 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The importance of water exclusion: an effective design strategy for detection of Al3+ ions with high sensitivity†‡
Joydev Hatai, Mousumi Samanta, V. Siva Rama Krishna, Suman Pal and Subhajit Bandyopadhyay*
Indian Institute of Science Education and Research (IISER) Kolkata, BCKV Main Campus PO, Mohanpur, Nadia WB 741252, India. E-mail: sb1@iiserkol.ac.in
First published on 5th September 2013
Abstract
Incorporation of hydrophobic units in a chemosensor for detection of ions in aqueous media seems counterintuitive. However, this work demonstrates that a sensor designed by incorporating local hydrophobicity around a multidentate receptor to achieve desolvation of a strongly hydrated Al3+ ion results in efficient binding of the ion, and consequently elicits a strong fluorescence response. The simultaneous detection of Fe3+ in water was suppressed by using the differential behaviour of the sensor in a methanol–water mixture. The sensor shows a remarkable detection limit of 57 nM for Al3+ which is among one of the lowest reported so far.
Introduction
Aluminium is the third most abundant metal ion in the Earth’s crust, yet it is toxic to biological systems.1 It is used in our daily lives, often as packaging material. Aluminium hydroxide combined with calcium hydroxide is used as over-the-counter medication for gastric hyperacidity. Aluminium chlorohydrate is used in deodorants as an antiperspirant. Aluminium salts are neurotoxic and are suspected to induce Parkinsons' disease1 and senile dementia, commonly known as Alzheimer's disease.2 It was also found that aluminium induces DNA damage and inhibits the repair of radiation-induced lesions in human peripheral blood lymphocytes.3 The aluminium ion is therefore a biologically important analyte and its monitoring is vital. However, in aqueous solution aluminium is strongly hydrated and consequently its coordination ability is poor.4 Development of optical sensors for aluminium that can detect this metal ion in aqueous media has hence been challenging. This problem is compounded by the fact that detection of the aluminium ion is often interfered by other ions and pH conditions.5 Consequently, compared to transition metal ions, there are drastically fewer fluorescent chemosensors for Al3+. Most Al3+ sensors work in organic media and this restricts their practical applications.6 The number of reports of chemosensors that can detect Al3+ in aqueous media is few.5a,b,7 Thus, the development of new ratiometric sensors for Al3+ with improved detection limits in the presence of water is desirable. Here we present a novel design strategy for the synthesis of a ratiometric fluorescent sensor for Al3+ in aqueous media with high sensitivity.Measures to overcome the key problem for the detection of Al3+ in water, namely, its strong hydration and weak coordination have been addressed in the design. This includes the introduction of local hydrophobicity and steric crowding around the metal binding sites by incorporation of multiple aryl units in the molecule which, at the same time, serve as the fluorophore. In addition, multiple donor centres in the molecule were introduced to ensure a strong chelate effect. It was envisioned that a combination of these two elements in the ligand would synergetically work towards exclusion of water8 from the metal and would ensure a high sensitivity for Al3+. Use of steric bulk to stabilize sensitive hydrido and chloro derivatives of aluminium is well known from the work of Power and coworkers, where the bulky substituents around the metal centres prevented further coordination.9 Sterically encumbered groups were used for desolvation of metal ions such as Eu and Yb by Hauber and Niemeyer.10 However, there is hardly any work extending this concept to design chemosensors for analytical detection of Al3+, perhaps because of the low solubility of such ligands in aqueous media. Given the fact that bulky ligands have low solubility in aqueous samples, it might seem counterintuitive to introduce hydrophobicity to desolvate water molecules from a hydrated aluminium ion. However, in this work we demonstrate that it can indeed be done. When the chemosensor is used with 1% DMSO as a cosolvent, it can efficiently detect Al3+ in aqueous media. In addition to the detection of Al3+, the sensor also detects Fe3+ in water. Recently, modulation of selectivity for metal ions was achieved with a change in solvent.11 Exploiting the same principle, i.e., differential behavior of the sensor towards the two ions in two different media, the simultaneous detection of Fe3+ in water was achieved. The selectivity of the sensor in MeOH–water is remarkably higher for Al3+. Thus, the addition of MeOH to an aqueous sample displays a ratiometric behavior with turn-on of fluorescence only if Al3+ ions are present. It may be noted that the naphthoyl group is not an ideal fluorophore for bioimaging because of its excitation and emission in the UV region, however it does not pose any problems for the in vitro detection of metal ions.
Results and discussion
Synthesis and fluorescence studies in MeOH and water
The synthesis of 1 was achieved following Scheme 1 (details in the Experimental section). Compound 2 was obtained by nucleophilic attack of o-phenylene diamine with bromomethyl compound 3 (ref. 12) in a single step. The reaction of 2 with naphthoyl chloride in the presence of triethyl amine afforded compound 1, which on purification gave a white solid in 60% yield.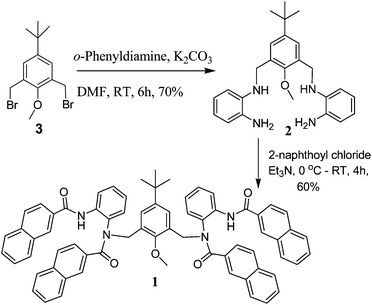 | ||
| Scheme 1 Preparation of chemosensor 1. | ||
The metal binding properties of chemosensor 1 (10 μM) were initially studied with fluorescence spectroscopy in MeOH with 1% DMSO as a cosolvent (Fig. 1). The spectra were recorded on addition of various alkali, alkaline earth and transition metal ions (Cr3+, Mn2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ca2+, Cd2+, Li+, Ba2+, Na+, Hg2+, Pb2+, Ag+ and Al3+). The emission behaviour was investigated upon excitation of the naphthoyl fluorophore at 290 nm (excitation and emission slits of 3 nm). Among the ions mentioned above, chemosensor 1 displayed a significant enhancement of the emission band, with a visual change from colourless to bright blue in the presence of Al3+ (inset, Fig. 1) under a 366 nm handheld UV light. For the other metal ions there was either a small enhancement of fluorescence (Cu2+, Cr3+) or a small quenching effect (Ni2+, Co2+). As shown in Fig. 1, no ratiometric behaviour of fluorescence was observed when MeOH was used as the solvent. The fluorescence behaviour of chemosensor 1 with Al3+ was then investigated with different proportions of MeOH and H2O. As the proportion of water was increased to 40% (H2O–MeOH, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v, with 1% DMSO as a cosolvent), the emission spectrum of 1 (10 μM, λex = 290 nm, slit widths = 3/3) showed three weak fluorescence bands at 335, 355 and 369 nm (Fig. 2). The quantum yield (Φ) of the sensor 1 under those conditions was found to be 0.074.
:3, v/v, with 1% DMSO as a cosolvent), the emission spectrum of 1 (10 μM, λex = 290 nm, slit widths = 3/3) showed three weak fluorescence bands at 335, 355 and 369 nm (Fig. 2). The quantum yield (Φ) of the sensor 1 under those conditions was found to be 0.074.
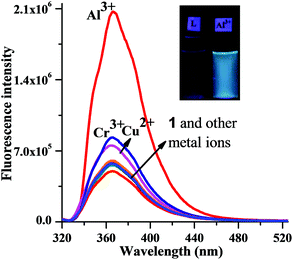 | ||
| Fig. 1 Change in the fluorescence intensity (λex = 290 nm, slit widths = 3/3) of receptor 1 (10 μM) upon addition of different metal salts (20 μM) in MeOH with 1% DMSO as a cosolvent at 25 °C. Inset: visual image taken under 366 nm UV-light of chemosensor 1 (50 μM) with Al3+ (50 μM). | ||
![Fluorescent titrations (λex = 290 nm, slit widths 3/3) of chemosensor 1 (10 μM) with Al3+ (0 to 50 μM) in mixed aqueous–organic media (H2O–MeOH, 2 : 3, v/v, 1% DMSO as a cosolvent, pH = 7.0) at 25 °C. Inset A: ratiometric fluorescence intensity [F375/F335] as a function of Al3+ added. Inset B: a close-up of the isoemissive point.](/image/article/2013/RA/c3ra42043d/c3ra42043d-f2.gif) | ||
| Fig. 2 Fluorescent titrations (λex = 290 nm, slit widths 3/3) of chemosensor 1 (10 μM) with Al3+ (0 to 50 μM) in mixed aqueous–organic media (H2O–MeOH, 2:3, v/v, 1% DMSO as a cosolvent, pH = 7.0) at 25 °C. Inset A: ratiometric fluorescence intensity [F375/F335] as a function of Al3+ added. Inset B: a close-up of the isoemissive point. | ||
Upon addition of Al3+ the emission band at 335 nm gradually decreased, while the bands at 355 and 367 nm underwent a red shift to 375 nm with a clear isoemissive point at 340 nm, along with a fivefold increase in fluorescence intensity. The value of Φ increased from 0.074 to 0.404. The ratio of intensity (F375/F335) at two wavelengths, 375 and 335 nm, steadily increased up to the addition of one equivalent of Al3+and then reached a saturation value with 4 equivalents of the metal ion (inset A, Fig. 2).
For practical applications, particularly for the analysis of contaminated water samples, it is crucial for the sensor to work under aqueous conditions. The solubility of sensor 1 in pure water (Milli-Q) was low. Organic ligands at neutral pH often possess poor solubility in water and hence their aqueous solutions are made by dilution from a stock solution prepared in an organic solvent. Thus, an optically transparent aqueous solution of chemosensor 1 was prepared by dilution of a 1 mM DMSO stock solution with Milli-Q water (H2O–DMSO, 99:1, v/v).
The fluorescence behaviour of chemosensor 1 (10 μM, λex = 290 nm, slit width 4/4) with Al3+ in the aqueous media (99:1 H2O–DMSO, v/v) was measured in the presence of TRIS, buffered at pH 7.0. On addition of Al3+, the value of Φ under these conditions increased from 0.06 to 0.20. The results shown in Fig. 3B indicate that the sensor also displays a ratiometric behaviour in aqueous media. The fluorescence intensity of the 340 nm band gradually decreased with addition of Al3+ whereas the intensity of the red shifted band at 375 nm increased with an isoemissive point at 352 nm.
![(A) Change in fluorescence intensity of receptor 1 (10 μM) upon addition of various metal salts (400 μM) in H2O (pH = 7.0) with 1% DMSO as a cosolvent at 25 °C (λex = 290 nm, slit width = 4/4). (B) Fluorescent titrations of chemosensor 1 (10 μM) with Al3+ in H2O, (buffered at pH = 7.0) with 1% DMSO as a cosolvent at 25 °C (λex = 290 nm, slit width 4/4). Inset: ratiometric fluorescence intensity [F375/F340] as function of Al3+ added.](/image/article/2013/RA/c3ra42043d/c3ra42043d-f3.gif) | ||
| Fig. 3 (A) Change in fluorescence intensity of receptor 1 (10 μM) upon addition of various metal salts (400 μM) in H2O (pH = 7.0) with 1% DMSO as a cosolvent at 25 °C (λex = 290 nm, slit width = 4/4). (B) Fluorescent titrations of chemosensor 1 (10 μM) with Al3+ in H2O, (buffered at pH = 7.0) with 1% DMSO as a cosolvent at 25 °C (λex = 290 nm, slit width 4/4). Inset: ratiometric fluorescence intensity [F375/F340] as function of Al3+ added. | ||
The emission intensity of 1 as well as the fluorescence enhancement upon addition of Al3+ was lower in the aqueous medium compared to that in either methanol or mixed aqueous–organic media, however, the increase in F375/F335 ratio in aqueous media had a linear behaviour up to a higher concentration of Al3+ (Fig. 3B, inset), making it suitable for quantitative estimation of Al3+ over a larger range of concentrations. In addition to Al3+, Fe3+ also showed an enhancement of fluorescence in water (Fig. 3A), a phenomenon that is often encountered with chemosensors for Al3+.5e Interestingly in reports of Al3+ sensors, the response to Fe3+ or Cr3+ often goes unreported.13 The fluorescence response of Cr3+ in water, although observed in our case, was less pronounced (Fig. 3A).
Competitive experiments and optimum pH range
A common limitation of the detection of Al3+ is interference by other cationic and anionic species.5,14 To test the selectivity and suitability of 1 for practical applications as a chemosensor for Al3+, competing experiments were carried out in the presence of various metal ions. For this purpose, emission spectra were recorded with solutions of 1 (10 μM) containing 5 equivalents of either Cr3+, Mn2+, Fe2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ca2+, Cd2+, Li+, Na+, Ba2+, Na+, Hg2+, Pb2+ or Ag+ followed by the addition of Al3+ (20 μM) in aqueous media (H2O, 1% DMSO v/v, as a cosolvent, pH = 7.0). Fig. 4A clearly demonstrates that detection of Al3+ is possible without any significant interference in the presence of the other metal ions.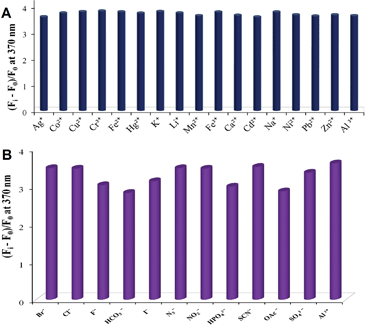 | ||
| Fig. 4 (A) Change in the ratio (Fi − F0)/F0 of fluorescence intensity (λex = 290 nm, slit widths = 3/3) of chemosensor 1 (10 μM) upon addition of Al3+ (20 μM) in the presence of competitive metal ions (50 μM) in aqueous media (H2O with 1% DMSO v/v, pH = 7.0). (B) Relative change in fluorescence intensity of chemosensor 1 (10 μM) at 370 nm with Al3+ (20 μM) in the presence of sodium salts of various anions (20 μM) in aqueous media buffered with TRIS at pH = 7. | ||
Classically, in qualitative inorganic analysis, several anions such as fluoride, acetate and phosphates are known as interfering ions since in their presence metal ions cannot be detected.15 The activity of the sensor 1 towards Al3+ was tested in the presence of various anions including the interfering ones.
The emission spectrum of 1 (10 μM) after the addition of Al3+ (20 μM) was recorded in the presence of Cl−, Br−, I−, SO42−, HCO3−, SCN−, N3−, H2PO4−, F−, OAc−, NO2−, NO3−, and ClO4−(20 μM) in the aqueous media at 25 °C. The results shown in Fig. 4B indicate that a small decrease (∼20%) in the emission intensity was observed with fluoride, biphosphate and acetate. Presence of other anions hardly affected the fluorescence intensity.
These experiments ascertain that chemosensor 1 can be used as a fluorescence sensor for Al3+ in the presence of the most of the interfering and various environmentally relevant anions. Photo-induced electron transfer (PET) processes have vastly been exploited in fluorescence based ion-sensing since its inception by Czarnick and de Silva.16 Akin to most of the reported cases with nitrogen containing ligands,6a,c,11 it was thought that the weak fluorescence of sensor 1 in the absence of Al3+ was due to a PET process from the nitrogen of the benzene-1,2-diamine moiety to the electron deficient naphthoyl fluorophore. The chelation of Al3+ with the donor atoms restricts the PET process, and consequently restores the fluorescence of the fluorophore.5f The small red shift was also consistent with the PET-inhibition mechanism.
Binding mode
For practical applications, and to gain mechanistic insight of the fluorescence enhancement, pH dependence of the chemosensor 1 was carried out. Sensor 1 can detect Al3+ effectively from pH 5 to 8. At lower pH (Fig. 5), enhancement of fluorescence was observed which is typical for a PET process with a proton sensitive donor centre.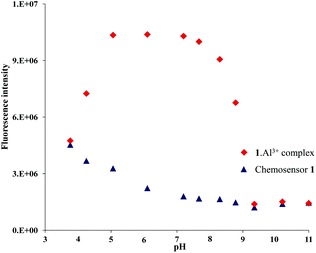 | ||
| Fig. 5 Change in the fluorescence intensity of the emission band at 370 nm of chemosensor 1 and the 1·Al3+ complex at different pH values. | ||
The enhancement at pH 3.8 with and without Al3+ was essentially the same. The enhancement in the fluorescence intensity of sensor 1 (10 μM) in the absence of any metal ions at pH 3.8 indicates that protonation at the donor atom of 1 is responsible for the PET process.5b,12 The fluorescence intensity of 1 with Al3+ at pH > 8.5 is low presumably because of the decomplexation of Al3+ from the [1·Al3+] complex by the OH− ions.17
A 1:1 stoichiometry was established from a Job's plot obtained from the fluorescence data at a total concentration of 8 μM of 1 and Al3+ (Fig. 6). Moreover, a peak at 1173 in the electrospray ionization mass spectrometry (ESI-MS) under these conditions provided additional evidence for the formation of a 1:1 complex of 1 with Al3+ (ESI, Fig. S2†). From the titration data, the dissociation constant (Kd) of 1 with Al3+ was found to be 1.7 × 10−6 M (Fig. 7).
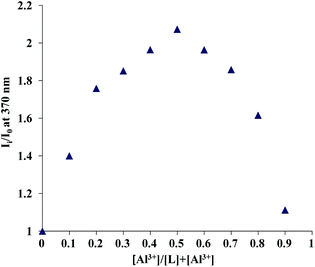 | ||
| Fig. 6 Job's plot for complexation of 1 with Al3+ plotted with emission data at 370 nm. The total concentration of the ligand and the metal was kept constant (8 μM) for each of the data sets. | ||
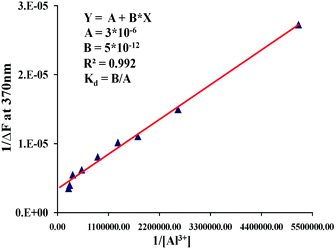 | ||
| Fig. 7 Benesi–Hildebrand plot for determination of binding constant Al3+ with chemosensor 1 in mixed aqueous–organic media (H2O–MeOH, 2:3, v/v, 1% DMSO as a cosolvent, pH = 7.0) at 25 °C. The dissociation constant (Kd) value is 1.7 × 10−6 M. | ||
To have an idea of the donor atom for the PET process, semiempirical ZINDO calculations were performed on the optimized geometry of the molecule 1.18 The orbital contour (Fig. 8) of the highest occupied molecular orbital suggests that the donor oxygen is most likely responsible for the PET process. The lowest unoccupied level is largely naphthalene based. Whereas the highest occupied level, contrary to our previous speculation of being N based, is rather localized on the oxygen of the central aryl unit. The difference in selectivity in the organic and the aqueous media is presumably because of the differential solvation of the metal ion. An additional reason for the fluorescence enhancement in aqueous media could be the enhanced solubility of the charged complexes in water which might circumvent the stacking and aggregation of the hydrophobic fluorophore, as recently observed by Fahrni and coworkers for fluorescent probes for Cu(I).19
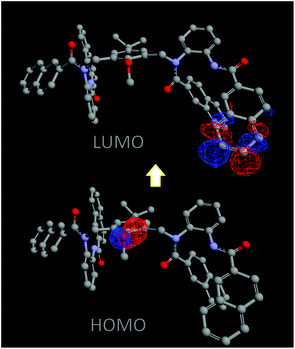 | ||
| Fig. 8 Calculated contour of the frontier orbitals. | ||
To elucidate the binding mode, 1H NMR titrations were carried out upon the addition of Al3+ to a solution of chemosensor 1 (1 mM) in CD3CN since the signals of the solutions in D2O were weak and had a low signal to noise ratio. Upon addition of Al3+ to the chemosensor 1, changes in the aromatic region of the 1H NMR spectra were difficult to decipher among the complex overlapping peaks (ESI, Fig. S6†).
However, clear changes were observed for the amide protons (Fig. 9A). The peaks at δ 8.59 and 8.42 of the chemosensor corresponding to the N–H protons shifted downfield to δ 8.81 and 8.78 respectively with addition of Al3+. This indicated the likely involvement of the amide moiety for the binding of Al3+. The OCH3 proton resonance at δ 3.98 also underwent a small downfield shift indicating the role of the oxygen of the anisole moiety in the complexation.20 Although the role of the amide was apparent from the NMR experiment, whether the coordination was through N or the O of the amide moiety was still inconclusive.
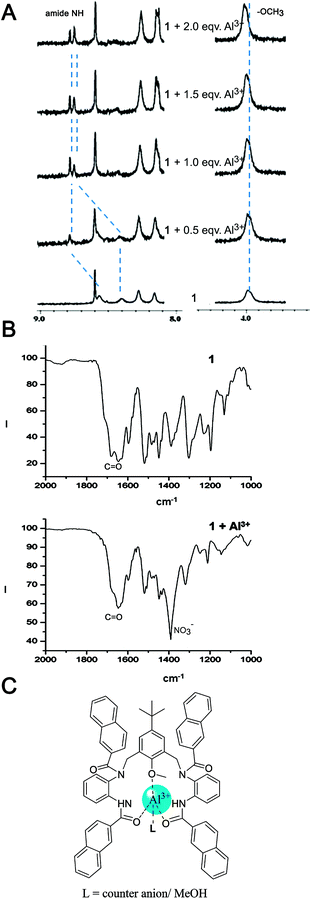 | ||
| Fig. 9 (A) 1H NMR spectra of chemosensor 1 upon addition of 0, 0.5, 1.0, 1.5, 2.0 equivalents of Al(NO3)3 in CD3CN; (B) IR spectra of chemosensor 1 in the presence and absence of Al3+; (C) putative binding mode based on the spectral data. | ||
To find an answer to that question, infrared spectroscopic studies of the free chemosensor and the chemosensor in the presence of Al3+ were carried out. In the IR spectra of the free chemosensor, multiple bands corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch were present due to the presence of a pair of secondary and a pair of tertiary amide groups (Fig. 9B). The CO band at 1681 cm−1 shifted by −36 cm−1 to 1645 cm−1 and merged with the bands in that region indicating coordination of the amide oxygen to the metal ion. An N-coordination would have shifted the band to a higher wavenumber. It might be noted that the intense band at ca. 1390 cm−1 is due to the presence of nitrate which was the counter anion of the aluminium salt. Thus, the combination of all the spectroscopic data coupled with the calculation suggest that the chelation involved multiple oxygen donors, namely the –OCH3 and CO of the secondary amide, but not through the amide nitrogens. Based on these data, a putative binding model has been presented in Fig. 9C.
O stretch were present due to the presence of a pair of secondary and a pair of tertiary amide groups (Fig. 9B). The CO band at 1681 cm−1 shifted by −36 cm−1 to 1645 cm−1 and merged with the bands in that region indicating coordination of the amide oxygen to the metal ion. An N-coordination would have shifted the band to a higher wavenumber. It might be noted that the intense band at ca. 1390 cm−1 is due to the presence of nitrate which was the counter anion of the aluminium salt. Thus, the combination of all the spectroscopic data coupled with the calculation suggest that the chelation involved multiple oxygen donors, namely the –OCH3 and CO of the secondary amide, but not through the amide nitrogens. Based on these data, a putative binding model has been presented in Fig. 9C.
Time course of fluorescence response, detection in water samples and the detection limit
The time course of the fluorescence response of sensor 1 was studied. The response was almost instantaneous. The response measured after 30 s of addition of Al3+ to the ligand in water (99%) showed a saturation of the fluorescence signal (ESI, Fig. S7†).The response in mixed aqueous–organic media was observed even when the concentration was less than 100 nM (Fig. S8†). The analytical detection limit, determined by standard methods,21 from the linear regression of the normalized fluorescence response curve at low concentration of Al3+ (Fig. 10) was found to be 57 nM in mixed aqueous–organic media which is far within the acceptable limit of 0.05 mg L−1 (1.85 μM) as suggested by the US-EPA for drinking water.22
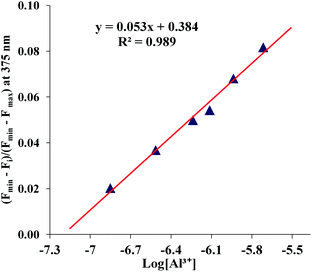 | ||
| Fig. 10 Normalized fluorescence intensity (λex = 290 nm, λem = 375 nm) of chemosensor 1 (10 μM) at each concentration of Al3+ in mixed aqueous–organic media (H2O–MeOH, 2:3, v/v, 1% DMSO as a cosolvent, pH = 7.0) at 25 °C. A linear curve was obtained from these normalized fluorescence intensity data. The analytical detection limit thus obtained was 5.7 × 10−8 M. | ||
It is also worth mentioning that there is only one other report of a fluorescence sensor that has a lower detection limit.7d
To test the practical use of our system, aluminium nitrate was added to water samples collected directly from the tap and these were subsequently used for fluorescence studies. The response was unaffected and was akin to the spectra provided in Fig. 3B.
However, under similar conditions (10 μM of 1 in 99% water, v/v), determination of Al3+ in seawater samples did not work, possibly because of the presence of heavy ions in high concentrations (e.g., ∼0.5 M chloride) that acted as quenchers (ESI, Fig. S9†).
To test whether the fluorescence enhancement of the chemosensor was indeed a result of the complexation with the metal ion and not a chemical reaction or a photoactivation of the probe, a strong metal ion chelator, disodium EDTA (100 μM), was found to revert the enhancement of the fluorescence of a solution of chemosensor 1 (10 μM) containing Al3+ (Fig. 11).23
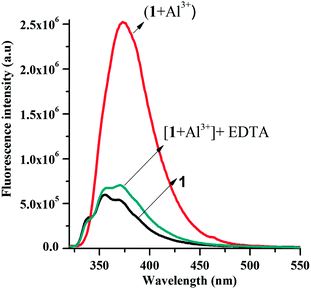 | ||
| Fig. 11 Fluorescence studies for establishing the reversibility in binding of 1 to Al3+ ion: black line: receptor 1 (10 μM); red line: 1 (10 μM) with Al3+ (40 μM); dark green line: 1 (10 μM) with Al3+ (40 μM) followed by addition of EDTA (100 μM). | ||
Conclusion
In conclusion, we have presented a rational design and synthesis of a fluorescent chemosensor for Al3+ that can detect this ion in aqueous media. In water, Fe3+ also responds to the sensor – the differentiation of the two metal ions can be done simply by addition of methanol to the sample, since the selectivity of the sensor in MeOH–H2O (3:2) is significantly higher for Al3+. Another advantage of the chemosensor is the low detection limit (57 nM), one of the lowest reported so far, is well within the US-EPA mandated value for Al3+ in drinking water. Although common interfering ions are known to hinder the detection of Al3+, chemosensor 1 can be used in the presence of most interfering basic and various environmentally relevant anions. However, in seawater samples detection of Al3+ was not possible. Experiments were performed to ensure that the detection of the ion by the chemosensor was indeed a result of the coordination of Al3+ to the chemosensor and the deterrence of the PET process.Experimental
General information
All reactants and reagents were commercially available and were used without further purification unless otherwise indicated. Solvents used were purified and dried using standard methods. The structures of the compounds were determined by nuclear magnetic resonance (NMR). 1H and 13C NMR spectra were recorded using 400 MHz (Jeol) and 500 MHz (Bruker) instruments, respectively. Chemical shifts are reported in δ values relative to an internal reference of tetramethylsilane (TMS) for 1H NMR and the solvent peak for 13C NMR except where noted. The solvents for the spectroscopy experiments were distilled from spectroscopy grade solvents, and were free from any fluorescent impurities. The solutions of metal ions were prepared from Al(NO3)3·9H2O, LiClO4·3H2O, NaClO4, KClO4, Ba(NO3)2·4H2O, Mn(ClO4)2, Fe(ClO4)2·xH2O, Co(ClO4)2·6H2O, Cd(NO3)2, AgNO3, Hg(NO3)2, Pb(ClO4)2, Ca(ClO4)2·4H2O, Cu(ClO4)2·6H2O, Ni(ClO4) and Zn(ClO4)2·6H2O, in MeOH and H2O. IR data were obtained with a FT-IR spectrometer. UV-vis spectra were recorded with a Hitachi U-4100 spectrophotometer. Fluorescence measurements were carried out with a Fluoromax-3 (Horiba Jobin Yvon). Mass data were obtained from an Acquity™ ultra performance LC. pH values were recorded with a Satorious Basic Meter PB-11 calibrated at pH 4, 7 and 10. Reactions were monitored by thin layer chromatography using Merck plates (TLC Silica Gel 60 F254). Developed TLC plates were visualized with UV light (254 nm and 366 nm). Silica gel (100–200 mesh, Merck) was used for column chromatography. Yields refer to the chemical yields of chromatographically and spectroscopically pure compounds.N,N′-(5-tert-Butyl-2-methoxy-1,3-phenylene)bis(methylene)dibenzene-1,2-diamine (5)
Dibromomethyl compound 3 (ref. 8) (0.20 g, 0.57 mmol) dissolved in DMF (5 mL) was added dropwise over a period of 30 min to a mixture of o-phenylenediamine (1.85 g, 17.14 mmol) and K2CO3 (1.58 g, 11.42 mmol) was dissolved in DMF (10 mL) at 0 °C. The reaction mixture was kept at 25 °C and monitored with TLC until the disappearance of the spots for the starting materials was observed. Water (200 mL) was added to the reaction mixture and vigorously stirred for 30 min, extracted with methylene chloride (30 mL × 3) and washed with brine (20 mL × 2). The organic layer was dried over Na2SO4 and the volatiles were evaporated. The residue after chromatography (hexane–ethylacetate) yielded 2 as a brown solid (0.16 g, 70%). 1H NMR (400 MHz, CDCl3) δ 7.34 (s, 2H, ArH), 6.79 (m, 4H, ArH), 6.71 (m, 4H, ArH), 4.32 (s, 4H, ArCH2), 3.84 (s, 3H, –OCH3), 1.27 (s, 9H, tBu); ESI-MS m/z calcd for C25H32N4O: 404, found 405 (M + H+).Chemosensor 1
2-Naphthoyl chloride (0.19 g, 1.0 mmol) dissolved in methylene chloride (3 mL) was added to a mixture of compound 2 (0.10 g, 0.25 mmol) and Et3N (0.20 mL, 1.6 mmol) in methylene chloride (5 mL) over a period of 10 minutes at 0 °C. The reaction mixture was stirred at 25 °C for 4 h, extracted with methylene chloride (15 mL × 2) and volatiles were removed under reduced pressure. The residue on chromatography (DCM–CH3CN, 95:5, v/v) yielded 1 as a white solid (0.15 g, 59%, mp 156–157 °C). 1H NMR (500 MHz, DMSO-d6) δ 9.99 (s, 2H, –NHCO), 8.63 (s, 2H, –ArH), 8.47 (m, 2H, –ArH), 8.13–6.81 (m, 36H, –ArH), 5.64 (m, 2H, –ArCH2), 4.94 (m, 2H, –ArCH2) 3.77 (s, 3H, –OCH3) 1.08 (s, 9H, –tBu). 13C (125 MHz, DMSO-d6) δ 169.74, 167.43, 165.87, 153.77, 145.94, 136.33, 134.91, 134.36, 133.40, 133.36, 133.12, 133.05, 132.13, 132.04, 131.79, 130.50, 129.25, 128.97128.29, 128.14, 128.06, 128.02, 127.86, 127.69, 126.90, 126.78, 126.49, 125.14, 124.45, 61.35, 46.46, 33.81, 30.90. FT-IR (KBr, cm−1): 3413, 3057, 2960, 1681, 1645, 1629, 1519, 1507, 1449, 1390, 1303, 1195, 774, 757. λabs in MeOH (nm, ε): 233 (88000), 281 (17000), 330 (1500). ESI-MS m/z calcd for C69H56N4O5K+: 1059.3888, found 1059.3902.General procedure for the UV-Vis and fluorescence studies
For absorption and emission spectra, stock solutions of metal perchlorate salts (1.0 mM) and the free chemosensor 1 (10 μM) were prepared in MeOH, MeOH–H2O (3:2, v/v) and in H2O respectively with 1% DMSO as the cosolvent. The test solutions were prepared by placing the chemosensor 1 (10 μM) and appropriate metal stock solution in a cuvette with a fixed volume of 2.6 mL. The resulting solution was shaken well before absorption and emission spectra were recorded. Quantum yield data reported here were measured relative to fluorescein in 0.1 N NaOH (Φ = 0.95).24 The integration of the emission spectra were obtained from the Fluoromax-3 instrument directly.Determination of the dissociation constant
The dissociation constant (Kd) was determined from the fluorescence titration experiment using the following equation:25 (Fi − F0) = ΔF = [Al3+](Fmax − F0)/(Kd + [Al3+]), where Fi is the observed fluorescence with varying amounts of Al3+, F0 is the fluorescence for the free chemosensor 1, Fmax is the saturation value of the fluorescence intensity for the Al3+complexes. To obtain a linear equation y = ax + b, reciprocal of the ΔF was plotted against the reciprocal of the concentrations of Al3+. Kd was calculated from the ratio a/b.Determination of the detection limit
The detection limits for the ion were calculated from the titration experiment following the reported method.21 The fluorescence intensity data at the emission maxima were normalised between the minimum intensity found at zero equivalents of Al3+, and the maximum enhanced intensity on addition of the Al3+. A linear curve was obtained from these normalised fluorescence intensity data. The detection limit was obtained by the extrapolation of the straight line on the x axis. Thus the value obtained for Al3+ was found to be 57 nM.Acknowledgements
We thank CSIR for a fellowship to JH, UGC for a fellowship to MS, IISER for SRFs to SP and VSRK. This work has been supported by a research grant from CSIR (01(2717)/13/EMR).Notes and references
- D. P. Perl and A. R. Brody, Science, 1980, 208, 297 CAS.
- D. P. Perl, D. C. Gajdusek, R. M. Garruto, R. T. Yanagihara and C. J. Gibbs, Science, 1982, 217, 1053 CAS.
- A. Lankoff, A. Banasik, A. Duma, D. Ochniak, H. Lisowska, T. Kuszewski, S. Góźdź and A. Wojcik, Toxicol. Lett., 2006, 161, 27 CrossRef CAS PubMed.
- K. Soroka, R. S. Vithanage, D. A. Phillips, B. Walker and P. K. Dasgupta, Anal. Chem., 1987, 59, 629 CrossRef CAS.
- (a) S. Kim, J. Y. Noh, K. Y. Kim, J. H. Kim, H. K. Kang, S.-W. Nam, S. H. Kim, S. Park, C. Kim and J. Kim, Inorg. Chem., 2012, 51, 3597 CrossRef CAS PubMed; (b) Y. Lu, S. Huang, Y. Liu, S. He, L. Zhao and X. Zeng, Org. Lett., 2011, 13, 5274 CrossRef CAS PubMed; (c) M. Arduini, F. Felluga, F. Mancin, P. Rossi, P. Tecilla, U. Tonellato and N. Valentinuzzi, Chem. Commun., 2003, 1606 RSC; (d) X.-H. Jiang, B.-D. Wang, Z.-Y. Yang, Y.-C. Liu, T.-R. Li and Z.-C. Liu, Inorg. Chem. Commun., 2011, 47, 1224 CrossRef PubMed; (e) S. B. Maity and P. K. Bharadwaj, Inorg. Chem., 2013, 52, 1161 CrossRef CAS PubMed; (f) A. Sahana, A. Banerjee, S. Das, S. Lohar, D. Karak, B. Sarkar, S. Mukhopadhyay, A. Mukherjee and D. Das, Org. Biomol. Chem., 2011, 9, 5523 RSC; (g) S. Das, A. Sahana, A. Banerjee, S. Lohar, D. A. Safin, M. G. Babashkina, M. Bolte, Y. Garcia, I. Hauli, S. K. Mukhopadhyay and D. Das, Dalton Trans., 2013, 42, 4757 RSC.
- (a) S. H. Kim, H. S. Choi, J. Kim, S. J. Lee, D. T. Quang and J. S. Kim, Org. Lett., 2010, 12, 560 CrossRef CAS PubMed; (b) D. Maity and T. Govindaraju, Chem. Commun., 2010, 46, 4499 RSC; (c) X. Sun, Y.-W. Wang and Y. Peng, Org. Lett., 2012, 14, 3420 CrossRef CAS PubMed; (d) W. Lin, L. Yuan and J. Feng, Eur. J. Org. Chem., 2008, 3821 CrossRef CAS; (e) A. B. Othman, J. W. Lee, Y. D. Huh, R. Abidi, J. S. Kim and J. Vicens, Tetrahedron, 2007, 63, 10793 CrossRef PubMed; (f) Y.-W. Wang, M.-X. Yu, Y.-H. Yu, Z.-P. Bai, Z. Shen, F.-Y. Li and X.-Z. You, Tetrahedron Lett., 2009, 50, 6169 CrossRef CAS PubMed; (g) D. Maity and T. Govindaraju, Inorg. Chem., 2010, 49, 7229 CrossRef CAS PubMed.
- (a) D. Maity and T. Govindaraju, Chem. Commun., 2012, 48, 1039 RSC; (b) B. K. Datta, C. Kar, A. Basu and G. Das, Tetrahedron Lett., 2013, 54, 771 CrossRef CAS PubMed; (c) K. K. Upadhyay and A. Kumar, Org. Biomol. Chem., 2010, 8, 4892 RSC; (d) L. Ang, W. Qin, X. Tang, W. Dou, W. Liu, Q. Teng and X. Yao, Org. Biomol. Chem., 2010, 8, 3751 RSC; (e) S. Goswami, A. Manna, S. Paul, K. Aich, A. K. Das and S. Chakraborty, Dalton Trans., 2013, 42, 8078 RSC.
- R. Breslow, S. Bandyopadhyay, M. Levine and W. Zhou, ChemBioChem, 2006, 7, 1491 CrossRef CAS PubMed.
- For example see: (a) R. J. Wehmschulte and P. P. Power, J. Am. Chem. Soc., 1996, 118, 791 CrossRef CAS; (b) R. J. Wehmschulte and P. P. Power, Inorg. Chem., 1996, 35, 2717 CrossRef CAS; (c) R. J. Wehmschulte and P. P. Power, Inorg. Chem., 1996, 35, 3262 CrossRef CAS PubMed.
- S. O. Hauber and M. Niemeyer, Inorg. Chem., 2005, 44, 8644 CrossRef CAS PubMed.
- (a) J. Hatai, S. Pal, G. P. Jose, T. Sengupta and S. Bandyopadhyay, RSC Adv., 2012, 2, 7033 RSC; (b) J. Hatai, S. Pal and S. Bandyopadhyay, RSC Adv., 2012, 2, 10941 RSC.
- J. Hatai, S. Pal, G. P. Jose and S. Bandyopadhyay, Inorg. Chem., 2012, 51, 10129 CrossRef CAS PubMed.
- (a) T.-H. Ma, M. Dong, Y.-M. Dong, Y.-W. Wang and Y. Peng, Chem.–Eur. J., 2010, 16, 10313 CrossRef CAS PubMed; (b) W. Lin, L. Yuan and J. Feng, Eur. J. Org. Chem., 2008, 3821 CrossRef CAS.
- A. Shokrollahi, M. Ghaedi, M. S. Niband and H. R. Rajabi, J. Hazard. Mater., 2008, 151, 642 CrossRef CAS PubMed and references therein.
- (a) A. I. Vogel, in Vogel's Qualitative Inorganic Analysis, ed. G. Svehla, Longman, Harlow, 1996 Search PubMed; (b) C. M. Andren and E. Rydin, J. Environ. Monit., 2009, 11, 1639 RSC; (c) K. K. Upadhyay and A. Kumar, Talanta, 2010, 82, 845 CrossRef CAS PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed; (b) D. H. Vance and A. W. Czarnik, J. Am. Chem. Soc., 1994, 116, 9397 CrossRef CAS; (c) M. E. Huston, E. U. Akkaya and A. W. Czarnik, J. Am. Chem. Soc., 1989, 111, 8735 CrossRef CAS; (d) Chemosensors: Principles, Strategies, and Applications, ed. B. Wang and E. V. Anslyn, John Wiley & Sons Inc., Hoboken, NJ, 2011 Search PubMed.
- (a) L. Wang, W. Qin, X. Tang, W. Dou, W. Liu, Q. Teng and X. Yao, Org. Biomol. Chem., 2010, 8, 3751 RSC; (b) J. Hatai, S. Pal and S. Bandyopadhyay, Tetrahedron Lett., 2012, 53, 4357 CrossRef CAS PubMed.
- (a) M. C. Zerner, G. H. Loew, R. F. Kirchner and U. T. Mueller-Westerhoff, J. Am. Chem. Soc., 1980, 102, 589 CrossRef CAS; (b) M. C. Zerner, J. E. Ridley, A. D. Bacon, W. D. Edwards, J. D. Head, J. McKelvey, J. C. Cuberson, P. Knappe, M. G. Cory, B. Weiner, J. D. Baker, W. A. Parkinson, D. Kannis, J. Yu, N. Roesch, M. Kotzian, T. Tamm, M. M. Karelson, X. Zheng, G. Pearl, A. Broo, K. Albert, J. M. Cullen, J. Li, G. D. Hawkins, J. D. Thompson, C. P. Kelly, D. A. Liotard, A. V. Marenich, C. J. Cramer and D. G. Truhlar, ZINDO-MN, version 2011, Quantum Theory Project, University of Florida, Gainesville, and Department of Chemistry, University of Minnesota, Minneapolis, 2011 Search PubMed.
- M. T. Morgan, P. Bagchi and C. J. Fahrni, J. Am. Chem. Soc., 2011, 133, 15906 CrossRef CAS PubMed.
- F. A. Miller and C. H. Wilkins, Anal. Chem., 1952, 24, 1253 CrossRef CAS.
- (a) A. Caballero, R. Martinez, V. Lloveras, I. Ratera, J. Vidal-Gancedo, K. Wurst, A. Tarranga, P. Molina and J. Veciana, J. Am. Chem. Soc., 2005, 127, 15666 CrossRef CAS PubMed; (b) M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414 CrossRef CAS.
- National Secondary Drinking Water Regulations of the US EPA: Maximum Contaminant Levels, http://water.epa.gov/drink/contaminants/secondarystandards.cfm, accessed 24 March 2013.
- M. S. Nasir, C. J. Fahrni, D. A. Suhy, K. J. Kolodsick, C. P. Singer and T. V. O'Halloran, JBIC, J. Biol. Inorg. Chem., 1999, 4, 775 CrossRef CAS.
- E. Coronado, J. R. Galan-Mascaros, C. Marti-Gastaldo, E. Palomares, J. R. Durrant, R. Vilar, M. Gratzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2005, 127, 12351 CrossRef CAS PubMed.
- (a) J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536 CrossRef CAS PubMed; (b) S. Mizukami, S. Okada, S. Kimura and K. Kikuchi, Inorg. Chem., 2009, 48, 7630 CrossRef CAS PubMed; (c) Z. Xu, K.-H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of characterization data for the products and spectroscopic methods. See DOI: 10.1039/c3ra42043d |
| ‡ This paper is dedicated to Prof. R. N. Mukherjee on his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2013 |