceptor substrate, contributing one ‘half’ of the pyrrolizidine core. Coupling of the prolinal derivative with DHAP catalysed by L-rhamnulose-1-phosphate aldolase (RhuA) from E. coli proceeded with excellent diastereoselectivity, but the following catalytic hydrogenation afforded mixtures of C3-epimeric products.128 Fortunately, the diastereomers proved separable by ion-exchange chromatography, allowing the isolation of the four hyacinthacine stereoisomers 220, 221, 223 and 224 in 9–23% yield from 218 (Scheme 26b). In a later study, further isomers and some closely related pyrrolizidines were prepared in the same manner, using an engineered variant of L-fuculose-1-phosphate aldolase (FucA) as biocatalyst.129 Finally, the concept has also been extended to indolizidines and quinolizidines, obtained via FucA- or RhuA-catalysis from 2-piperidyl-substituted aldehydes.130
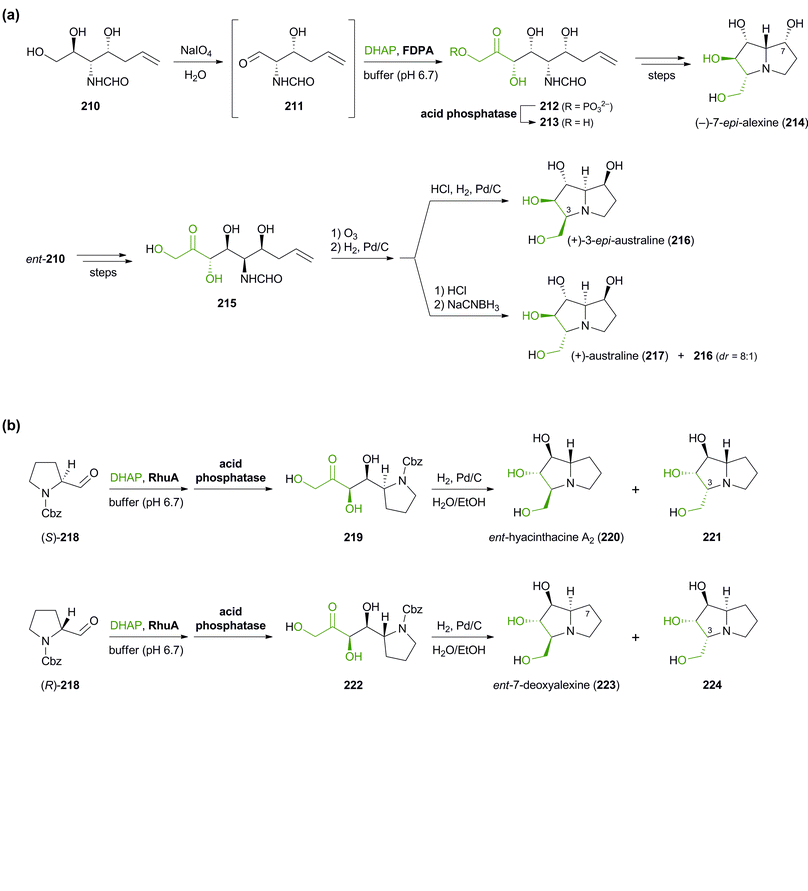 |
| | Scheme 26 Aldolase-catalysed C–C bond formation in the synthesis of pyrrolizidine alkaloids: (a) chemo-enzymatic preparation of (−)-7-epi-alexine (214), (+)-3-epi-australine (216), and (+)-australine (217) using D-fructose-1,6-diphosphate aldolase (FDPA), and (b) chemo-enzymatic preparation of hyacinthacine isomers 220, 221, 223 and 224 using L-rhamnulose-1-phosphate aldolase (RhuA). | |
4.2 Transaminases
Recent years have witnessed the ‘coming of age’ of ω-transaminases, which have been intensively studied as biocatalysts for the asymmetric synthesis or kinetic resolution of a wide range of chiral amines,103b,131 and which have also found industrial application.132 The prospect of gaining direct access to chiral primary amines from the corresponding ketones in high enantiomeric excess makes ω-transaminases also promising biocatalysts for the asymmetric synthesis of alkaloids, but their potential in this respect has only been started to be explored. Kroutil and co-workers have recently reported an application of ω-transaminases in the synthesis of piperidine alkaloids:133 The authors demonstrated that the reductive amination of δ-diketones 226 catalysed by various ω-transaminases proceeds exclusively at the less hindered carbonyl group (only in one case were minor amounts of the regioisomeric product detected). The amino ketones thus formed cyclised spontaneously to the corresponding cyclic imines, which upon catalytic hydrogenation afforded cis-2,6-disubstituted piperidines as single stereoisomers (Scheme 27). Since both (R)- and (S)-selective ω-transaminases could be employed in this chemo-enzymatic sequence, it provided access to both enantiomers of the final products. Thus, natural (+)-(2R,6S)-dihydropinidine (48) and its non-natural enantiomer were obtained in 65% and 72% overall yield, respectively, and only three steps from pyranone 225. In a recent follow-up study, the authors have also succeeded in establishing a trans-selective reduction protocol (LiAlH4 reduction at low temperature in the presence of Et3Al as a Lewis acid), which provided epi-dihydropinidine (ent-73) in good, but not perfect diastereoselectivity (dr = 18:82 syn/anti).134 Furthermore, the transamination/hydrogenation sequence has recently been applied in the asymmetric synthesis of the fire ant venom alkaloid isosolenopsin, a longer-chain analogue of dihydropinidine.135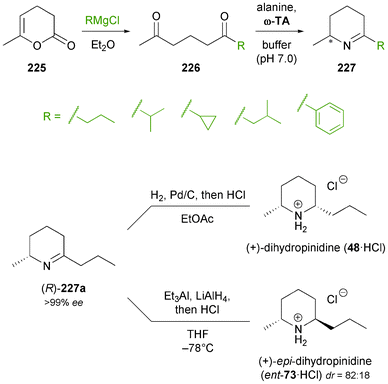 |
| | Scheme 27 Regioselective asymmetric mono-amination of 1,5-diketones 226 catalysed by ω-transaminases, and its application to the chemo-enzymatic synthesis of dihydropinidine (48) and epi-dihydropinidin (ent-73). | |
A combination of enzymatic transamination and subsequent imine reduction has also been used in the synthesis of the 1,3-disubstituted tetrahydroisoquinoline 231 and a second, structurally related compound (Scheme 28).136 Closure of the tetrahydroisoquinoline ring was achieved by a Bischler–Napieralski cyclisation, and catalytic hydrogenation again proceeded with perfect diastereoselectivity, affording (1S,3R)-231 in 30% overall yield over six steps from ketone 228.
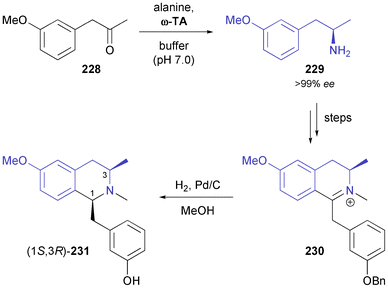 |
| | Scheme 28 Synthesis of isoquinoline alkaloid 231via a combination of enzymatic transamination and catalytic imine hydrogenation. | |
4.3 Pictet–Spenglerases
In recent years, two C–C bond-forming enzymes involved in alkaloid biosynthesis have attracted increased attention from biocatalysis researchers: norcoclaurine synthase (NCS, EC 4.2.1.78) and strictosidine synthase (STR, EC 4.3.3.2) both catalyse asymmetric Pictet–Spengler reactions and are hence referred to as ‘Pictet–Spenglerases’.137 As such, they both join two relatively simple molecules–a 2-arylethylamine and an aldehyde–to form a nitrogen heterocycle with excellent stereo-chemical control of the resulting chiral centre (Scheme 29). Since asymmetric Pictet–Spengler reactions are still difficult to achieve with chemo-catalytic methods,137 NCS and STR offer the chance to establish novel, otherwise non-viable synthetic routes towards benzylisoquinoline and indole alkaloids.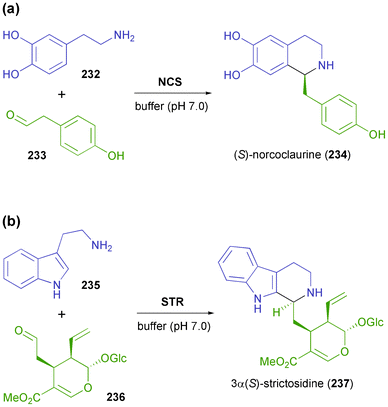 |
| | Scheme 29 The natural reactions of (a) norcoclaurine synthase and (b) strictosidine synthase, which involve C–N bond formation and stereoselective C–C bond formation through an asymmetric Pictet–Spengler reaction. | |
4.3.1 Norcoclaurine synthase. Norcoclaurine synthase is a plant enzyme that catalyses the first committed step in the synthesis of benzylisoquinoline alkaloids–the condensation of dopamine (232) and 4-hydroxyphenylacetaldehyde (233) forming (S)-norcoclaurine (234) as product (Scheme 29a).138 The enzyme has first been isolated from plant tissue and partially characterised in 2001,139 and soon afterwards the cloning and heterologous expression in E. coli have been reported.140 Mechanistic studies141 and functional analysis142 followed, and soon the potential of NCS as a biocatalyst for asymmetric synthesis was recognised: Bonamore et al. have described the one-pot chemo-enzymatic synthesis of (S)-norcoclaurine (234) using NCS from Thalictrum flavum and in situ generation of 233via NaOCl-oxidation of tyrosine (238, Scheme 30a).143 The two steps were carried out sequentially in phosphate buffer as reaction medium, and (S)-234 was obtained in 81% yield and 93% ee. Later investigations by Pesnot et al. revealed that the choice of the right reaction setup is crucial for stereoselectivity, since a background activity of phosphate buffer was detected.144 The same authors have developed a fluorescamine-based screening assay which was used to elucidate the substrate spectrum of NCS from Coptis japonica.145 Variations of the substitution patterns of both reaction partners were tested, revealing a rather limited scope of amine derivatives, while more than 10 structurally diverse aldehydes were accepted. Preparative biotransformations afforded (S)-tetrahydroisoquinolines, including the non-natural compounds 239–242 (Scheme 30b), in good to excellent yield (56–99%) and with an excellent ee of >95%. Interestingly, using heptanal as substrate gave rise to both the expected para-coupling product (S)-240 and the rather unusual ortho-isomer 241.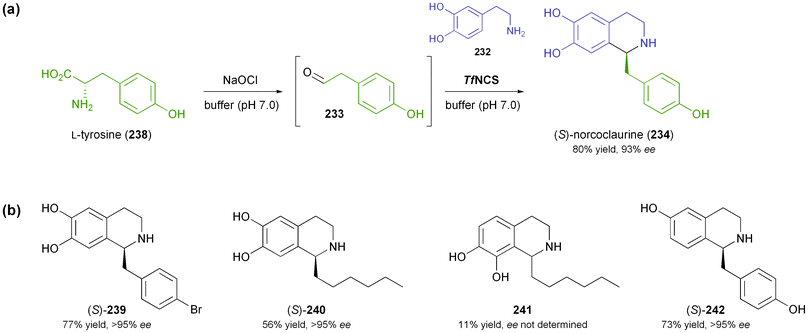 |
| | Scheme 30 Application of norcoclaurine synthase in the asymmetric synthesis of tetrahydroisoquinolines: (a) one-pot chemo-enzymatic synthesis of (S)-norcoclaurine (234) using NCS from Thalictrum flavum, (b) non-natural tetrahydroisoquinolines 239–242 prepared using NCS from Coptis japonica. | |
The substrate scope of NCS from Thalictrum flavum has also been investigated recently, focusing on variation of the aldehyde.146 Fifteen non-natural derivatives were found to be accepted, but unfortunately the enantiomeric purity of the reaction products was not investigated.
4.3.2 Strictosidine synthase. The second Pictet–Spenglerase with application in biocatalysis is strictosidine synthase. In fact, STR was the first Pictet–Spenglerase to be found in nature, isolated and characterised. Its natural reaction is the condensation of tryptamine (235) and the monoterpenoid aldehyde secologanin (236) to form 3α(S)-strictosidine (237, Scheme 29b), representing the first committed step in the synthesis of monoterpenoid indole alkaloids.137 The first preparative application of this biotransformation dates back to 1982, when Pfitzner & Zenk immobilised a partially purified STR obtained from Catharanthus roseus cell cultures on CNBr-activated sepharose and used the immobilised enzyme for the gram-scale synthesis of 3α(S)-strictosidine (237).147More recently, the STRs from Catharanthus roseus, Rauvolfia serpentina and Ophiorrhiza pumila have been heterologously expressed and studied with respect to their substrate scope and their potential as biocatalysts, and the X-ray crystal structure of the Rauvolfia enzyme has been solved.137,148 In contrast to NCS, strictosidine synthases have generally been found to accept a broader range of amines, including the 1-oxa and 1-thia analogues of tryptamine, 7-azatryptamine (243), and various methylated or fluorinated derivatives.137,148d,149 The scope of aldehyde substrates was shown to be limited to secologanin (236) and some close analogues thereof for the two highly homologous STRs from Catharanthus and Rauvolfia.149,150 The Ophiorrhiza enzyme, on the other hand, accepts several simple aldehydes, e.g. hexanal or phenylacetaldehyde, as substrates, forming the corresponding tryptoline (tetrahydro-β-carboline) products in >98% ee.151 However, the observed rate constants (kobs) were in the low ‘per hour’-range, and the Km-values for the non-natural aldehydes were estimated to lie beyond the solubility limits of the investigated compounds. Because of these limitations, preparative-scale applications of strictosidine synthase have thus far been restricted to using secologanin (236) as aldehyde substrate. Nevertheless, structurally diverse products can be accessed by using chemo-enzymatic approaches, as has recently been demonstrated by Stöckigt, Yu, and co-workers:152 The authors describe the use of His-tagged Rauvolfia STR immobilised on a nickel-NTA column for the production of strictosidine (237) and its 12-aza-analogue 244, both of which were converted further into related indole alkaloids. For instance, treatment with 2 M sulfuric acid resulted in the formation of (+)-nacycline (245) and (+)-12-azanacycline (246), while enzymatic deglycosylation153 followed by NaBH4 reduction provided (−)-tetrahydroalstonine (247) and the corresponding 12-aza-compound 248 (Scheme 31a). Overall yields of 18–60% (from 235/243) were attained for these products.
![Biocatalytic application of strictosidine synthase: (a) chemo-enzymatic synthesis of (+)-nacycline (245), (−)-tetrahydroalstonine (247), and their aza-analogues, and (b) preparation of piperazino[1,2-a]indole alkaloids 250 using indole-1-ethanamine derivatives 249 as substrates.](/image/article/2013/RA/c3ra42123f/c3ra42123f-s31.gif) |
| | Scheme 31 Biocatalytic application of strictosidine synthase: (a) chemo-enzymatic synthesis of (+)-nacycline (245), (−)-tetrahydroalstonine (247), and their aza-analogues, and (b) preparation of piperazino[1,2-a]indole alkaloids 250 using indole-1-ethanamine derivatives 249 as substrates. | |
Very recently, it has been discovered that Rauvolfia STR can also form heterocycles with an entirely different skeletal structure, as it can couple secologanin (236) with indole-1-ethanamine derivatives 249 to give piperazino[1,2-a]indole alkaloids 250 (Scheme 31b).154 Three products were isolated from small preparative-scale transformations (50 μmol substrate converted) and characterised by NMR, but no yields were reported. Still, this study demonstrates that also non-tryptoline alkaloids can be synthesised with the help of strictosidine synthase. The range of accessible products may be further expanded by using engineered STR variants,155 as has already been shown by studies in planta.156
5. Conclusions and outlook
Over the last decades, chemo-enzymatic alkaloid synthesis has developed into a prospering field of research that complements the established options for alkaloid production–isolation, semi-synthesis and total organic synthesis. For many years, research in the field has been dominated by chiral building block approaches relying mainly on lipases, esterases and toluene dioxygenase as biocatalysts, and due to its flexibility this research branch is still well represented today. However, the role of biocatalysis in the asymmetric synthesis of alkaloids has also been significantly extended in recent years: Kinetic resolution and deracemisation of entire alkaloids, as well as biocatalytic key C–C bond formation have gained importance (Fig. 7), and the scope of used enzymes has become a lot broader. An important factor in these developments is the exploitation of enzymes involved in alkaloid biosynthesis, such as norcoclaurine synthase, strictosidine synthase, and berberine bridge enzyme, which are able to carry out asymmetric transformations that are not yet possible by purely chemical means. Indeed, one third of the reports on chemo-enzymatic alkaloid syntheses published in the last three years (11 out of 33, 2010–2012) use enzymes from plant secondary metabolism.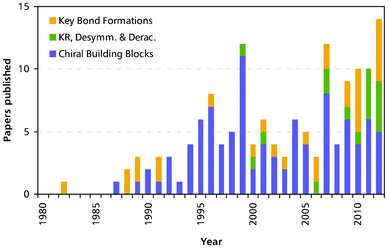 |
| | Fig. 7 Chemo-enzymatic total syntheses of alkaloids published per year. The colours indicate the different chemo-enzymatic approaches as discussed in this review: (a) biocatalytic preparation of chiral building blocks (blue), (b) biocatalytic kinetic resolution, desymmetrisation, and deracemisation of alkaloids (green), and (c) chemo-enzymatic syntheses that use enzymes for key asymmetric C–C and/or C–N bond formation (orange). | |
Furthermore, the development of novel chemo-enzymatic strategies for alkaloid synthesis, in which biocatalysis takes a more central role, has led to shorter routes requiring less protective group chemistry than the classical building block approaches. This fact can be illustrated by comparing the ω-transaminase-based preparation of (+)-dihydropinidine (48, Scheme 27) recently reported by Simon et al. with earlier chemo-enzymatic syntheses of the same compound (Table 2). The transaminase route is not only the shortest one by far, it also gives a more than three times higher yield than the second-best alternative. In addition, this synthesis also compares favourably with purely chemical asymmetric syntheses of dihydropinidine.133
Table 2 Comparison of different chemo-enzymatic syntheses of (+)-dihydropinidine (48)
| Article | Starting material | Steps | Biocatalytic step | Overall yield | Ref. |
|---|
| Since no yields are reported for the last steps, an exact value can not be calculated. |
|---|
| Momose et al. 1992 |  | 24 | lipase-catalysed desymmetrisation | <1%a | 45a |
| Chênevert & Dickman 1996 | 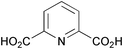 | 14 | lipase-catalysed desymmetrisation | 18% | 48b |
| Yamauchi et al. 2004 | 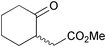 | 17 | baker's yeast reduction | 0.7% | 159 |
| Simon et al. 2012 | 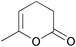 | 3 | transaminase-catalysed reductive amination | 65% | 133 |
The picture is similar when comparing different methods (Table 3) for the production of D-fagomine (207, Scheme 25), an iminosugar first isolated from buckwheat seeds in 1974,157 which is being discussed as a functional food ingredient due to its hypoglycaemic properties.158 Extraction of the alkaloid from natural sources gives only limited yields and requires at least three purification steps. State-of-the-art organic asymmetric syntheses provide 207 in up to 34% yield and require 6–9 steps; however, it must be taken into account that the reported ‘starting materials’ are not commercially available and therefore also need to be synthesised. The chemo-enzymatic route based on an asymmetric aldol reaction catalysed by D-fructose-6-phosphate aldolase (FSA) affords the target compound in only two steps and 51–65% yield from dihydroxyacetone and a Cbz-protected aminoaldehyde, which in turn can be easily prepared in 69% yield from 3-amino-1-propanol.
Table 3 Comparison of different production methods for D-fagomine (207)
| Article | Starting material | Steps | Key steps | Overall yield | Ref. |
|---|
| Purification steps. |
|---|
| Kato et al. 1997 | leaves and roots of Xanthocercis zambesiaca | 3a | extraction (50% aq. MeOH), ion exchange chromatography | leaves: 0.03% | 160 |
| roots: 0.13% |
| | | | | | |
| Asano et al. 2001 | leaves of Morus alba | 4a | extraction (50% aq. EtOH), ion exchange chromatography | 0.02% | 161 |
| | | | | | |
| Kato et al. 2003 | seeds of Castanospermum australis | 3a | extraction (50% aq. MeOH), ion exchange chromatography | 0.04% | 162 |
| | | | | | |
| Banba et al. 2001 | 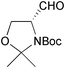 | 6 | ring-closing metathesis, epoxidation, epoxide hydrolysis | 12% | 163 |
| Takahata et al. 2003 |
| Kumari et al. 2009 | 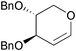 | 7 | chloroamidation, intramolecular reductive amination | 34% | 164 |
| Kim et al. 2011 | 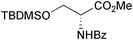 | 9 | diastereoselective ketone reduction, Pd-catalysed intramolecular allylic substitution | 19% | 165 |
| Kundu & Ghosh 2011 | 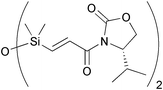 | 6 | intramolecular reductive C–C coupling | 11% | 166 |
| Castillo et al. 2006 | 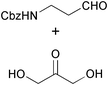 | 2 | biocatalytic asymmetric aldol reaction, intramolecular reductive amination | Castillo: 51% Sugiyama: 65% | 125a,b |
| Sugiyama et al. 2007 |
Examples like these demonstrate that the strategic use of biocatalytic key transformations in chemo-enzymatic sequences can lead to truly practicable routes for the asymmetric total synthesis of alkaloids. The possibilities for further exploration of this concept in the future are vast: For instance, Pictet–Spenglerases other than norcoclaurine synthase and strictosidine synthase are known137 and might be applied in a biocatalytic context. Asymmetric reductive amination catalysed by ω-transaminases is likely to provide efficient access to further alkaloids or enable novel routes towards advanced intermediates in alkaloid synthesis. The broad applicability of chemo-enzymatic deracemisation using monoamine oxidase to complex, bulky amines–including several alkaloids–has just recently been demonstrated, and further developments in this field can be expected. Finally, the biosynthetic pathways that give rise to the incredible number and structural diversity of alkaloids found in nature represent an almost infinite source of novel enzymes that has just been started to be explored.
We believe that future developments along these lines–in combination with the modern possibilities of enzyme discovery and engineering,167 and recent efforts aimed at a better integration of biocatalytic and chemical transformations168–will continue to establish a more central role of biocatalysis in the asymmetric synthesis of alkaloids.
Abbreviations
| Ac | acetyl |
| AIBN | azobisisobutyronitrile |
| Alloc | allyloxycarbonyl |
| BBE | berberine bridge enzyme |
| Bn | benzyl |
| Boc | tert-butoxycarbonyl |
| BMIM | 1-butyl-3-methylimidazolium |
| Bz | benzoyl |
| CAL-B | lipase B from Candida antarctica |
| Cbz | carboxybenzyl |
| CDH | catechol dehydrogenase |
| CPDMO | cyclopentadecanone monooxygenase |
| CPMO | cyclopentanone monooxygenase |
| dba | dibenzylideneacetone |
| DEAD | diethyl azodicarboxylate |
| DHAP | dihydroxyacetone phosphate |
| DIBAL | diisobutylaluminium hydride |
| DMAP | 4-dimethylaminopyridine |
| DMF | N,N-dimethylformamide |
| DMS | dimethyl sulphide |
| dppe | 1,2-bis(diphenylphosphino)ethane |
| dppf | 1,1′-bis(diphenylphosphino)ferrocene |
| dr | diastereomeric ratio |
| ee | enantiomeric excess |
| FDPA | D-fructose-1,6-diphosphate aldolase |
| FucA | L-fuculose-1-phosphate aldolase |
| FSA | D-fructose-6-phosphate aldolase |
| Km | Michaelis constant |
| MAO | monoamine oxidase |
| MAO-N | monoamine oxidase from Aspergillus niger |
| MTBE | methyl tert-butyl ether |
| Ms | mesyl (methanesulfonyl) |
| NADPH | β-nicotinamide adenine dinucleotide phosphate |
| NCS | norcoclaurine synthase |
| NMM | N-methylmorpholine |
| NMO | N-methylmorpholine N-oxide |
| NMR | nuclear magnetic resonance |
| NTA | nitrilotriacetic acid |
| PLE | pig liver esterase |
| PPL | porcine pancreatic lipase |
| RhuA | L-rhamnulose-1-phosphate aldolase |
| SN | nucleophilic substitution |
| STR | strictosidine synthase |
| ω-TA | ω-transaminase |
| TBDMS | tert-butyldimethylsilyl |
| TBDPS | tert-butyldiphenylsilyl |
| TDO | toluene dioxygenase |
| TEMPO | 2,2,6,6-tetramethylpiperidine 1-oxyl |
| Tf | triflyl (trifluoromethanesulfonyl) |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TMS | trimethylsilyl |
| Ts | tosyl (p-toluenesulfonyl) |
Acknowledgements
The authors would like to thank the Austrian Science Fund (FWF) for financial support (FWF projects J3244-N17 and J3292-B12), as well as Dr Michael Fuchs, Dipl.-Chem. Florian Mayer and Prof. Wolfgang Kroutil for their critical and helpful comments on the manuscript.Notes and references
-
(a) W. C. Evans, in Trease and Evans' Pharmacognosy, ed. W. C. Evans, Elsevier, Edinburgh, 16th edn, 2009 Search PubMed;
(b) T. Aniszewski, Alkaloids—Secrets of Life, Elsevier, Amsterdam, 2007 Search PubMed;
(c) M. Hesse, Alkaloids: Nature's Curse or Blessing, Wiley-VHC, Weinheim, 2002 Search PubMed;
(d) M. F. Roberts and M. Wink (ed.), Alkaloids: Biochemistry, Ecology, and Medicinal Applications, Plenum Press, New York, 1998 Search PubMed.
- W. Meissner, J. Chem. Phys., 1819, 25, 377–381 Search PubMed.
- S. W. Pelletier, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Wiley, New York, 1983, vol. 1, pp. 1–31 Search PubMed.
- J. Buckingham, K. H. Baggaley, A. D. Roberts and L. F. Szabó (ed.), Dictionary of Alkaloids, Second Edition with CD-ROM, CRC Press, Boca Raton, 2010 Search PubMed.
-
(a) E. Fattorusso and O. Taglialatela-Scafati (ed.), Modern Alkaloids: Structure, Isolation, Synthesis and Biology, Wiley-VCH, Weinheim, 2008 Search PubMed;
(b) P. J. Facchini, Annu. Rev. Plant Physiol. Plant Mol. Biol., 2001, 52, 29–66 CrossRef CAS.
- T. Efferth, P. C. H. Li, V. S. B. Konkimalla and B. Kaina, Trends Mol. Med., 2007, 13, 353–361 CrossRef CAS.
- I. Raskin, D. M. Ribnicky, S. Komarnytsky, N. Ilic, A. Poulev, N. Borisjuk, A. Brinker, D. A. Moreno, C. Ripoll, N. Yakoby, J. M. O'Neal, T. Cornwell, I. Pastor and B. Fridlender, Trends Biotechnol., 2002, 20, 522–531 CrossRef CAS.
- A. M. Walji and D. W. C. MacMillan, Synlett, 2007, 1477–1489 CAS.
-
(a) P. A. Wender, N. F. Badham, S. P. Conway, P. E. Floreancig, T. E. Glass, C. Gränicher, J. B. Houze, J. Jänichen, D. Lee, D. G. Marquess, P. L. McGrane, W. Meng, T. P. Mucciaro, M. Mühlebach, M. G. Natchus, H. Paulsen, D. B. Rawlins, J. Satkofsky, A. J. Shuker, J. C. Sutton, R. E. Taylor and K. Tomooka, J. Am. Chem. Soc., 1997, 119, 2755–2756 CrossRef CAS;
(b) P. A. Wender, N. F. Badham, S. P. Conway, P. E. Floreancig, T. E. Glass, J. B. Houze, N. E. Krauss, D. Lee, D. G. Marquess, P. L. McGrane, W. Meng, M. G. Natchus, A. J. Shuker, J. C. Sutton and R. E. Taylor, J. Am. Chem. Soc., 1997, 119, 2757–2758 CrossRef CAS.
-
(a) H. Tabata, Adv. Biochem. Eng. Biotechnol., 2004, 87, 1–23 CrossRef CAS;
(b) United States Pat., 2011/0086397 A1, 2011 Search PubMed.
- US Environmental Protection Agency, 2004 Greener Synthetic Pathways Award, http://www.epa.gov/greenchemistry/pubs/pgcc/winners/gspa04.html (accessed 31.03.2013) Search PubMed.
- J. Ziegler and P. J. Facchini, Annu. Rev. Plant Biol., 2008, 59, 735–769 CrossRef CAS.
- E. Leonard, W. Runguphan, S. O'Connor and K. J. Prather, Nat. Chem. Biol., 2009, 5, 292–300 CrossRef CAS.
- A. Nakagawa, H. Minami, J.-S. Kim, T. Koyanagi, T. Katayama, F. Sato and H. Kumagai, Nat. Commun., 2011, 2, 326 CrossRef.
- H. Minami, J.-S. Kim, N. Ikezawa, T. Takemura, T. Katayama, H. Kumagai and F. Sato, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7393–7398 CrossRef CAS.
- K. M. Hawkins and C. D. Smolke, Nat. Chem. Biol., 2008, 4, 564–573 CrossRef CAS.
-
(a) A. M. Hailes and N. C. Bruce, Appl. Environ. Microbiol., 1993, 59, 2166–2170 CAS;
(b) N. C. Bruce, C. E. French, A. M. Hailes, M. T. Long and D. A. Rathbone, Trends Biotechnol., 1995, 13, 200–205 CrossRef CAS;
(c) C. E. French, A. M. Hailes, D. A. Rathbone and N. C. Bruce, Ann. N. Y. Acad. Sci., 1996, 799, 97–101 CrossRef CAS;
(d) B. Boonstra, D. A. Rathbone, C. E. French, E. H. Walker and N. C. Bruce, Appl. Environ. Microbiol., 2000, 66, 5161–5166 CrossRef CAS;
(e) B. Boonstra, D. A. Rathbone and N. C. Bruce, Biomol. Eng., 2001, 18, 41–47 CrossRef CAS;
(f) A. J. Walker and N. C. Bruce, Tetrahedron, 2004, 60, 561–568 CrossRef CAS.
- F. Sagui, C. Chirivì, G. Fontana, S. Nicotra, D. Passarella, S. Riva and B. Danieli, Tetrahedron, 2009, 65, 312–317 CrossRef CAS.
- C. Chirivi, G. Fontana, D. Monti, G. Ottolina, S. Riva and B. Danieli, Chem.–Eur. J., 2012, 18, 10355–10361 CrossRef CAS.
- Z. S. Deng, J. X. Li, P. Teng, P. Li and X. R. Sun, Org. Lett., 2008, 10, 1119–1122 CrossRef CAS.
- Z. S. Deng, D. Zhao, Y. Hu, J. X. Li, K. Zou and J. Z. Wang, Chin. Chem. Lett., 2012, 23, 321–324 CrossRef CAS.
- S. Hanessian, J. Org. Chem., 2012, 77, 6657–6688 CrossRef CAS ; and references cited therein.
- For a general review on biocatalytically prepared chiral building blocks in organic synthesis, see:
(a) T. Fischer and J. Pietruszka, Top. Curr. Chem., 2010, 297, 1–43 CrossRef CAS.
- M. Heinrich and H. Lee Teoh, J. Ethnopharmacol., 2004, 92, 147–162 CrossRef CAS.
- World Health Organization, WHO Model Lists of Essential Medicines, http://www.who.int/medicines/publications/essentialmedicines/en/ (accessed 31.03.2013) Search PubMed.
- R. Riva, L. Banfi and B. Danieli, J. Chem. Soc., Chem. Commun., 1987, 4, 299–300 RSC.
-
(a) K. Laumen and M. Schneider, Tetrahedron Lett., 1985, 26, 2073–2076 CrossRef CAS;
(b) G. Guanti, L. Banfi, E. Narisano, R. Riva and S. Thea, Tetrahedron Lett., 1986, 27, 4639–4642 CrossRef CAS.
- In this and the following schemes, atoms directly derived from the biocatalytically prepared building block are highlighted in blue, and enantiotopic groups recognised by the used enzyme are highlighted by a green circle.
- B. Danieli, G. Lesma, M. Mauro, G. Palmisano and D. Passarella, J. Org. Chem., 1995, 60, 2506–2513 CrossRef CAS.
- Both enantiomers of lactone 3 can also be obtained via enzymatic Baeyer–Villiger oxidation of the corresponding symmetrical ketone. Cyclopentanone monooxygenase (CPMO) from Comamonas sp. NCIMB 9872 (previously classified as Pseudomonas) and cyclopentadecanone monooxygenase (CPDMO) from Pseudomonas sp. HI-70 have been identified as suitable enzymes for this transformation, and the E. coli expression host has been exploited for regeneration of the required NADPH cofactor. However, lactone 3 prepared by this method has not been employed in the synthesis of alkaloids. See:
(a) M. D. Mihovilovic, B. Müller, M. M. Kayser and P. Stanetty, Synlett, 2002, 700–702 CrossRef CAS;
(b) M. D. Mihovilovic, F. Rudroff, B. Müller and P. Stanetty, Bioorg. Med. Chem. Lett., 2003, 13, 1479–1482 CrossRef CAS;
(c) D.
V. Rial, D. A. Bianchi, P. Kapitanova, A. Lengar, J. B. van Beilen and M. D. Mihovilovic, Eur. J. Org. Chem., 2008, 1203–1213 CrossRef CAS;
(d) M. J. Fink, T. C. Fischer, F. Rudroff, H. Dudek, M. W. Fraaije and M. D. Mihovilovic, J. Mol. Catal. B: Enzym., 2011, 73, 9–16 CrossRef CAS
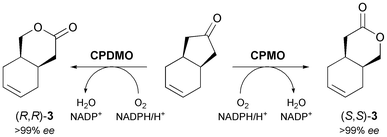 .
. -
(a) G. L. Adams, P. J. Carroll and A. B. Smith III, J. Am. Chem. Soc., 2012, 134, 4037–4040 CrossRef CAS;
(b) G. L. Adams, P. J. Carroll and A. B. Smith III, J. Am. Chem. Soc., 2013, 135, 519–528 CrossRef CAS.
- B. Danieli, G. Lesma, M. Mauro, G. Palmisano and D. Passarella, Tetrahedron: Asymmetry, 1990, 1, 793–800 CrossRef CAS.
- B. Danieli, G. Lesma, M. Mauro, G. Palmisano and D. Passarella, Tetrahedron, 1994, 50, 8837–8852 CrossRef CAS.
- P. Celestini, B. Danieli, G. Lesma, A. Sacchetti, A. Silvani, D. Passarella and A. Virdis, Org. Lett., 2002, 4, 1367–1370 CrossRef CAS.
- The following commercial lipase preparations are mentioned in this review (source organism in parentheses): from Amano Enzyme Inc.: lipase PS (Burkholderia cepacia, previously classified as Pseudomonas cepacia), lipase CE (Humicola lanuginosa), lipase AK (Pseudomonas fluorescens); from Meito Sangyo Co., Ltd.: lipase QLM (Alcaligenes sp.); from Novozymes A/S: Novozym 435 (lipase B from Candida antarctica); from Roche Applied Science: Chirazyme L-2 (lipase B from Candida antarctica).
- G. Lesma, S. Crippa, B. Danieli, D. Passarella, A. Sacchetti, A. Silvani and A. Virdis, Tetrahedron, 2004, 60, 6437–6442 CrossRef CAS.
- G. Carrea, B. Danieli, G. Palmisano, S. Riva and M. Santagostino, Tetrahedron: Asymmetry, 1992, 3, 775–784 CrossRef CAS.
- G. Palmisano, P. D'Anniballe and M. Santagostino, Tetrahedron, 1994, 50, 9487–9494 CrossRef CAS.
- G. Palmisano, M. Santagostino, S. Riva and M. Sisti, Tetrahedron: Asymmetry, 1995, 6, 1229–1232 CrossRef CAS.
- T. Yamane and K. Ogasawara, Synlett, 1996, 925–926 CrossRef CAS.
- H. Nakashima, M. Sato, T. Taniguchi and K. Ogasawara, Synthesis, 2000, 817–823 CrossRef CAS.
-
(a) K. Tanaka, T. Taniguchi and K. Ogasawara, Tetrahedron Lett., 2001, 42, 1049–1052 CrossRef CAS;
(b) M. Hayashi, K. Motosawa, A. Satoh, M. Shibuya, K. Ogasawara and Y. Iwabuchi, Heterocycles, 2009, 77, 855–863 CrossRef CAS.
- M. Hayashi, T. Unno, M. Takahashi and K. Ogasawara, Tetrahedron Lett., 2002, 43, 1461–1464 CrossRef CAS.
- H. Koizumi, S. Yokoshima and T. Fukuyama, Chem.–Asian J., 2010, 5, 2192–2198 CrossRef CAS.
-
(a) T. Momose, N. Toyooka and M. Jin, Tetrahedron Lett., 1992, 33, 5389–5390 CrossRef CAS;
(b) T. Momose, N. Toyooka and M. Jin, J. Chem. Soc., Perkin Trans. 1, 1997, 2005–2014 RSC.
- T. Momose and N. Toyooka, Tetrahedron Lett., 1993, 34, 5785–5786 CrossRef CAS.
-
(a) T. Momose and N. Toyooka, J. Org. Chem., 1994, 59, 943–945 CrossRef CAS;
(b) N. Toyooka, K. Tanaka, T. Momose, J. W. Daly and H. M. Garraffo, Tetrahedron, 1997, 53, 9553–9574 CrossRef CAS.
-
(a) R. Chênevert and M. Dickman, Tetrahedron: Asymmetry, 1992, 3, 1021–1024 CrossRef;
(b) R. Chênevert and M. Dickman, J. Org. Chem., 1996, 61, 3332–3341 CrossRef.
- R. Chênevert, G. M. Ziarani and M. Dasser, Heterocycles, 1999, 51, 593–598 CrossRef.
-
(a) R. Chênevert, G. M. Ziarani, M. P. Morin and M. Dasser, Tetrahedron: Asymmetry, 1999, 10, 3117–3122 CrossRef;
(b) G. Lesma, A. Colombo, N. Landoni, A. Sacchetti and A. Silvani, Tetrahedron: Asymmetry, 2007, 18, 1948–1954 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella and A. Silvani, Tetrahedron: Asymmetry, 1996, 7, 345–348 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella and A. Silvani, J. Org. Chem., 1998, 63, 3492–3496 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella, A. Silvani and N. Viviani, Tetrahedron, 1999, 55, 11871–11878 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella, A. Sacchetti, A. Silvani and A. Virdis, Org. Lett., 2004, 6, 493–496 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella, A. Sacchetti and A. Silvani, Tetrahedron Lett., 2005, 46, 7121–7123 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella and A. Silvani, Tetrahedron Lett., 2000, 41, 3489–3492 CrossRef CAS.
- M. Angoli, A. Barilli, G. Lesma, D. Passarella, S. Riva, A. Silvani and B. Danieli, J. Org. Chem., 2003, 68, 9525–9527 CrossRef CAS.
- D. Passarella, S. Riva, G. Grieco, F. Cavallo, B. Checa, F. Arioli, E. Riva, D. Comi and B. Danieli, Tetrahedron: Asymmetry, 2009, 20, 192–197 CrossRef CAS.
- D. Passarella, A. Barilli, F. Belinghieri, P. Fassi, S. Riva, A. Sacchetti, A. Silvani and B. Danieli, Tetrahedron: Asymmetry, 2005, 16, 2225–2229 CrossRef CAS.
- A. Barilli, F. Belinghieri, D. Passarella, G. Lesma, S. Riva, A. Silvani and B. Danieli, Tetrahedron: Asymmetry, 2004, 15, 2921–2925 CrossRef CAS.
-
(a) C.-H. Tan, T. Stork, N. Feeder and A. B. Holmes, Tetrahedron Lett., 1999, 40, 1397–1400 CrossRef CAS;
(b) C.-H. Tan and A. B. Holmes, Chem.–Eur. J., 2001, 7, 1845–1854 CrossRef CAS.
- S. Vrielynck, M. Vandewalle, A. M. García, J. L. Mascareñas and A. Mouriño, Tetrahedron Lett., 1995, 36, 9023–9026 CrossRef CAS.
- H. Ooi, A. Urushibara, T. Esumi, Y. Iwabuchi and S. Hatakeyama, Org. Lett., 2001, 3, 953–955 CrossRef CAS.
- S. Akai, K. Tanimoto, Y. Kanao, S. Omura and Y. Kita, Chem. Commun., 2005, 2369–2371 RSC.
- H. Nemoto, K. Tanimoto, Y. Kanao, S. Omura, Y. Kita and S. Akai, Tetrahedron, 2012, 68, 7295–7301 CrossRef CAS.
- T. Hudlicky and J. Reed, Synlett, 2009, 685–703 CrossRef CAS.
-
(a) D. T. Gibson, J. R. Koch and R. E. Kallio, Biochemistry, 1968, 7, 2653–2662 CrossRef CAS;
(b) D. T. Gibson, J. R. Koch, C. L. Schuld and R. E. Kallio, Biochemistry, 1968, 7, 3795–3802 CrossRef CAS;
(c) D. T. Gibson, M. Hensley, H. Yoshioka and T. J. Mabry, Biochemistry, 1970, 9, 1626–1630 CrossRef CAS.
- T. Hudlicky, H. Luna, J. D. Price and F. Rulin, J. Org. Chem., 1990, 55, 4683–4687 CrossRef CAS.
-
(a) T. Hudlicky, J. Rouden, H. Luna and S. Allen, J. Am. Chem. Soc., 1994, 116, 5099–5107 CrossRef CAS;
(b) C. R. Johnson, A. Golebiowski, H. Sundram, M. W. Miller and R. L. Dwaihy, Tetrahedron Lett., 1995, 36, 653–654 CrossRef CAS;
(c) M. G. Banwell, X. Ma, N. Asano, K. Ikeda and J. N. Lambert, Org. Biomol. Chem., 2003, 1, 2035–2037 RSC.
-
(a) T. Hudlicky, ARKIVOC, 2006, 7, 276–291 CrossRef;
(b) U. Rinner and T. Hudlicky, Synlett, 2005, 365–387 CAS;
(c) L. Ingrassia, F. Lefranc, V. Mathieu, F. Darro and R. Kiss, Transl. Oncol., 2008, 1, 1–13 CrossRef.
- T. Hudlicky and H. F. Olivo, J. Am. Chem. Soc., 1992, 114, 9694–9696 CrossRef CAS.
- X. Tian, T. Hudlicky and K. Königsberger, J. Am. Chem. Soc., 1995, 117, 3643–3644 CrossRef CAS.
-
(a) X. Tian, R. Maurya, K. Königsberger and T. Hudlicky, Synlett, 1995, 1125–1126 CrossRef CAS;
(b) T. Hudlicky, X. Tian, K. Königsberger, R. Maurya, J. Rouden and B. Fan, J. Am. Chem. Soc., 1996, 118, 10752–10765 CrossRef CAS.
-
(a) D. Gonzalez, T. Martinot and T. Hudlicky, Tetrahedron Lett., 1999, 40, 3077–3080 CrossRef CAS;
(b) T. Hudlicky, U. Rinner, D. Gonzalez, H. Akgun, S. Schilling, P. Siengalewicz, T. A. Martinot and G. R. Pettit, J. Org. Chem., 2002, 67, 8726–8743 CrossRef CAS.
- U. Rinner, P. Siengalewicz and T. Hudlicky, Org. Lett., 2002, 4, 115–117 CrossRef CAS.
-
(a) U. Rinner, T. Hudlicky, H. Gordon and G. R. Pettit, Angew. Chem., Int. Ed., 2004, 43, 5342–5346 CrossRef CAS;
(b) T. Hudlicky, U. Rinner, K. J. Finn and I. Ghiviriga, J. Org. Chem., 2005, 70, 3490–3499 CrossRef CAS.
-
(a) J. Collins, M. Drouin, X. Sun, U. Rinner and T. Hudlicky, Org. Lett., 2008, 10, 361–364 CrossRef CAS;
(b) J. Collins, U. Rinner, M. Moser, T. Hudlicky, I. Ghiviriga, A. E. Romero, A. Kornienko, D. Ma, C. Griffin and S. Pandey, J. Org. Chem., 2010, 75, 3069–3084 CrossRef CAS;
(c) S. Vshyvenko, J. Scattolon, T. Hudlicky, A. E. Romero and A. Kornienko, Bioorg. Med. Chem. Lett., 2011, 21, 4750–4752 CrossRef CAS;
(d) S. Vshyvenko, J. Scattolon, T. Hudlicky, A. E. Romero, A. Kornienko, D. Ma, I. Tuffley and S. Pandey, Can. J. Chem., 2012, 90, 932–943 CrossRef CAS.
-
(a) U. Rinner, H. L. Hillebrenner, D. R. Adams, T. Hudlicky and G. R. Pettit, Bioorg. Med. Chem. Lett., 2004, 14, 2911–2915 CrossRef CAS;
(b) V. De La Sovera, A. Bellomo and D. Gonzalez, Tetrahedron Lett., 2011, 52, 430–433 CrossRef CAS;
(c) M. Moser, X. Sun and T. Hudlicky, Org. Lett., 2005, 7, 5669–5672 CrossRef CAS;
(d) T. Hudlicky, M. Moser, S. C. Banfield, U. Rinner, J. C. Chapuis and G. R. Pettit, Can. J. Chem., 2006, 84, 1313–1337 CrossRef.
- Oxidation of these compounds by whole cells of Pseudomonas putida had been reported earlier; see:
(a) C. C. R. Allen, D. R. Boyd, H. Dalton, N. D. Sharma, I. Brannigan, N. A. Kerley, G. N. Sheldrake and S. C. Taylor, J. Chem. Soc., Chem. Commun., 1995, 117–118 RSC.
- H. Akgün and T. Hudlicky, Tetrahedron Lett., 1999, 40, 3081–3084 CrossRef.
- M. Matveenko, O. J. Kokas, M. G. Banwell and A. C. Willis, Org. Lett., 2007, 9, 3683–3685 CrossRef CAS.
- M. Matveenko, M. G. Banwell and A. C. Willis, Tetrahedron, 2008, 64, 4817–4826 CrossRef CAS.
-
(a) U. Rinner and T. Hudlicky, Top. Curr. Chem., 2012, 309, 33–66 CrossRef CAS;
(b) J. Zezula and T. Hudlicky, Synlett, 2005, 388–405 CAS;
(c) B. H. Novak, T. Hudlicky, J. W. Reed, J. Mulzer and D. Trauner, Curr. Org. Chem., 2000, 4, 343–362 CrossRef CAS.
-
(a) G. Butora, T. Hudlicky, S. P. Fearnley, A. G. Gum, M. R. Stabile and K. Abboud, Tetrahedron Lett., 1996, 37, 8155–8158 CrossRef CAS;
(b) G. Butora, T. Hudlicky, S. P. Fearnley, M. R. Stabile, A. G. Gum and D. Gonzalez, Synthesis, 1998, 665–681 CrossRef CAS.
-
(a) G. Butora, A. G. Gum, T. Hudlicky and K. A. Abboud, Synthesis, 1998, 275–278 CrossRef CAS;
(b) J. Zezula, T. Hudlicky and I. Ghiviriga, Collect. Czech. Chem. Commun., 2001, 66, 1269–1286 CrossRef CAS.
-
(a) D. A. Frey, C. Duan and T. Hudlicky, Org. Lett., 1999, 1, 2085–2087 CrossRef CAS;
(b) D. A. Frey, C. Duan, I. Ghiviriga and T. Hudlicky, Collect. Czech. Chem. Commun., 2000, 65, 561–569 CrossRef CAS;
(c) J. Zezula, K. C. Rice and T. Hudlicky, Synlett, 2007, 2863–2867 CAS.
- H. Leisch, A. T. Omori, K. J. Finn, J. Gilmet, T. Bissett, D. Ilceski and T. Hudlicky, Tetrahedron, 2009, 65, 9862–9875 CrossRef CAS.
- J. Duchek, T. G. Piercy, J. Gilmet and T. Hudlicky, Can. J. Chem., 2011, 89, 709–728 CrossRef CAS.
-
(a) M. G. Banwell, N. Y. Gao, B. D. Schwartz and L. V. White, Top. Curr. Chem., 2012, 309, 163–202 CrossRef CAS;
(b) M. G. Banwell, N. Y. Gao, X. Ma, L. Petit, L. V. White, B. D. Schwartz, A. C. Willis and I. A. Cade, Pure Appl. Chem., 2012, 84, 1329–1339 CrossRef CAS;
(c) M. G. Banwell, A. L. Lehmann, R. S. Menon and A. C. Willis, Pure Appl. Chem., 2011, 83, 411–423 CrossRef CAS.
-
(a) M. G. Banwell, O. J. Kokas and A. C. Willis, Org. Lett., 2007, 9, 3503–3506 CrossRef CAS;
(b) O. J. Kokas, M. G. Banwell and A. C. Willis, Tetrahedron, 2008, 64, 6444–6451 CrossRef CAS;
(c) M. Matveenko, M. G. Banwell and A. C. Willis, Org. Lett., 2008, 10, 4693–4696 CrossRef CAS.
-
(a) A. D. Findlay and M. G. Banwell, Org. Lett., 2009, 11, 3160–3162 CrossRef CAS;
(b) M. T. Jones, B. D. Schwartz, A. C. Willis and M. G. Banwell, Org. Lett., 2009, 11, 3506–3509 CrossRef CAS.
-
(a) L. V. White, B. D. Schwartz, M. G. Banwell and A. C. Willis, J. Org. Chem., 2011, 76, 6250–6257 CrossRef CAS;
(b) B. D. Schwartz, M. G. Banwell and I. A. Cade, Tetrahedron Lett., 2011, 52, 4526–4528 CrossRef CAS.
- B. D. Schwartz, M. T. Jones, M. G. Banwell and I. A. Cade, Org. Lett., 2010, 12, 5210–5213 CrossRef CAS.
- For a single example of the use of an isolated alcohol dehydrogenase in alkaloid synthesis, see:
(a) M. De Amici, C. De Micheli, G. Molteni, D. Pitrè, G. Carrea, S. Riva, S. Spezia and L. Zetta, J. Org. Chem., 1991, 56, 67–72 CrossRef CAS.
- N. Toyooka, Y. Yoshida and T. Momose, Tetrahedron Lett., 1995, 36, 3715–3718 CrossRef CAS.
- N. Toyooka, Y. Yoshida, Y. Yotsui and T. Momose, J. Org. Chem., 1999, 64, 4914–4919 CrossRef CAS.
-
(a) N. Toyooka, Y. Yotsui, Y. Yoshida and T. Momose, J. Org. Chem., 1996, 61, 4882–4883 CrossRef CAS;
(b) N. Toyooka, M. Okumura and H. Takahata, J. Org. Chem., 1999, 64, 2182–2183 CrossRef CAS;
(c) N. Toyooka, M. Okumura, H. Takahata and H. Nemoto, Tetrahedron, 1999, 55, 10673–10684 CrossRef CAS.
-
(a) B. Das and P. Madhusudhan, Tetrahedron, 1999, 55, 7875–7880 CrossRef CAS;
(b) B. Das and P. Madhusudhan, J. Chem. Res. (S), 2000, 476–477 CrossRef CAS.
- T. Choshi, Y. Uchida, Y. Kubota, J. Nobuhiro, M. Takeshita, T. Hatano and S. Hibino, Chem. Pharm. Bull., 2007, 55, 1060–1064 CrossRef CAS.
-
(a) A. Kamal, K. V. Ramana and M. V. Rao, J. Org. Chem., 2001, 66, 997–1001 CrossRef CAS;
(b) C.-Y. Yu and O. Meth-Cohn, Tetrahedron
Lett., 1999, 40, 6665–6668 CAS;
(c) C. Rusconi, N. Vaiana, M. Casagrande, N. Basilico, S. Parapini, D. Taramelli, S. Romeo and A. Sparatore, Bioorg. Med. Chem., 2012, 20, 5980–5985 CrossRef CAS.
- R. Chênevert and P. Morin, Bioorg. Med. Chem., 2009, 17, 1837–1839 CrossRef.
- E. Forró, L. Schönstein and F. Fülöp, Tetrahedron: Asymmetry, 2011, 22, 1255–1260 CrossRef.
-
(a) N. J. Turner, Curr. Opin. Chem. Biol., 2010, 14, 115–121 CrossRef CAS;
(b) M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef;
(c) N. J. Turner, Curr. Opin. Chem. Biol., 2004, 8, 114–119 CrossRef CAS.
- In this section, only the deracemisation or stereoinversion of alkaloids using MAO is discussed. However, the synthesis of non-natural isoquinoline alkaloids via a combination of MAO-catalysed oxidative desymmetrisation of meso-pyrrolidines, an Ugi-type multicomponent reaction, and a Pictet–Spengler cyclisation has also been reported. See:
(a) A. Znabet, J. Zonneveld, E. Janssen, F. J. J. De Kanter, M. Helliwell, N. J. Turner, E. Ruijter and R. V. A. Orru, Chem. Commun., 2010, 46, 7706–7708 RSC.
- C. J. Dunsmore, R. Carr, T. Fleming and N. J. Turner, J. Am. Chem. Soc., 2006, 128, 2224–2225 CrossRef CAS.
- K. R. Bailey, A. J. Ellis, R. Reiss, T. J. Snape and N. J. Turner, Chem. Commun., 2007, 3640–3642 RSC.
- I. Rowles, K. J. Malone, L. L. Etchells, S. C. Willies and N. J. Turner, ChemCatChem, 2012, 4, 1259–1261 CrossRef CAS.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS.
- S. Wallner, C. Dully, B. Daniel and P. Macheroux, in Handbook of Flavoproteins, ed. R. Hille, S. M. Miller and B. Palfey, Walter de Gruyter, Berlin/Boston, 2012, pp. 1–30 Search PubMed.
-
(a) A. Winkler, A. Lyskowski, S. Riedl, M. Puhl, T. M. Kutchan, P. Macheroux and K. Gruber, Nat. Chem. Biol., 2008, 4, 739–741 CrossRef CAS;
(b) H. M. Gaweska, K. M. Roberts and P. F. Fitzpatrick, Biochemistry, 2012, 51, 7342–7347 CrossRef CAS.
- A. Winkler, K. Motz, S. Riedl, M. Puhl, P. Macheroux and K. Gruber, J. Biol. Chem., 2009, 284, 19993–20001 CrossRef CAS.
- A. Winkler, F. Hartner, T. M. Kutchan, A. Glieder and P. Macheroux, J. Biol. Chem., 2006, 281, 21276–21285 CrossRef CAS.
- V. Resch, J. H. Schrittwieser, S. Wallner, P. Macheroux and W. Kroutil, Adv. Synth. Catal., 2011, 353, 2377–2383 CrossRef CAS.
-
(a) J. H. Schrittwieser, V. Resch, J. H. Sattler, W. D. Lienhart, K. Durchschein, A. Winkler, K. Gruber, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2011, 50, 1068–1071 CrossRef CAS;
(b) J. H. Schrittwieser, V. Resch, S. Wallner, W. D. Lienhart, J. H. Sattler, J. Resch, P. Macheroux and W. Kroutil, J. Org. Chem., 2011, 76, 6703–6714 CrossRef CAS.
- V. Resch, H. Lechner, J. H. Schrittwieser, S. Wallner, K. Gruber, P. Macheroux and W. Kroutil, Chem.–Eur. J., 2012, 18, 13173–13179 CrossRef CAS.
- H.-X. Ge, J. Zhang, Y. Dong, K. Cui and B.-Y. Yu, Chem. Commun., 2012, 48, 6127–6129 RSC.
- The formation of these glycosides by fermentation with Gliocladium deliquescens has been reported earlier:
(a) H.-X. Ge, J. Zhang, C. Kai, J.-H. Liu and B.-Y. Yu, Appl. Microbiol. Biotechnol., 2012, 93, 2357–2364 CrossRef CAS.
-
(a) R. L. Pederson, M.-J. Kim and C.-H. Wong, Tetrahedron Lett., 1988, 29, 4645–4648 CrossRef CAS;
(b) T. Ziegler, A. Straub and F. Effenberger, Angew. Chem., Int. Ed. Engl., 1988, 27, 716–717 CrossRef;
(c) C. H. Von der Osten, A. J. Sinskey, C. F. Barbas, R. L. Pederson, Y. F. Wang and C. H. Wong, J. Am. Chem. Soc., 1989, 111, 3924–3927 CrossRef CAS;
(d) R. L. Pederson and C. H. Wong, Heterocycles, 1989, 28, 477–480 CrossRef CAS.
-
(a) T. Kajimoto, K. K. C. Liu, R. L. Pederson, Z. Zhong, Y. Ichikawa, J. A. Porco and C. H. Wong, J. Am. Chem. Soc., 1991, 113, 6187–6196 CrossRef CAS;
(b) M. Mitchell, L. Qaio and C. H. Wong, Adv. Synth. Catal., 2001, 343, 596–599 CrossRef CAS;
(c) M. L. Mitchell, L. V. Lee and C. H. Wong, Tetrahedron Lett., 2002, 43, 5691–5693 CrossRef CAS.
-
(a) K. K. C. Liu, T. Kajimoto, L. Chen, Z. Zhong, Y. Ichikawa and C. H. Wong, J. Org. Chem., 1991, 56, 6280–6289 CrossRef CAS;
(b) J. Calveras, M. Egido-Gabás, L. Gómez, J. Casas, T. Parella, J. Joglar, J. Bujons and P. Clapés, Chem.–Eur. J., 2009, 15, 7310–7328 CrossRef CAS.
- F. Morís-Varas, X.-H. Qian and C.-H. Wong, J. Am. Chem. Soc., 1996, 118, 7647–7652 CrossRef.
-
(a) L. Espelt, T. Parella, J. Bujons, C. Solans, J. Joglar, A. Delgado and P. Clapés, Chem.–Eur. J., 2003, 9, 4887–4899 CrossRef CAS;
(b) L. Espelt, J. Bujons, T. Parella, J. Calveras, J. Joglar, A. Delgado and P. Clapés, Chem.–Eur. J., 2005, 11, 1392–1401 CrossRef CAS.
- For a review, see:
(a) M. Schümperli, R. Pellaux and S. Panke, Appl. Microbiol. Biotechnol., 2007, 75, 33–45 CrossRef. For recent examples with connection to iminocyclitol synthesis, see:
(b) L. Babich, L. J. C. van Hemert, A. Bury, A. F. Hartog, P. Falcicchio, J. van der Oost, T. van Herk, R. Wever and F. P. J. T. Rutjes, Green Chem., 2011, 13, 2895–2900 RSC;
(c) L. Babich, A. F. Hartog, L. J. C. van Hemert, F. P. J. T. Rutjes and R. Wever, ChemSusChem, 2012, 5, 2348–2353 CrossRef CAS.
-
(a) M. Schürmann and G. A. Sprenger, J. Biol. Chem., 2001, 276, 11055–11061 CrossRef;
(b) M. Schürmann and G. A. Sprenger, J. Mol. Catal. B: Enzym., 2002, 19–20, 247–252 CrossRef.
-
(a) J. A. Castillo, J. Calveras, J. Casas, M. Mitjans, M. P. Vinardell, T. Parella, T. Inoue, G. A. Sprenger, J. Joglar and P. Clapés, Org. Lett., 2006, 8, 6067–6070 CrossRef CAS;
(b) M. Sugiyama, Z. Hong, P.-H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 14811–14817 CrossRef CAS;
(c) A. L. Concia, C. Lozano, J. A. Castillo, T. Parella, J. Joglar and P. Clapés, Chem.–Eur. J., 2009, 15, 3808–3816 CrossRef CAS.
- A. Romero and C.-H. Wong, J. Org. Chem., 2000, 65, 8264–8268 CrossRef CAS.
- J. Calveras, J. Casas, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2007, 349, 1661–1666 CrossRef CAS.
- Please note that in the original article (Calveras et al. 2007, ref. 127) the incorrect (1S)-configuration was assigned to compounds 221 and 224. Based on this assignment, the aldol reaction was thought to proceed with low diastereoselectivity. The correct stereochemistry of 221 and 224 was unequivocally established in a later study (Garrabou et al. 2010, ref. 129) by reassessment of the NMR spectra and comparison with those of the products obtained by fuculose-1-phosphate aldolase catalysis. Thus, it was demonstrated that both aldolases are fully stereoselective in the reactions under investigation, while the reductive amination proceeded with low selectivity, affording C3-epimers.
- X. Garrabou, L. Gómez, J. Joglar, S. Gil, T. Parella, J. Bujons and P. Clapés, Chem.–Eur. J., 2010, 16, 10691–10706 CrossRef CAS.
- L. Gómez, X. Garrabou, J. Joglar, J. Bujons, T. Parella, C. Vilaplana, P. J. Cardona and P. Clapés, Org. Biomol. Chem., 2012, 10, 6309–6321 Search PubMed.
-
(a) S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS;
(b) D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS.
- R. C. Simon, F. Zepeck and W. Kroutil, Chem.–Eur. J., 2013, 19, 2859–2865 CrossRef CAS.
- R. C. Simon, C. S. Fuchs, H. Lechner, F. Zepeck and W. Kroutil, Eur. J. Org. Chem., 2013, 2013, 3397–3402 CrossRef CAS.
- A. A. Orden, J. H. Schrittwieser, V. Resch, F. G. Mutti and W. Kroutil, Tetrahedron: Asymmetry, 2013, 24, 744–749 CrossRef CAS.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef.
- N. Samanani and P. J. Facchini, J.
Biol. Chem., 2002, 277, 33878–33883 CrossRef CAS.
- N. Samanani and P. J. Facchini, Planta, 2001, 213, 898–906 CrossRef CAS.
- N. Samanani, D. K. Liscombe and P. J. Facchini, Plant J., 2004, 40, 302–313 CrossRef CAS.
- L. Y. P. Luk, S. Bunn, D. K. Liscombe, P. J. Facchini and M. E. Tanner, Biochemistry, 2007, 46, 10153–10161 CrossRef CAS.
- H. Minami, E. Dubouzet, K. Iwasa and F. Sato, J. Biol. Chem., 2007, 282, 6274–6282 CrossRef CAS.
- A. Bonamore, I. Rovardi, F. Gasparrini, P. Baiocco, M. Barba, C. Molinaro, B. Botta, A. Boffi and A. Macone, Green Chem., 2010, 12, 1623–1627 RSC.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Chem. Commun., 2011, 47, 3242–3244 RSC.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS.
- B. M. Ruff, S. Bräse and S. E. O'Connor, Tetrahedron Lett., 2012, 53, 1071–1074 CrossRef CAS.
- U. Pfitzner and M. H. Zenk, Planta Med., 1982, 46, 10–14 CrossRef CAS.
-
(a) Y. Yamazaki, H. Sudo, M. Yamazaki, N. Aimi and K. Saito, Plant Cell Physiol., 2003, 44, 395–403 CrossRef CAS;
(b) T. M. Kutchan, N. Hampp, F. Lottspeich, K. Beyreuther and M. H. Zenk, FEBS Lett., 1988, 237, 40–44 CrossRef CAS;
(c) C. A. Roessner, R. Devagupta, M. Hasan, H. J. Williams and A. I. Scott, Protein Expression Purif., 1992, 3, 295–300 CrossRef CAS;
(d) X. Ma, S. Panjikar, J. Koepke, E. Loris and J. Stöckigt, Plant Cell, 2006, 18, 907–920 CrossRef CAS;
(e) L. Yang, H. Zou, H. Zhu, M. Ruppert, J. Gong and J. Stöckigt, Chem. Biodiversity, 2010, 7, 860–870 CrossRef CAS.
-
(a) J. F. Treimer and M. H. Zenk, Eur. J. Biochem., 1979, 101, 225–233 CrossRef CAS;
(b) E. McCoy, M. C. Galan and S. E. O'Connor, Bioorg. Med. Chem. Lett., 2006, 16, 2475–2478 CrossRef CAS.
- P. Bernhardt and S. E. O'Connor, Tetrahedron Lett., 2009, 50, 7118–7120 CrossRef CAS.
- P. Bernhardt, A. R. Usera and S. E. O'Connor, Tetrahedron Lett., 2010, 51, 4400–4402 CrossRef CAS.
- H. B. Zou, H. J. Zhu, L. Zhang, L. Q. Yang, Y. P. Yu and J. Stöckigt, Chem.–Asian J., 2010, 5, 2400–2404 CrossRef CAS.
- It has also been shown that strictosidine formation and deglycosylation can be achieved in a cascade fashion using E. coli or S. cerevisiae cells that co-express strictosidine synthase and strictosidine β-glucosidase. However, these bi-enzymatic reactions have not been performed on preparative scale. See:
(a) A. Geerlings, F. Redondo, A. Contin, J. Memelink, R. van der Heijden and R. Verpoorte, Appl. Microbiol. Biotechnol., 2001, 56, 420–424 CrossRef CAS;
(b) J. Stöckigt, B. Hammes and M. Ruppert, Nat. Prod. Res., 2010, 24, 759–766 CrossRef.
- F. Wu, H. Zhu, L. Sun, C. Rajendran, M. Wang, X. Ren, S. Panjikar, A. Cherkasov, H. Zou and J. Stöckigt, J. Am. Chem. Soc., 2012, 134, 1498–1500 CrossRef CAS.
-
(a) P. Bernhardt, E. McCoy and S. E. O'Connor, Chem. Biol., 2007, 14, 888–897 CrossRef CAS;
(b) S. Chen, M. C. Galan, C. Coltharp and S. E. O'Connor, Chem. Biol., 2006, 13, 1137–1141 CrossRef CAS;
(c) E. A. Loris, S. Panjikar, M. Ruppert, L. Barleben, M. Unger, H. Schübel and J. Stöckigt, Chem. Biol., 2007, 14, 979–985 CrossRef CAS.
-
(a) W. Runguphan and S. E. O'Connor, Nat. Chem. Biol., 2009, 5, 151–153 CrossRef CAS;
(b) W. Runguphan, X. Qu and S. E. O'Connor, Nature, 2010, 468, 461–464 CrossRef CAS.
- M. Koyama and S. Sakamura, Agric. Biol. Chem., 1974, 38, 1111–1112 CrossRef CAS.
-
(a) N. Asano, Curr. Top. Med. Chem., 2003, 3, 471–484 CrossRef CAS;
(b) L. Gómez, E. Molinar-Toribio, M. Á. Calvo-Torras, C. Adelantado, M. E. Juan, J. M. Planas, X. Cañas, C. Lozano, S. Pumarola, P. Clapés and J. L. Torres, Br. J. Nutr., 2012, 107, 1739–1746 CrossRef.
- S. Yamauchi, S. Mori, Y. Hirai and Y. Kinoshita, Biosci., Biotechnol., Biochem., 2004, 68, 676–684 CrossRef CAS.
- A. Kato, N. Asano, H. Kizu, K. Matsui, A. A. Watson and R. J. Nash, J. Nat. Prod., 1997, 60, 312–314 CrossRef CAS.
- N. Asano, T. Yamashita, K. Yasuda, K. Ikeda, H. Kizu, Y. Kameda, A. Kato, R. J. Nash, H. S. Lee and K. S. Ryu, J. Agric. Food Chem., 2001, 49, 4208–4213 CrossRef CAS.
- A. Kato, E. Kano, I. Adachi, R. J. Molyneux, A. A. Watson, R. J. Nash, G. W. J. Fleet, M. R. Wormald, H. Kizu, K. Ikeda and N. Asano, Tetrahedron: Asymmetry, 2003, 14, 325–331 CrossRef CAS.
-
(a) Y. Banba, C. Abe, H. Nemoto, A. Kato, I. Adachi and H. Takahata, Tetrahedron: Asymmetry, 2001, 12, 817–819 CrossRef CAS;
(b) H. Takahata, Y. Banba, H. Ouchi, H. Nemoto, A. Kato and I. Adachi, J. Org. Chem., 2003, 68, 3603–3607 CrossRef CAS.
- N. Kumari, B. G. Reddy and Y. D. Vankar, Eur. J. Org. Chem., 2009, 160–169 CAS.
- J.-Y. Kim, Y. Mu, X. Jin, S.-H. Park, V.-T. Pham, D.-K. Song, K.-Y. Lee and W.-H. Ham, Tetrahedron, 2011, 67, 9426–9432 CrossRef CAS.
- P. K. Kundu and S. K. Ghosh, Tetrahedron: Asymmetry, 2011, 22, 1090–1096 CrossRef CAS.
-
(a) G. A. Behrens, A. Hummel, S. K. Padhi, S. Schätzle and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2191–2215 CrossRef CAS;
(b) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS.
-
(a) H. C. Hailes, P. A. Dalby and J. M. Woodley, J. Chem. Technol. Biotechnol., 2007, 82, 1063–1066 CrossRef CAS;
(b) A. C. Marr and S. Liu, Trends Biotechnol., 2011, 29, 199–204 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a