 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biphenyl dioxygenase-catalysed cis-dihydroxylation of tricyclic azaarenes: chemoenzymatic synthesis of arene oxide metabolites and furoquinoline alkaloids†
Derek R. Boyd*a, Narain D. Sharmaa, Jonathan G. Carrolla, Pui L. Lokea, Colin R. O'Dowda and Christopher C. R. Allenb
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, UK BT9 5AG. E-mail: dr.boyd@qub.ac.uk; Tel: +44 (0) 28 90974421
bSchool of Biological Sciences, Queen's University Belfast, Belfast, UK BT9 5AG
First published on 3rd May 2013
Abstract
Biotransformation of acridine, dictamnine and 4-chlorofuro[2,3-b]quinolone, using whole cells of Sphingomonas yanoikuyae B8/36, yielded five enantiopure cyclic cis-dihydrodiols, from biphenyl dioxygenase-catalysed dihydroxylation of the carbocyclic rings. cis-Dihydroxylation of the furan ring in dictamnine and 4-chlorofuro[2,3-b]quinoline, followed by ring opening and reduction, yielded two exocyclic diols. The structures and absolute configurations of metabolites have been determined by spectroscopy and stereochemical correlation methods. Enantiopure arene oxide metabolites of acridine and dictamnine have been synthesised, from the corresponding cis-dihydrodiols. The achiral furoquinoline alkaloids robustine, γ-fagarine, haplopine, isohaplopine-3,3′-dimethylallylether and pteleine have been obtained, from either cis-dihydrodiol, catechol or arene oxide metabolites of dictamnine.
Introduction
Polycyclic azaarenes are ubiquitous in the environment as atmospheric pollutants, resulting from incomplete combustion of nitrogen-containing molecules present in fossil fuels or tobacco and also as plant alkaloids.1a,b Some larger members of the family of aza-polycyclic aromatic hydrocarbons (APAHs) present a significant hazard to human health, resulting from the mutagenicity/carcinogenicity of their mammalian metabolites.1c–e The mineralization of APAHs and alkaloids containing azaaromatic rings by soil bacteria, via non-mutagenic/non-carcinogenic metabolites can, therefore, play a useful role in reducing this problem. Earlier bacterial studies from these laboratories have focused on the toluene dioxygenase (TDO)-catalysed biodegradation of bicyclic heterocycles including quinolines,2a,b benzo[b]thiophenes2c and benzo[b]furans,2c using the UV4 mutant strain of Pseudomonas putida (Schemes 1a and 1b). Regioselective cis-dihydroxylation of the carbocyclic and the heterocyclic rings in the quinolines (5,6 and/or 7,8 and/or 2,3 bonds), benzo[b]thiophenes (4,5 and/or 2,3 bonds) and benzo[b]furans (6,7 and/or 2,3 bonds), occurred to give the corresponding cis-dihydrodiol metabolites. The 3-hydroxyquinoline and anthranilic acid metabolites of quinoline were assumed to be derived from the undetected heterocyclic cis-3,4-dihydrodiol intermediate (Scheme 1a).2a,b Further metabolism of the benzo[b]furan 2,3-cis-diols involved spontaneous ring opening and enzyme-catalysed carbonyl reduction to give exocyclic phenolic diol products (Scheme 1b).2c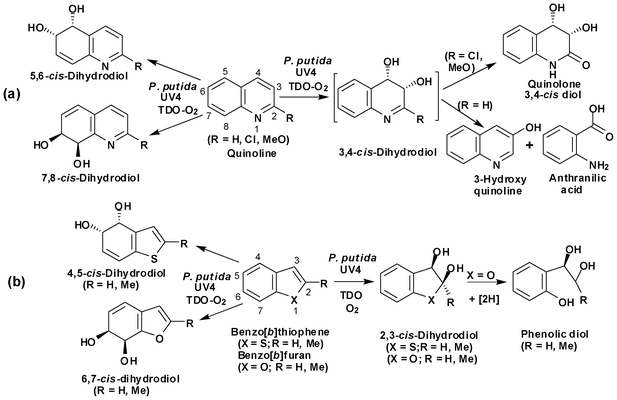 | ||
| Scheme 1 | ||
Dihydroxylation of the 3,4-bond in the electron-deficient pyridine ring of the quinoline substrates was found to yield only minor metabolites in comparison with its carbocyclic 5,6- and 7,8-bonds. However, when benzo[b]thiophene and benzo[b]furan substrates, containing electron-rich heterocyclic rings, were used as substrates, dihydroxylation of the 2,3-bond revealed a more favourable metabolic route (Schemes 1a and 1b).
The steric dimensions of the active site in TDO, expressed in P. putida UV4, limited the acceptable size of substrates to mono- or bi-cyclic arenes (Schemes 1a and 1b). However, the biphenyl dioxygenase (BPDO) enzyme, present in the B8/36 mutant strain of Sphingomonas yanoikuyae, has a larger active site and was able to accept tri-, and tetra-cyclic arenes (e.g. benzo[f]quinoline, benzo[h]quinoline, phenanthridine,3a benzo[c]phenanthridine,3bScheme 2) as substrates. It is noteworthy that in these examples a marked regioselective preference for cis-dihydroxylation was found at a bond within the bay-region.
 | ||
| Scheme 2 | ||
As part of an earlier programme3b,c to investigate the ability of BPDO to catalyse the cis-tetrahydroxylation of larger polycyclic aromatic rings, it was found that bis-cis-dihydrodiols were formed as further metabolites of the initial cis-dihydrodiols derived from larger carbocyclic (e.g. anthracene, chrysene, benz[a]anthracene) and heterocyclic (e.g. acridine, phenazine, benzo[b]naphtha[2,1-d]thiophene) substrates. The similarity in size and shape of the linear tricyclic arenes, anthracene and acridine, and their acceptability as substrates for the BPDO enzyme,3c,d prompted this comparative biotransformation study of acridine with furo[2,3-b]quinoline substrates. Following our earlier reports on the isolation and synthesis4a–e of quinoline alkaloids, from plants of the Rutaceae family, e.g. Choisya ternata, and Skimmia japonica, linear furoquinolines (4-chlorofuro[2,3-b]quinoline and dictamnine) were briefly examined as potential substrates, using whole cells of S. yanoikuyae B8/36 expressing BPDO enzyme.4d
In our preliminary studies of the biotransformations of acridine and dictamnine, using S. yanoikuyae B8/36, we had reported3d,4d the presence of the corresponding cis-dihydrodiol metabolites. This comprehensive study now provides full structural and stereochemical characterization of all new bacterial metabolites and shows how they can be utilized in the chemoenzymatic synthesis of a wider range of animal and plant metabolites, e.g. arene oxides and furoquinoline alkaloids.
Results and discussion
(i) Biotransformation of acridine 1
The mammalian metabolism and mutagenicity of acridine 1 have been studied over many years using dog, rabbit and rat liver cells.5a–d The major metabolites were found to be 2-hydroxyacridine, 9-acridone, 2-hydroxy-9-acridone and trans-1,2-dihydroxy-1,2-dihydroacridine 3 (Scheme 3). It is probable that trans-dihydrodiol 3 and 2-hydroxyacridine were derived from the undetected acridine 1,2-oxide 2. The identification of these mammalian metabolites of acridine 1, which could be accounted for, mainly, by monooxygenase-catalysed oxidation, prompted the preliminary3d and current study of its dioxygenase-catalysed metabolism. | ||
| Scheme 3 | ||
Biotransformation of acridine 1, using S. yanoikuyae B8/36 under similar conditions to those used for other azaarene substrates,3a–d followed by extraction (EtOAc) and column chromatography, yielded cis-dihydrodiol 4 ([α]D +71) in acceptable yield (42%). The structure of cis-diol 4 was determined by NMR, MS and elemental microanalysis. The enantiomeric excess value (ee) was estimated as >98% by reaction with (R)-(+)- and (S)-(−)-2-(1-methoxyethyl)phenylboronic acid (MPBA) and 1H-NMR analysis of the resulting boronates.
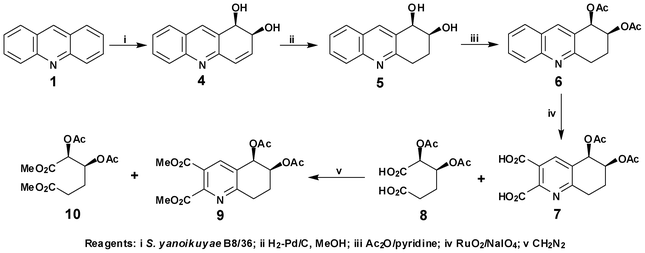
The absolute configuration of cis-dihydrodiol 4 was initially assigned as (1R,2S), based on the well established 1H-NMR pattern previously observed for MPBA derivatives from other polycyclic arene cis-dihydrodiol metabolites (e.g. from naphthalene, anthracene, phenanthrene and their aza-analogues).2a,b,3a,b The observation of a larger chemical shift value (δH 3.18) for the MeO group protons, using the (R)-(+)-MPBA compared with the value obtained using (S)-(−)-MPBA (δH 3.11), was again assumed to be consistent with a benzylic (R) and an allylic (S) configuration for cis-dihydrodiol 4. The reliability of the MPBA method for the linear azaarene cis-dihydrodiol 4 was confirmed by an unequivocal stereochemical correlation sequence similar to that used for other polycyclic arene cis-dihydrodiols (Scheme 4).3a,6 The sequence involved a catalytic hydrogenation (H2,Pd/C) to yield cis-tetrahydrodiol 5 followed by bis-acetylation (Ac2O, pyridine) to give cis-diacetate 6. In the final step, an oxidative ring opening reaction (RuO2/NaIO4) gave a mixture of dicarboxylic acid products (7/8). It was assumed that the bicyclic dicarboxylic acid 7 was formed initially and then a part of it degraded to acyclic dicarboxylic acid 8via a further oxidative ring opening reaction. The mixture of dicarboxylic acids 7 and 8 was methylated (CH2N2) to yield dimethylesters 9 and 10 which were separated by column chromatography. The minor component, dimethyl(2,3-diacetoxy)adipate 10 ([α]D −14) was of established (2S,3S) configuration6 and thus the (1R,2S) configuration was unequivocally assigned to (+)-cis-dihydrodiol 4.
 | ||
| Scheme 4 | ||
It has been proposed that the mutagenicity/carcinogenicity associated with some larger PAHs and APAHs results from: (i) a monooxygenase-catalysed epoxidation of a carbocyclic ring to yield an arene oxide (cf. compound 2), (ii) an epoxide hydrolase-catalysed hydrolysis of the arene oxide to yield a trans-dihydrodiol (cf. compound 3), (iii) a monooxygenase-catalysed epoxidation of the alkene bond in the trans-dihydrodiol to yield diastereoisomeric trans-diol epoxides and (iv) nucleophilic attack of DNA on the epoxide ring within a bay region to yield a covalent adduct.1b–f Although the corresponding acridine trans-diol epoxides from metabolite 3 could, in principle, also be mutagens, their synthesis and mutagenicity has not yet been reported.
(ii) Biotransformation of furoquinolines 11–13
In common with acridine 1, the mammalian metabolism and mutagenicity of dictamnine 12 and other furoquinoline alkaloids, e.g. γ-fagarine, had been reported earlier.7a–e In a more recent study, from these laboratories, the furoquinoline alkaloid skimmianine 13 was found to be the major compound present in C. ternata,4a and was thus available as a potential substrate for the current biotransformation studies. However, dictamnine 12, another furoquinoline alkaloid required as a potential substrate, was not isolated from C. ternata. Thus, a five-step chemical synthesis of dictamnine 12 was carried out, starting from aniline and using the literature procedure8a–c which involved 4-chlorofuroquinoline 11 as precursor. Furoquinolines 11 and 12 were thus also available as possible substrates for BPDO.Furoquinolines 11–13 were added, individually, as substrates to S. yanoikuyae B8/36, under the conditions used previously for the successful biotransformation of acridine 1. The results, shown in Scheme 5, indicate that while 4-chlorofuro[2,3-b]quinoline 11 and dictamnine 12 each yielded a mixture of two cis-dihydrodiol products 14–17, resulting from BPDO-catalysed cis-dihydroxylation of the 5,6 and 7,8 bonds of the carbocyclic ring, skimmiamine 13 was not an acceptable substrate. The mixtures of metabolites (14/15 and 16/17) were separated into individual cis-dihydrodiols by PLC purification. The structures of diol metabolites (14–17) were established by analyses of NMR and MS data while the ee values (>98%) and absolute configurations were again determined by formation of the corresponding diastereomeric MPBA esters and their analysis by 1H-NMR spectroscopy. As found for the acridine cis-dihydrodiol 4, the larger chemical shift values (δH) for the exocyclic MeO group of cis-dihydrodiols 14–17, (δH 3.23–3.25) using the (R)-(+)-MPBA compared with (S)-(−)-MPBA 14–17, (δH 3.19–3.20) were consistent with benzylic (R) and allylic (S) configurations in each case.
 | ||
| Scheme 5 | ||
While cis-dihydroxylation had occurred exclusively at the 1,2 bond of acridine 1, similar regioselectivity for the equivalent (5,6) bond in furoquinolines 11 and 12 was not found. A modest preference (38% yield) was observed for BPDO-catalysed cis-dihydroxylation at the 7,8-bond to form cis-diol 15 compared with the 5,6-bond (10% yield) to give cis-diol 14, when 4-chlorofuro[2,3-b]quinoline 11 was the substrate. A stronger preference for oxidation of the 7,8-bond was found with dictamnine 12 as the substrate, which resulted in cis-diol 17 being the major metabolite (20–30% yield) relative to cis-diol 16 (1–3% yield). The combined isolated yields (21–33%) of dictamnine cis-dihydrodiols (16 and 17) were slightly lower than 4-chlorofuroquinoline cis-dihydrodiols (14 and 15, 48%); no cis-dihydrodiol metabolites were detected from skimmianine 13 as substrate. These observations suggest that the presence of substituents at C-4, C-7 and C-8 and the overall steric requirements of the substrate within the active site of the BPDO enzyme are important factors. Based on isolated yields, it appears that cis-dihydroxylation occurred preferentially at the less sterically hindered 7,8-bond and that the best yields resulted from the use of the smaller substrates (11 and 12). As the largest substrate, skimmianine 13, did not yield cis-diol metabolites, this is consistent with its failure to be accommodated within the BPDO active site. However, alternative factors, including aqueous solubility, toxicity and further metabolism, could influence the isolated yields of bioproducts.
The most polar metabolites, formed from 4-chlorofuro[2,3-b]quinoline 11 and dictamnine 12, were found to be exocyclic diols (compounds 20 and 23) but were isolated in very low yields (1–2%, Scheme 5). While the structures of optically active diols 20 and 23 were assigned by NMR and MS spectroscopic analysis, their ee values and absolute configurations were not determined. It was assumed, that the exocyclic diols 20 and 23, resulted from: (a) BPDO-catalysed cis-dihydroxylation at the 2,3-bond to give transient intermediates 18 and 21, (b) reversible ring opening of these hemiacetals (cf. mutarotation) to yield the undetected aldehydes 19 and 22, (c) epimerization, following reversible ring closure, to yield a mixture of the corresponding cis- and trans-dihydrodiols, and (d) carbonyl reductase-catalysed (CRED) reduction of the aldehyde group in intermediates 19 and 22. A similar sequence of TDO-catalysed cis-dihydroxylation of the furan ring of benzo[b]furans, spontaneous equilibration via a reversible ring opening process to yield the corresponding phenolic aldehydes and CRED-catalysed reduction of the resulting aldehyde group was earlier assumed to account for the isolation of the exocyclic diols shown in Scheme 1 (b).2c
The origin of mutagenicity associated with dictamnine 13 has not yet been rigorously established.7a–e However, it has been proposed that, in common with other naturally occurring mutagenic furans, e.g. aflatoxin B1 and 8-methoxypsoralen, the corresponding transient furan epoxides,7f formed as initial mammalian metabolites via monooxygenase-catalysed epoxidation, e.g. arene oxide 24 (Scheme 5) may be responsible for their mutagenicity. It has been proposed that the mutagenicity results from the ability of furan epoxides to form covalently bound adducts following nucleophilic ring-opening reactions with DNA.7e,f
(iii) Application of acridine cis-dihydrodiol 4 in the synthesis of arene oxide 2
As part of an earlier study of the mammalian metabolism and mutagenicity/carcinogenicity of PAHs and APAHs, (1R,2S)-arene oxide 2 was obtained via an eight stage chemical synthesis, involving a chemical resolution of MTPA esters, with an overall yield of ca. 13%.9 Alkaline hydrolysis (KOH, t-BuOH) of (1R,2S)-arene oxide 2 gave the mammalian metabolite (1R,2R)-trans-1,2-dihydroacridine-1,2-diol 3.9 In the current study, the possibility of a much shorter synthesis of acridine 1,2-oxide 2 was examined (Scheme 6), using the readily available bacterial metabolite, (1R,2S)-cis-1,2-dihydroacridine-1,2-diol 4. Treatment of diol 4 with 1-bromocarbonyl-1-methylethyl acetate, in acetonitrile solution, gave a mixture of bromoacetates 25/26 whose structures were determined from 1H-NMR and MS data. Due to their instability, during attempted separation, the mixture of bromoacetates 25 and 26 in Et2O solution was reacted directly with NaOMe. Using this two step method, the relatively stable (1R,2S)-arene oxide 2 was synthesised from cis-dihydrodiol 4 in 66% yield. Despite the stability of arene oxide 2, it was not detected during mammalian metabolism, probably due to its further metabolism via a rapid epoxide hydrolase-catalysed conversion to the corresponding trans-dihydrodiol 3.5a–e A preliminary study3d later showed that when the stable acridine cis-dihydrodiol 4 was used as a substrate for S. yanoikuyae B8/36, it was also further metabolised and formed a bis-cis-dihydrodiol bioproduct.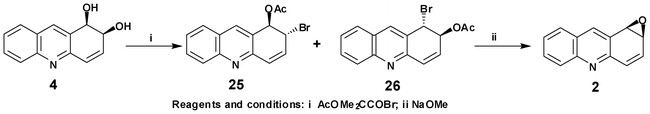 | ||
| Scheme 6 | ||
(iv) Application of dictamnine cis-dihydrodiols 16 and 17 as precursors in the synthesis of furoquinoline alkaloids
The potential of dictamnine cis-dihydrodiol metabolites 16 and 17 in the biomimetic synthesis of furoquinoline alkaloids, including the proposed arene oxide intermediate 27, was of biosynthetic interest (Schemes 7 and 8). Possible biosynthetic pathways to furoquinoline alkaloids occurring in Rutaceaeous plants, e.g. Skimmia japonica and Choisya ternata, have been studied using 14C-labelled precursors.10a These labelling studies showed that enzyme-catalysed hydroxylation could occur on the benzene ring of dictamnine 12 to yield a wider range of furoquinoline alkaloids e.g. skimmianine 13 and possibly also robustine 30 and γ-fagarine 31 (Scheme 7). It was proposed that skimmianine 13 could be formed via a monooxygenase-catalysed epoxidation of dictamnine 12, to yield the transient arene oxide 27, followed by epoxide hydrolase-catalysed hydrolysis to yield trans-dihydrodiol 28.10a The possibility of an alternative dioxygenase-catalysed cis-dihydroxylation of dictamnine 12 to yield cis-dihydrodiol 17 was also discussed.10a The enzyme-catalysed oxidations of trans- and cis-dihydrodiols, to yield catechols followed by O-methylation, are well established metabolic steps1a and, when allied to the earlier labelling studies,10a either type of enzymatic oxidation could account for the formation of catechol 29 and skimmianine 13. To date, none of the potential biosynthetic intermediates 17, 27–29 have been detected by the labelling studies using Choisya ternata10a or found among the furoquinoline alkaloids recently isolated from this4a or other plants in the Rutaceae family.10b | ||
| Scheme 7 | ||
 | ||
| Scheme 8 | ||
As expected, the B8/36 mutant strain of S. yanoikuyae did not yield catechol metabolites e.g. compound 29 from dictamnine 12 (Scheme 7). The biphenyl cis-diol dehydrogenase (DD) activity required to catalyse the dehydrogenation of cis-dihydrodiols to yield catechols, was blocked in the B8/36 strain. However, when the wild type strain of S. yanoikuyae (B1), expressing both BPDO and DD enzymes, was used with dictamnine 12, the only metabolite identified and isolated was cis-dihydrodiol 16, albeit in low yield (8%). This observation is consistent with both cis-dihydrodiols 16 and 17 being formed but the major metabolite (17) being further metabolized preferentially.
The E. coli narB recombinant strain, expressing naphthalene DD, has been used successfully to produce catechols in good yields from the corresponding monocyclic arene cis-dihydrodiols.11 Using E. coli narB and cis-dihydrodiol 15 as substrate, catechol 35 was detected by 1H-NMR spectroscopy but in low yield. Surprisingly, the required catechol metabolite 29, derived from dictamnine cis-dihydrodiol 17, could not be obtained using this method. However, it was possible to obtain catechol 29 in good yield (85%) using boron tribromide for the selective O-demethylation of skimmianine 13, isolated earlier4a from Choisya ternata (Scheme 8).
Convincing evidence of monooxygenase-catalysed epoxidation of dimethylallyl groups, and hydrolysis of the resulting epoxides to yield vicinal diols, is available from biosynthetic studies of quinoline alkaloids from plants of the Rutaceae family.10a,b Furthermore, monooxygenase-catalysed epoxidation of azaarenes, to yield the corresponding arene oxides, e.g. quinoline 5,6-oxide from quinoline, using liver microsomes with inhibition of epoxide hydrolase activity,12a,b provides a precedent for the formation of the elusive dictamnine arene oxide 27 and trans-dihydrodiol 28 metabolites. While the dioxygenase-catalysed cis-dihydroxylation of polycyclic azaarenes in bacteria, e.g. S. yanoikuyae B8/36, is well established (Schemes 1, 2 and 4), there appears to be little evidence of this pathway occurring in plants. Consequently, the monooxygenase-catalysed epoxidation sequence, shown in Scheme 7, is currently favoured over the dioxygenase pathway. Epoxidation, as an initial step, can readily account for the formation of both monophenols (e.g. robustine 30), catechols (e.g. 7,8-dihydroxydictamnine 29), and their methylated derivatives (e.g. γ-fagarine 31 and skimmianine 13).
Our attempt to synthesise the proposed dictamnine arene oxide metabolite 27 from cis-dihydrodiol 17, via a two-step process similar to that used earlier for acridine arene oxide 2 (Scheme 6), was unsuccessful. This was due to compound 17 being less stable under the reaction conditions and more readily dehydrated under acid conditions to yield phenols (e.g. robustine 30). An alternative approach (Scheme 8) was adopted involving the catalytic hydrogenation (H2, Pd–C) of compound 17 to yield the stable cis-tetrahydrodiol 32 (76% yield). Treatment of diol 32 with 1-bromocarbonyl-1-methylethyl acetate gave trans-bromoacetate 33 in good yield (90%). Benzylic bromination of bromoacetate 33 (NBS, CCl4) gave an inseparable mixture of diastereoisomers 34 which was immediately treated with sodium methoxide in THF, to yield the proposed dictamnine arene oxide metabolite 27 (60% yield from compound 33). Initial attempts to purify this elusive arene oxide by PLC resulted in its aromatization to give the furoquinoline alkaloid robustine 30. Purification of (7S,8R)-dictamnine oxide 27 was achieved by careful crystallization. A sample of oxide 27 was found to survive in CDCl3 solution without decomposition, at ambient temperature over a 24 h period.
In the final phase of this study, cis-dihydrodiols 16 and 17, arene oxide 27 and catechol 29, as confirmed or proposed metabolites of dictamnine 12, were utilized as synthetic precursors of other furoquinoline alkaloids, using biomimetic methods (Scheme 8). While robustine 30 was obtained by isomerisation of arene oxide 27 under acidic conditions, the acid-catalysed dehydration of cis-dihydrodiol 17 was the preferred route. Methylation of robustine 30 with diazomethane yielded the alkaloid γ-fagarine 31. Under similar conditions, methylation of catechol 29 occurred mainly at C-8, to yield the alkaloid haplopine 36. Treatment of catechol 29 in acetone with 1-chloro-3-methylbut-2-ene in presence of K2CO3 resulted in the preferential prenylation at C-8 to yield phenol 37, which on methylation yielded the alkaloid, isohaplopine-3,3′-dimethylallylether 38. Acid-catalysed dehydration of cis-dihydrodiol 16, to form phenol 39, followed by methylation, yielded the furoquinoline alkaloid pteleine 40.
Conclusion
The bacterial cis-dihydroxylation of acridine 1 and furoquinolines 11 and 12, catalysed by BPDO, yielded five carbocyclic cis-dihydrodiols, (4, 14–17) and two exocyclic diols (20 and 23), derived from the transient heterocyclic diols 18 and 21. The structures and absolute configurations of most of the isolated metabolites were established by spectroscopic analysis and stereochemical correlation methods. cis-Dihydrodiols 4 and 17 were used in the synthesis of the corresponding arene oxides, 2 and 27, which had been proposed as intermediates in mammalian metabolism of acridine 1 and dictamnine 12. cis-Dihydrodiol 16 and 17 and arene oxide 27, as derivatives of dictamnine 12, have been used in the synthesis of a wide range of furoquinoline alkaloids including robustine 30, γ-fagarine 31, haplopine 36, isohaplopine-3,3′-dimethylallylether 38 and pteleine 40.Experimental
1H and 13C NMR spectra were recorded on Bruker Avance 400, DPX-300 and DRX-500 instruments. Chemical shifts (δ) are reported in ppm relative to SiMe4 and coupling constants (J) are given in Hz. Mass spectra were run at 70 eV, on a VG Autospec Mass Spectrometer, using a heated inlet system. Accurate molecular weights were determined by the peak matching method, with perfluorokerosene as the standard. CD spectra were recorded in spectroscopic grade acetonitrile using a JASCO J-720 instrument. A PerkinElmer 341 polarimeter was used for optical rotation ([α]D) measurements (ca. 20 °C). Flash column chromatography and preparative layer chromatography (PLC) were performed on Merck Kieselgel type 60 (250–400 mesh) and PF254/366 plates respectively. Merck Kieselgel type 60F254 analytical plates were employed for TLC. Authentic samples of 4-chlorofuro[2,3-b]quinoline 11 and skimmianine 13 were available from earlier studies.4a–e As reported,2bSphingomonas yanoikuyae B8/36 was grown on minimal salts medium with 0.5% sodium succinate and 0.5 g L−1 of yeast extract. Biphenyl dioxygenase (BPDO) was induced, during the exponential phase of growth, by the addition of m-xylene (1 cm3 L−1) every 0.5 h for 7 h. Substrate concentration was 0.5 mg cm−3.Synthesis of dictamnine 12
To a solution of 4-chlorofuro[2,3-b]quinoline 11 (3 g, 14.7 mmol) in dry methanol (120 cm3) was added sodium methoxide (4.05 g, 75 mmol) and the mixture refluxed (2 h) under nitrogen. After removal of methanol from the reaction mixture, under reduced pressure, the brown residue obtained was treated with ice cold water and the aqueous mixture extracted with chloroform (3 × 100 cm3). The combined organic extract was dried (Na2SO4), concentrated under reduced pressure, and the crude product purified by flash chromatography (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, EtOAc:hexane) to afford dictamnine 12 as a yellow solid (1.65 g, 56%); Rf 0.5, (1:1 EtOAc/hexane); m.p. 133–134 °C (lit.13a 132–133 °C); δH (300 MHz, CDCl3) 4.45 (3 H, s, Me), 7.08 (1 H, d, J3,2 2.8, 3-H), 7.45 (1 H, dd, J6,5 9.1, J6,7 7.0 6-H), 7.62 (1 H, d, J2,3 2.8, 2-H), 7.69 (1 H, dd, J7,8 8.4, J7,6 7.0 J 7.0, 7-H), 8.0 (1 H, d, J8,7 8.4, 8-H), 8.26 (1 H, d, J5,6 9.1, 5-H).
:1, EtOAc:hexane) to afford dictamnine 12 as a yellow solid (1.65 g, 56%); Rf 0.5, (1:1 EtOAc/hexane); m.p. 133–134 °C (lit.13a 132–133 °C); δH (300 MHz, CDCl3) 4.45 (3 H, s, Me), 7.08 (1 H, d, J3,2 2.8, 3-H), 7.45 (1 H, dd, J6,5 9.1, J6,7 7.0 6-H), 7.62 (1 H, d, J2,3 2.8, 2-H), 7.69 (1 H, dd, J7,8 8.4, J7,6 7.0 J 7.0, 7-H), 8.0 (1 H, d, J8,7 8.4, 8-H), 8.26 (1 H, d, J5,6 9.1, 5-H).(a) Biotransformation of acridine 1
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1421.O).O).
O), 1421.O).O).(b) Biotransformation of dictamnine 12
A small scale biotransformation of dictamnine 12 (60 mg), using S. yanoikuyae B8/36, followed by extraction with EtOAc yielded a mixture of two dihydrodiol metabolites, (5R,6S)-4-methoxy-5,6-dihydrofuro[2,3-b]quinoline-5,6-diol 16 and (7S,8R)-4-methoxy-7,8-dihydrofuro-[2,3-b]quinoline-7,8-diol 17. These diols were separated by PLC (7% MeOH/CHCl3). A repeat larger scale biotransformation of dictamnine 12 (0.6 g, 3.02 mmol), using S. yanoikuyae B8/36, yielded a mixture of three compounds, diol 16 (8 mg, 1.1%), diol 17 (0.143 g, 20%) and 3-(1,2-dihydroxyethyl)-4-methoxy-1,2-dihydro-2-quinolinone 23 (7 mg, 1.0%).(c) Biotransformation of 4-chlorofuro[2,3-b]quinoline 11
A small-scale biotransformation on 4-chlorofuro[2,3-b]quinoline 11 (0.8 g, 3.93 mmol), using S. yanoikuyae B8/36, followed by concentration of the aqueous portion under reduced pressure and extraction of the concentrate by ethyl acetate yielded a mixture of products. PLC purification of the mixture (6% MeOH/CHCl3) gave (7S,8R)-4-chloro-7,8-dihydrofuro[2,3-b]quinoline-7,8-diol 15 (0.35 g, 38%); Rf 0.6 (6% MeOH/CHCl3), (5R,6S)-4-chloro-5,6-dihydrofuro[2,3-b]quinoline-5,6-diol 14 (93 mg, 10%); Rf 0.5 (6% MeOH/CHCl3) and 4-chloro-3-(1,2-dihydroxyethyl)-1,2-dihydro-2-quinolinone 20 (23 mg, 2.5%); Rf 0.4 (6% MeOH/CHCl3).(d) Chemoenzymatic synthesis of (1R,2S)-1,2-epoxy-1,2-dihydroacridine (acridine 1,2-oxide) 2
To a stirred solution of cis-dihydrodiol 4 (0.1 g, 0.47 mmol) in dry MeCN (4 cm3) was added, at 0 °C, 1-bromocarbonyl-1-methylethyl acetate (0.109 g, 0.52 mmol). The reaction mixture was stirred (1.5 h) at room temperature, diluted with Et2O (30 cm3) and the solution washed thoroughly with 5% aqueous NaHCO3 solution (15 cm3). The organic layer was dried (Na2SO4) and concentrated to yield an inseparable mixture of bromoacetates 25 and 26 in which bromoacetate 25 was the major component (0.12 g, 81%); (Found: M+ 317.0061. C15H12NO279Br requires 317.0051).(1R,2R)-1-Acetoxy-2-bromo-1,2-dihydroacridine25. δH (500 MHz, CDCl3) 1.99 (3 H, s, OCOMe), 4.93 (1 H, dd, J2,1 2.5, J2,3 5.7, 2-H), 6.35 (1 H, d, J1,2 2.5, 1-H), 6.62 (1 H, dd, J3,2 5.7, J3,4 9.8, 3-H), 7.01 (1 H, d, J4,3 9.8, 4-H), 7.55–8.09 (4 H, m, Aromatic), 8.27 (1 H, s, 9-H). (1S,2S)-1-Bromo-2-acetoxy-1,2-dihydroacridine26. δH (500 MHz, CDCl3) 2.00 (3 H, s, OCOMe), 5.51 (1 H, d, J1,2 2.5, 1-H), 5.73(1 H, dd, J2,1 2.5, J2,3 5.4, 2-H), 6.58 (1 H, dd, J3,2 5.4, J3,4 9.7, 3-H), 7.16 (1 H, d, J4,3 9.7, 4-H), 7.54–8.09 (4 H, m, Aromatic), 8.13 (1 H, s, 9-H); m/z (EI): 317 (M+, 79Br, 41%), 238 (80), 196 (100).
A cooled solution (0 °C) of bromoacetates 25 and 26 (0.1 g, 0.31 mmol) in dry Et2O (10 cm3) was treated with NaOMe (24 mg, 0.44 mmol). The reaction mixture was stirred overnight at room temperature. After addition of water (10 cm3) to the reaction mixture, it was extracted with Et2O (3 × 15 cm3), the combined ether extract dried (Na2SO4) and the solvent evaporated under reduced pressure to yield (1R,2S)-acridine-1,2-oxide 2 as a light brown solid (51 mg, 83%); m.p. 136–137 °C (CHCl3/hexane) (lit.9 136–138 °C); [α]D +30 (c 0.49, CHCl3) (lit.9 +22, CHCl3); (Found: M+ 195.0689. C13H9NO requires 195.0684); δH (500 MHz, CDCl3) 4.08 (1 H, dd, J2,1 3.8, J2,3 6.1, 2-H), 4.55 (1 H, d, J1,2 3.8, 1-H), 6.78 (1 H, dd, J3,2 6.1, J3,4 10.0, 3-H), 7.03 (1 H, d, J4,3 10.0, 4-H), 7.51 (1 H, dd, J6,7 7.0, J6,5 8.2, 6-H), 7.67 (1 H, dd, J7,6 7.0, J7,8 8.6, 7-H), 7.78 (1 H, J8,7 8.6, 8-H), 8.02 (1 H, J5,6 8.2, 5-H), 8.30 (1 H, s, 9-H); δC (125 MHz, CDCl3) 52.4, 56.7, 126.1, 127.4, 127.8, 127.9, 129.8, 130.1, 131.9, 134.5, 137.9, 148.6, 151.8; m/z (EI): 195 (M+, 84%), 167 (100).
(e) Chemoenzymatic synthesis of dictamnine 7,8-oxide 27 and 4-methoxyfuro[2,3-b]quinoline-7,8-diol 29
The synthesis of dictamnine 7,8-oxide 27 from the crude dibromoacetate 34 (0.15 g, 0.36 mmol) was carried out following the second step of the method described for the synthesis of arene oxide 2, using dry THF (3 cm3) instead of Et2O as a solvent. The crude sample of 7,8-oxide 27 crystallized out of ether/hexane solution to give a pure sample as a light brown solid (43 mg, 56%); m.p. 125–127 °C (from CHCl3/hexane); (Found: M+ 215.0586. C12H9NO3 requires 215.0582); [α]D +93.1 (c 0.36, CHCl3); δH (500 MHz, CDCl3) 4.18 (1 H, ddd, J7,8 3.71, J7,6 3.66, J7,5 1.84, 7-H), 4.30 (3 H, s, OMe), 4.65 (1 H, d, J8,7 3.84, 8-H), 6.37 (1 H, dd, J6,5 9.86, J6,7 3.66, 6-H), 7.01 (1 H, d, J3,2 2.57, 3-H), 7.20 (1 H, dd, J5,6 9.86, J5,7 1.8, 5-H), 7.62 (1 H, d, J2,3 2.57, 2-H); δC (125 MHz, CDCl3) 54.4, 58.4, 58.9, 104.6, 105.3, 106.6, 113.5, 122.6, 124.4, 143.0, 148.8, 157.4; m/z (EI): 215 (M+, 100%), 200 (57), 172 (30), 83 (27); ECD: λ/nm 322 (Δε 0.747), 288 (Δε 0.867), 241 (Δε 7.87), 203 (Δε −6.38).
(f) Chemoenzymatic synthesis of furoquinoline alkaloids 30, 31, 36, 37, 38 and 40
Acknowledgements
We thank the European Social Fund (JGC), Oxford Glycosciences (PL) and the Queen's University of Belfast (JGC, PL, CO'D) for financial support.References
- (a) B. L. Goodwin, in Handbook of biotransformations of aromatic compounds, CRC Press LLC, Boca Raton, 2005, pp. 109–124 Search PubMed; (b) M. Dong, I. Schmeltz, E. J. La Voie and D. Hoffmann, Carcinogenesis, in Polynuclear Aromatic Hydrocarbons, Raven Press, New York, 1978, vol. 3, p. 97 Search PubMed; (c) A. W. Wood, R. L. Chang, W. Levin, S. Kumar, S. Shirai, D. M. Jerina, R. E. Lehr and A. H. Conney, Cancer Res., 1986, 46, 2760 CAS; (d) M. I. Willems, G. Dubois, D. R. Boyd, R. J. H. Davies, L. Hamilton, J. J. McCullough and P. J. van Bladeren, Mutat. Res., Genet. Toxicol., 1992, 278, 227 CrossRef CAS; (e) S. Kumar, R. L. Chang, A. W. Wood, J. G. Xie, M. T. Huang, X. X. Cui, P. L. Cole, H. C. Sikka, S. K. Balani, A. H. Conney and D. M. Jerina, Carcinogenesis, 2001, 22, 951 CrossRef CAS; (f) H. Ling, J. M. Sayer, B. S. Plosky, H. Yagi, F. Boudsocq, R. Woodgate, D. M. Jerina and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2265 CrossRef CAS.
- (a) D. R. Boyd, N. D. Sharma, M. R. J. Dorrity, M. V. Hand, R. A. S. McMordie, J. F. Malone, H. P. Porter, J. Chima, H. Dalton and G. N. Sheldrake, J. Chem. Soc., Perkin Trans. 1, 1993, 1065 RSC; (b) D. R. Boyd, N. D. Sharma, L. V. Modyanova, J. G. Carroll, J. F. Malone, C. C. R. Allen, J. T. G. Hamilton, D. T. Gibson, R. E. Parales and H. Dalton, Can. J. Chem., 2002, 80, 589 CrossRef CAS; (c) D. R. Boyd, N. D. Sharma, I. N. Brannigan, T. A. Evans, S. A. Haughey, B. T. McMurray, J. F. Malone, P. B. A. McIntyre, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2012, 10, 7292 RSC.
- (a) D. R. Boyd, N. D. Sharma, G. P. Coen, F. Hempenstall, V. Ljubez, J. F. Malone, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2008, 6, 3957 RSC; (b) D. R. Boyd, N. D. Sharma, F. Hempenstall, M. A. Kennedy, J. F. Malone, S. M. Resnick and D. T. Gibson, J. Org. Chem., 1999, 64, 4005 CrossRef CAS; (c) D. R. Boyd, N. D. Sharma, T. Belhocine, P. L. Loke, J. F. Malone, S. McGregor and C. C. R. Allen, Chem. Commun., 2006, 4934 RSC; (d) D. R. Boyd, N. D. Sharma, J. G. Carroll, C. C. R. Allen, D. A. Clarke and D. T. Gibson, Chem. Commun., 1999, 1201 RSC.
- (a) D. R. Boyd, N. D. Sharma, P. L. Loke, J. F. Malone, W. C. McRoberts and J. T. G. Hamilton, Org. Biomol. Chem., 2007, 5, 2983 RSC; (b) D. R. Boyd, N. D. Sharma, S. A. Barr, J. G. Carroll and J. F. Malone, J. Chem. Soc., Perkin Trans. 1, 2000, 3397 RSC; (c) S. A. Barr, D. R. Boyd, N. D. Sharma and P. L. Loke, Heterocycles, 2009, 79, 831 CrossRef CAS; (d) D. R. Boyd, N. D. Sharma, C. R. O'Dowd, J. G. Carroll, P. L. Loke and C. C. R. Allen, Chem. Commun., 2005, 3989 RSC; (e) D. R. Boyd, N. D. Sharma, P. L. Loke, J. F. Malone, W. C. Mc Roberts and J. T. G. Hamilton, Chem. Commun., 2002, 3070 RSC.
- (a) K. D. McMurtrey and T. J. Knight, Mutat. Res. Lett., 1984, 140, 7 CrossRef CAS; (b) K. D. McMurtrey and C. J. Welch, J. Liq. Chromatogr., 1986, 9, 2749 CrossRef CAS; (c) G. M. Seixas, B. M. Audon, P. G. Hollingshead and W. G. Thilly, Mutat. Res., Genet. Toxicol., 1982, 102, 201 CrossRef CAS; (d) A. Matsuoka, K. Shudo, Y. Saito, T. Sofuni and M. Jr. Ishidate, Mutat. Res., Genet. Toxicol., 1982, 102, 275 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, R. Boyle, J. F. Malone, J. Chima and H. Dalton, Tetrahedron: Asymmetry, 1993, 4, 1307 CrossRef CAS.
- (a) M. Mizuta and H. Kanamori, Mutat. Res. Lett., 1985, 144, 221 CrossRef CAS; (b) H. Paulini, U. Eilert and O. Schimmer, Mutagenesis, 1987, 2, 271 CrossRef CAS; (c) F. Hafele and O. Schimmer, Mutagenesis, 1988, 3, 349 CrossRef CAS; (d) H. Paulini, R. Waibel and O. Schimmer, Mutat. Res. Lett., 1989, 227, 179 CrossRef CAS; (e) B. Klier and O. Schimmer, Mutagenesis, 1999, 14, 181 CrossRef CAS; (f) F. P. Guengerich, Arch. Biochem. Biophys., 2003, 409, 59 CrossRef.
- (a) H. Tuppy and F. Bohm, Monatsh. Chem., 1956, 87, 720 CrossRef CAS; (b) M. F. Grundon and N. J. McCorkindale, J. Chem. Soc., 1957, 2177 RSC; (c) S.-C. Kuo, S.-C. Huang, L.-J. Huang, H.-E. Cheng, T.-P. Lin, C.-H. Wu, K. Ishii and H. Nakamura, J. Heterocycl. Chem., 1991, 28, 955 CrossRef CAS.
- D. R. Boyd, M. R. J. Dorrity, L. Hamilton, J. F. Malone and A. Smith, J. Chem. Soc., Perkin Trans. 1, 1994, 2711 RSC.
- (a) M. F. Grundon, D. M. Harrison and C. G. Sypropoulos, J. Chem. Soc., Perkin Trans. 1, 1974, 2181 RSC; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (c) N. S. Narasimhan and R. S. Mali, Tetrahedron, 1974, 30, 4153 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, B. E. Byrne, S. D. Shepherd, V. Ljubez, C. C. R. Allen, L. A. Kulakov, M. J. Larkin and H. Dalton, Chem. Commun., 2002, 1914 RSC.
- (a) S. K. Agarwal, D. R. Boyd, H. P. Porter, W. B. Jennings, S. J. Grossman and D. M. Jerina, Tetrahedron Lett., 1986, 26, 4253 CrossRef; (b) S. K. Agarwal, D. R. Boyd, R. J. H. Davies, L. Hamilton, D. M. Jerina, J. J. McCullough and H. P. Porter, J. Chem. Soc., Perkin Trans. 1, 1990, 1969 RSC.
- (a) D. R. Boyd and M. F. Grundon, J. Chem. Soc. C, 1970, 556 RSC; (b) I. A. Bessonova, Chem. Nat. Compd., 1989, 1, 46 Search PubMed; (c) N. S. Narasimhan, M. V. Paradhaker and R. H. Alurker, Tetrahedron, 1971, 27, 1351 CrossRef CAS; (d) I. A. Bessonova, Chem. Nat. Compd., 1999, 35, 589 CrossRef CAS; (e) J. A. Grina, M. R. Ratcliff and F. R. Stermitz, J. Org. Chem., 1982, 47, 2648 CrossRef CAS; (f) F. Werny and P. J. Scheuer, Tetrahedron, 1963, 19, 1293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra42026d |
| This journal is © The Royal Society of Chemistry 2013 |