 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Piperolein B, isopiperolein B and piperamide C9:1(8E): total synthesis and cytotoxicities†
Francis
Kayamba‡
,
Christopher
Dunnill‡
,
David J.
Hamnett
,
Arantxa
Rodríguez
,
Nikolaos T.
Georgopoulos§
* and
Wesley J.
Moran§
*
Department of Chemical & Biological Sciences, School of Applied Sciences, University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: w.j.moran@hud.ac.uk; n.georgopoulos@hud.ac.uk; Fax: +44 (0)1484 47 2182
First published on 15th July 2013
Abstract
Total syntheses of the reported structures of piperolein B, isopiperolein B and piperamide C9:1(8E) have been achieved. The analytical data reported for piperolein B and piperamide C9:1(8E) match the synthetic values, however those for isopiperolein B do not. The cytotoxicities of these three structurally similar compounds against cancer cell lines of different tissue origins were evaluated and the results indicated that these compounds show differential effects on cancer cell viability.
Introduction
Studies of plants of the genus Piper have led to the isolation of 277 different amide alkaloids to date.1 Some species of Piper plants are widely used in folk medicine and the isolated amides have been shown to possess a wide range of biological activities. Amongst these, piperolein B, 1 which can be isolated from black pepper, Piper nigrum, has been reported to possess larvicidal,2 hepatoprotective3 and enzymatic inhibition activity (Fig. 1).4 It has also been shown to be a TRP agonist.5 Piperamide C9:1(8E), 2 was also tested for larvicidal activity and was found to be six times more toxic than piperolein B, 1. More recently, isopiperolein B, 3 was isolated.6 This was later shown to have antibacterial activity7 and cytotoxic activity towards the human cervical carcinoma cell line HeLa.8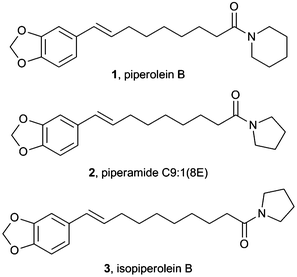 | ||
| Fig. 1 Reported structures for three Piper alkaloids. | ||
To date there have been few total syntheses of members of this family of amide alkaloids. However, Strunz and co-workers have synthesised over a dozen of these compounds using a multi-step aldol condensation–Grob-type fragmentation strategy,9 whilst several dimeric compounds have been prepared through cycloaddition processes.10 We were interested in developing a short synthetic route into piperolein B, 1 and the closely related molecules 2 and 3 and investigating their anti-cancer properties.
Results and discussion
A simple synthetic strategy was conceived for the preparation of these compounds (Scheme 1). Amidation of either 8-nonenoic acid or 9-decenoic acid with piperidine or pyrrolidine provided the amides 6, 7 and 8 in good yields. Subsequent Ru-catalyzed cross metathesis with styrene derivative 9 delivered the three natural products 1, 2 and 3 in moderate yields. These yields of isolated compounds were lower than expected due to difficulties in separating the natural products from the dimer of styrene 9.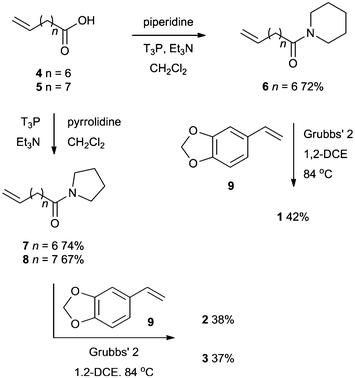 | ||
| Scheme 1 Synthesis of alkaloids 1, 2 and 3. | ||
The analytical data for piperolein B, 1 and piperamide C9:1(8E), 2 matched the literature values. However, the data for amide 3 did not match the literature values for isopiperolein B (Table 1). Moreover, the literature values are inconsistent with a pyrrolidine amide and more closely resemble the data for a piperidine amide. This difference can clearly be seen in the aliphatic region of the 1H NMR spectra of amides 1, 2 and 3 (Fig. 2). The reported data for isopiperolein B is more like 1 than 3. It remains unclear as to what the actual structure of this alkaloid is.
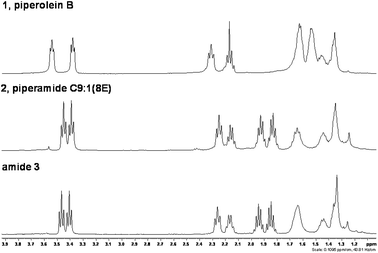 | ||
| Fig. 2 1H NMR spectra showing the clear difference between piperidine and pyrrolidine amides. | ||
| 1H lit. data | 1H amide 3 | 13C lit. data | 13C amide 3 |
|---|---|---|---|
| 1.35–1.50, 14H | 1.29–1.50, 8H | 24.5 | 24.8 |
| 1.56–1.71, 2H | 25.4 | 25.3 | |
| 1.85, 2H, pent | 26.5 | 26.5 | |
| 1.95, 2H, pent | 28.9 | 29.4 | |
| 2.15, 2H, q | 2.12–2.21, 2H | 29.3 | 29.7 |
| 2.30, 2H, t | 2.26, 2H, t | 29.7 | 29.8 |
| 3.40, 2H, t | 3.41, 2H, t | 32.8 | 33.3 |
| 3.55, 2H, t | 3.47, 2H, t | 33.4 | 35.2 |
| 5.90, 2H, s | 5.93, 2H, s | 42.5 | 45.9 |
| 5.95–6.15, 1H | 6.04, 1H, dt | 46.6 | 47.0 |
| 6.25, 1H, d | 6.28, 1H, d | 100.8 | 101.2 |
| 6.60–7.00, 3H | 6.71–6.89, 3H | 106.0 | 105.7 |
| 108.5 | 108.5 | ||
| 119.6 | 120.5 | ||
| 128.0 | 129.6 | ||
| 129.2 | 129.7 | ||
| 132.4 | 132.8 | ||
| 146.4 | 146.8 | ||
| 147.8 | 148.2 | ||
| 171.4 | 172.3 |
As the potential anti-proliferative effects of these compounds on carcinoma cells remain virtually unexplored, we were interested in comparing and contrasting the biological effects of these three structurally very similar amides on cancer cells. Importantly, unlike the majority of previously published studies that routinely involve testing compounds on single cell lines, which in most cases are not closely representative of the tumors of origin, we assessed the effect of compounds 1, 2, and 3 on the colorectal carcinoma cell line HCT116 and the urothelial (urinary bladder) carcinoma cell line EJ. We have previously reported that these cell lines are highly representative of the original tumors of origin and have characterized their responses to anti-proliferative (pro-apoptotic) responses in vitro.11 This allowed us not only to determine the effect of the compounds on relevant representative cell lines but also permitted cell type-specific distinctions to be made. In addition to examining cytotoxic effects on cell cultures of standard cell density, i.e. 5–6 × 103 cells per well,12 we also tested for biological effects using cultures of lower cell density (3 × 103 cells per well) in order to compare our findings to a previous report in which cytotoxicity detection assays were performed using relatively low density cultures of carcinoma (HeLa) cells to study the effects of isopiperolein B (Fig. 3).8
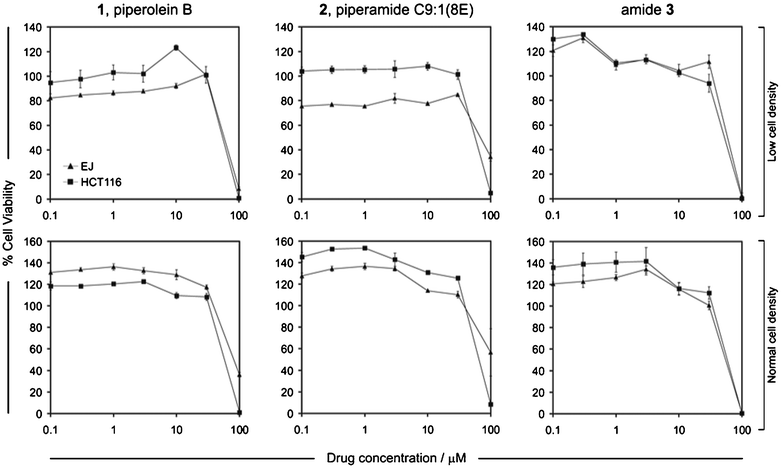 | ||
| Fig. 3 The effect of the synthetic compounds 1, 2 and 3 on carcinoma cell viability. HCT116 and EJ carcinoma cells were grown at 3 × 103 cells per well (low cell density) and 6 × 103 cells per well (normal cell density), before being treated with the indicated doses of compounds 1, 2 and 3. Cell viability (expressed as % in relation to controls) was assessed 48 h later as described in the experimental section and results are expressed as mean % values (± standard error of the mean). Results are mean values for six technical replicates and are representative of three independent experiments. | ||
Piperolein B, 1 and piperamide C9:1(8E), 2 showed little (if any) detectable cytotoxicity at concentrations below 10 μM. In comparison, amide 3 showed some cytotoxicity at 10 μM, more significant effects at 30 μM and it caused complete cellular death in all cultures at higher concentrations (100 μM). This was independent of the initial seeding density of the cell cultures. All compounds demonstrated cytotoxicity at concentrations over 30 μM but only compound 3 resulted in 100% loss of viability in both cell lines and was hence the most cytotoxic independent of cell culture density and cell type. Interestingly, treatment with compound 1 at 100 μM caused 100% death only in HCT116 cells but not in EJ cells, indicating a differential effect between the cell lines. This however was only noticeable when cells were cultured at normal (but not low) density. Such a cell type-dependent differential response was even more pronounced in the case of compound 2; at high doses, the compound showed almost 100% toxicity in HCT116 cells but the effect was much less pronounced in EJ cells which showed 60% cell viability.
Morphological observation of the cultures by routine, phase contrast microscopy indicated that compound 3 caused rapid and extensive cell death within less than 24 h after treatment and complete death in cultures of both cell lines by 48 h. The effect of the other two compounds was slower, and particularly in the case of compound 2 for EJ cells only a proportion of cells appeared fully apoptotic (dead) and a fraction of these remained alive.
Our findings indicate that the choice of initial cell density might be important if subtle cytotoxicity capacities and cell type-specific effects are to be revealed. The example of compound 1 exemplifies this point: the use of low cell density indicated complete cytotoxicity, yet the use of normal (standard) density revealed a dose and cell type-specific response. Thus the use of cultures that are of too low confluence might mask cell type specific responses and also suggest misleadingly high cytotoxic capacity.
It should also be noted that when treated at sub-lethal drug concentrations, most cultures showed an increased level of growth, i.e. over 100% cell viability in comparison to negative controls. This is not an anomalous observation that was specific to our experiments. Our recent in vitro findings (unpublished) and observations by others13 both in vitro and in vivo suggest that both normal and cancer epithelial cells exhibit a biphasic response to cytotoxic chemicals, consisting of increased growth at sub-cytotoxic drug concentrations, yet dramatic cytotoxicity at higher doses. This appears to be an inherent ability of epithelial cells to respond to such ‘insults’ as part of a repair response.13a
Collectively, our results demonstrate for the first time that all of the three compounds exhibit differential effects on carcinoma cells and these effects appear to be both dose- and cell type-dependent. Of the three compounds, 3, the reported structure of isopiperolein B, was the most cytotoxic and piperamide C9:1(8E), 2 was the least cytotoxic demonstrating that the length of the connecting alkyl chain has an impact on activity. Consideration of the structures and the observed differences in their cytotoxic capacities could be used as a basis for structure–activity studies as well as for detailed biological investigations in order to understand the structural basis for these activities. In this way, the design of compounds with improved activities and pharmacological properties for anticancer therapy should be possible.
Conclusion
A simple synthetic strategy to three structurally related Piper compounds has been developed and their cytotoxic properties studied. Critically, the assigned structure of isopiperolein B has been shown not to be that of amide 3, although compound 3 was the most cytotoxic compound of the three tested.Experimental
1H NMR spectra were recorded at 400 MHz. Chemical shifts are reported in ppm from tetramethylsilane with the solvent resonance as the internal standard (CDCl3: 7.26 ppm). 13C NMR were recorded with complete proton decoupling. Chemical shifts are reported in ppm from tetramethylsilane with the solvent as the internal standard (CDCl3: 77.4 ppm). Mass spectrometry (m/z) was performed in ESI mode, with only molecular ions being reported. Infrared (IR) spectra νmax are reported in cm−1. All purchased reagents were used as received without further purification. Petroleum ether refers to the fraction boiling at 40–60 °C. All reactions were performed under a N2 atmosphere.General procedure for amidation
1-Propanephosphonic acid anhydride solution (T3P; 50% in DMF, 1.39 mL, 4.8 mmol, 3 equiv.) was dissolved in CH2Cl2 (5 mL) and cooled to 0 °C. Triethylamine (1.3 mL, 9.6 mmol, 6 equiv.) and the carboxylic acid (1.6 mmol, 1 equiv.) were added. The reaction mixture was left to stir at 0 °C for 0.5 h, then piperidine or pyrrolidine (1.6 mmol, 1 equiv.) was added. After stirring overnight at room temperature the reaction was quenched with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The combined organics were dried (MgSO4), filtered and concentrated under vacuum to provide the corresponding amide. The crude products were deemed to be sufficiently pure and no further purification was performed.General procedure for cross metathesis
Amide (0.20 mmol, 1 equiv.) and 5-vinylbenzo[d][1,3]dioxole 9 (120 mg, 0.81 mmol, 4 equiv.) were dissolved in 1,2-DCE (5 mL). Grubbs' second generation catalyst (18 mg, 0.020 mmol, 0.1 equiv.) was added and the mixture was heated at reflux for 2 days. The reaction mixture was concentrated under vacuum and the residue purified by flash chromatography (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether–EtOAc).
:1 petroleum ether–EtOAc).
Assessment of compound cytotoxicity
The colorectal carcinoma HCT116 and bladder carcinoma EJ cell lines were used to assess the effect of the synthetic compounds on cancer cell viability. Cells were seeded in 96-well plates at low (3 × 103 cells per well) and normal (6 × 103 cells per well) density in culture medium, which was a 1:1 (v/v) mixture of DMEM and RPMI (SigmaAldrich) supplemented with 5% (v/v) fetal bovine serum. Cells were incubated at 37 °C in a humidified atmosphere containing 5% (v/v) CO2 and permitted to adhere overnight. Following synthesis, the compounds were aseptically reconstituted in DMSO at a final concentration of 20 mM. For testing, all compounds were diluted to concentrations 0.1, 0.3, 1, 3, 10 and 30 and 100 μM in culture medium. For each condition, six replicate samples were prepared. Solvent-alone treatments were also included for comparison (controls). Cells were then cultured for a period of 48 h before cell viability assays were performed. To assess cell viability, the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) was used, which is a colorimetric MTS viability assay based on the principle that viable cells have the ability to bioreduce the MTS tetrazolium compound into a coloured formazan product which is soluble in culture medium. Following drug treatment of cells for 48 h, CellTiter reagent was added to each well (as recommended by the manufacturer). After a 4-hour incubation at 37 °C, cell viability was determined by measuring absorbance at 492 nm on a FLUOStar OPTIMA plate reader (BMG LabTech). Absorbance readings were obtained following background subtraction and % cell viability was calculated for each drug concentration in comparison to controls.
Notes and references
- For reviews, see:
(a) J. C. do Nascimento, V. F. de Paula, J. M. David and J. P. David, Quim. Nova, 2012, 35, 2288 CrossRef CAS
; (b) V. S. Parmar, S. C. Jain, K. S. Bisht, R. Jain, P. Taneja, A. Jha, O. D. Tyagi, A. K. Prasad, J. Wengel, C. E. Olsen and P. M. Boll, Phytochemistry, 1997, 46, 597 CrossRef CAS
- F. Kiuchi, N. Nakamura, Y. Tsuda, K. Kondo and H. Yoshimura, Chem. Pharm. Bull., 1988, 36, 2452 CrossRef CAS
-
(a) H. Matsuda, K. Ninomiya, T. Morikawa, D. Yasuda, I. Yamaguchi and M. Yoshikawa, Bioorg. Med. Chem., 2009, 17, 7313 CrossRef CAS
-
(a) S. W. Lee, M.-C. Rho, H. R. Park, J.-H. Choi, J. Y. Kang, J. W. Lee, K. Kim, H. S. Lee and Y. K. Kim, J. Agric. Food Chem., 2006, 54, 9759 CrossRef CAS
- Y. Okumura, M. Narukawa, Y. Iwasaki, A. Ishikawa, H. Matsuda, M. Yoshikawa and T. Watanabe, Biosci., Biotechnol., Biochem., 2010, 74, 1068 CrossRef CAS
- P. V. Srinivas and J. M. Rao, Phytochemistry, 1999, 52, 957 CrossRef CAS
- S. V. Reddy, P. V. Srinivas, B. Praveen, K. H. Kishore, B. C. Raju, U. S. Murthy and J. M. Rao, Phytochemistry, 2004, 11, 697 CAS
- G.-H. Tang, D.-M. Chen, B.-Y. Qiu, L. Sheng, Y.-H. Wang, G.-W. Hu, F.-W. Zhao, L.-J. Ma, H. Wang, Q.-Q. Huang, J.-J. Xu, C.-L. Long and J. Li, J. Nat. Prod., 2011, 74, 45 CrossRef CAS
-
(a) G. M. Strunz and H. J. Finlay, Can. J. Chem., 1996, 74, 419 CrossRef CAS
-
(a) K. Wei, S. Wang, Z. Liu, Y. Du, X. Shi, T. Qi and S. Ji, Tetrahedron Lett., 2013, 54, 2264 CrossRef CAS
-
(a) N. T. Georgopoulos, A. Merrick, N. Scott, P. J. Selby, A. Melcher and L. K. Trejdosiewicz, Int. J. Cancer, 2007, 121, 1373 CrossRef CAS
- N. T. Georgopoulos, L. A. Kirkwood, D. C. Walker and J. Southgate, PLoS One, 2010, 5, e13621 Search PubMed
-
(a) R. Paus, I. S. Haslam, A. A. Sharov and V. A. Botchkarev, Lancet Oncol., 2013, 14, e50 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for compounds 1, 2, 3, 6, 7 and 8. See DOI: 10.1039/c3ra42060d |
| ‡ These authors contributed equally to this work. |
| § Joint senior authors. |
| This journal is © The Royal Society of Chemistry 2013 |