 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of inorganic aerogels via rapid gelation using chloride precursors†
Helmut
Schäfer
*a,
Barbara
Milow
b and
Lorenz
Ratke
*b
aUniversity of Osnabrck, Institute of Chemistry of New Materials, Physical Chemistry, Barbarastraße 7, 49069 Osnabrück, Germany. E-mail: helmut.schaefer@uos.de
bGerman Aerospace Center, Institute of Materials Physics in Space, Linder Höhe, 51147 Cologne, Germany. E-mail: lorenz.ratke@dlr.de
First published on 13th June 2013
Abstract
This paper describes the synthesis of silica, titania, zirconia and mixed-metal-oxide aerogels by a new facile approach. The corresponding low cost and non-toxic chlorides instead of the well established but more expensive and deleterious alkoxy precursors were used as starting materials. Hydrolysis of the precursors led to wet gels. The strong exothermic reaction in the case of TiCl4 or SiCl4 as a gel precursor can be controlled by both cooling and immersing the needle of the syringe in the aqueous solution. Under optimal conditions a rapid formation of silica- and zirconia based hydrogels could be achieved even without additional amounts of an auxiliary agent. It is shown that in all other cases the hydrolysis reaction and gel formation can be controlled by adding agents with crosslinking properties like propylene oxide, dilution with alcohols or starting from water glass. Water glass not only has the function of a cheap network former but also allows the strict adjustment of the pH value via neutralization of the acid forming hydrolysis reactions. Supercritical drying of the wet alkogel in CO2 delivered aerogels with BET surfaces up to 1390 m2 g−1, as determined by nitrogen adsorption measurements. In addition, the characterization includes XRD and SEM. These findings provide new physical insights into the hydrolysis of the gel precursors and lead to a significant reduction in production costs of inorganic aerogels.
A Introduction
Aerogels are highly porous materials with interesting properties including high specific surface area, extremely low density, low thermal conductivity and high thermal stability.1–5 Already more than 80 years back Kistler prepared the first aerogel, namely a silica aerogel by supercritical drying of silica jelly.6 Due to their unique properties aerogels are already used for a wide range of applications, including optical7–12 and electrical sensing,13,14 catalysis,15–17 as well as thermal18,19 and acoustic insulations.20,21The alkoxide route, characterized by sol–gel transition via hydrolysis and condensation reactions of metal-, semimetal- or non-metal-alkoxide precursors such as tetraethoxysilane (TEOS),22–24 tetramethoxysilane(TMOS),24–27,30 polyethoxydisiloxane (PEDS)24 in the case of silica aerogels, zirconium n-propoxide,28,29 zirconium i-propoxide,30 or zirconium n-butoxide31 in the case of zirconia aerogels and titanium n-butoxide,27,32,33 titanium i-propoxide,27,34 in the case of titania aerogels, presents a common synthesis route for achieving high quality aerogels. These precursors have some drawbacks as they are costly, toxic, and depending on the nature of the alkoxy precursor and the hydrolysis conditions they can cause large gelation times up to a tenth of an hour.28,29,31,35 Furthermore, careful handling of the water-sensitive compounds to prevent partial hydrolysis prior to the desired hydrolysis reaction is required.36–38 These drawbacks limit the scale-up of the synthesis procedures and pose a constraint on the use of these materials in fields where large amounts of relatively cheap materials are required (aerogelic binders, aerogel granules, foundry sand and as thermal insulating compounds in vehicles, pipelines, packaging and home building). Especially for large scale preparation of high quality aerogels the inorganic salt based synthesis routes are still challenging. Only in the case of silica aerogels a cost-effective and fast synthesis procedure could be developed by using water-glass instead of a TEOS or TMOS precursor.39 In comparison with silica aerogels for instance zirconia aerogels show even slightly better properties with respect to thermal conductivity40 or melting point.41
Many papers have been published dealing with the synthesis of aerogels via the alkoxide route whereas only a small number of papers are related to the use of inorganic salts for example as zirconium sources for the synthesis of zirconia aerogels.
Chen and Jiao reported in 2007 the generation of zirconia aerogels by electrolyzing ZrOCl2 solution followed by supercritical drying of the so formed gel.42 The groups of A. E. Gash,36,43–46 L. J. Hope-weeks,47,48 B. Zhou,49 K. Nakanishi et al.50–52 have done plenty of work regarding the development and investigation of synthesis procedures leading to diverse oxide aerogels by using inorganic salt and additional epoxide as precursor. Anyway chloride-derived silica, titania or zirconia aerogels were rarely reported. Already more than 10 years ago Gash et al. reported on the successful generation of a chromia aerogel starting from nitrates or chlorides.43 The general suitability of the described synthesis strategy in order to synthesize other transition metal oxide aerogels was mentioned in the paper, but only the synthesis and the properties of the corresponding chromia aerogels were shown. Because of the toxic properties of chromium in its hexavalent state engineers generally try to avoid the element chromium.53
The work described in this contribution aims on the development of synthesis procedures for silica-, titania- and zirconia-aerogels, as well as mixed-metal-oxide aerogels based on cheap and non-toxic chloride based sources54 or water glass, characterized by a rapid gel-formation. Via hydrolyses of TiCl4 titania aerogels could be achieved. Titanium tetrachloride is an important intermediate in the production of titanium metal and the pigment titanium dioxide.55–57 On the basis of the bulk FOB (free on board) price of 1900 $ per ton (99.9% purity),58 it can be considered as one of the cheapest titanium sources. The price of titanium alkoxides of this purity is at least five times higher.59 To the best of our knowledge, up to now no aerogels were synthesized using TiCl4 as a starting material. Titania particles60–63 showing different particle sizes or coatings64 have been achieved by sol–gel approaches using TiCl4.
We show that zirconia aerogels are accessible upon hydrolysis reaction starting from the corresponding chloride salt ZrCl4, a compound which can also be considered as cheap with a bulk FOB price of 11 $ kg−1.65 No reference could be found up to now confirming the usage of this compound in order to generate pure zirconia aerogel. Where useful, water glass was inserted in our synthesis strategy. Water glass is a common network former for the generation of silica gels and aerogels. As water-glass is an economical basic chemical, an industrial process based on this precursor was developed for some time by BASF.66 Zirconia aerogels were so far achieved by using other starting materials, respectively ZrCl4 was used for other purposes: via sol–gel synthesis ZrO2 aerogel was gained starting from Zr3(PO4)4.67 Kauzlarich et al. synthesized yttria-stabilized zirconia by a non-alkoxide sol–gel route via hydrolysis of ZrCl4 and YCl3.36 Richards et al. showed that iron-doped zirconia aerogels can be achieved by using zirconyl nitrate and iron nitrate as zirconium, respectively iron precursors via a solvothermal approach.68 Different high vacuum technologies like chemical vapour deposition (CVD),69 atomic layer deposition (ALD),70,71 and atomic layer chemical vapor deposition (ALCVD)72 have been used to generate zirconia thin films starting from ZrCl4.
We achieved mixed silica–titania aerogels through the reaction of water glass with TiCl4 whereas in the literature only the alkoxy route for the generation of theses compounds (based on a mixture of Si- and Ti-alkoxides4,73,74 or single source alkoxy precursors75–77) could be found. With respect to titania–zirconia aerogels, no reports in the literature could be found, indicating the access of this mixed oxide aerogels by using titanium and zirconium chlorides. Wang et al. reported in 2005 on the synthesis of titania–zirconia composite particles by a solvothermal procedure in an alcoholic solution at elevated temperatures involving chloride salts.78 Only one paper reported on the synthesis of this composite aerogel starting from titanium-zirconium salts (Ti(SO4)2 and ZrOCl2). The properties, especially the specific surface area with a value of 387 m2 g−1 at intensive heat treatment, were not satisfying.79 The alternative synthesis routes presented herein not only allow the reduction in the synthesis cost and the improvement in the scalability of the procedures, but also enable fine-tuning of the materials properties such as pore structure, constitution and density of the aerogels.
B Experimental methods
B1 Reagents
Zirconium(IV) chloride (99.5%, Aldrich), silicium(IV) chloride (99%, Aldrich), propylene oxide (99%, Aldrich), titanium(IV) chloride (98%, Fluka) and water glass (1.39 g cm−3, Aldrich) were used as received unless noted otherwise. The synthesized aerogel samples were sputtered with gold and the morphology of the aerogels was investigated with a scanning electron microscope LEO 1530 VP. For this purpose non treated aerogel bodies as well as sliced aerogel bodies were used. Powder diffraction analyses were performed with an X'Pert Pro Diffractometer (Panalytical). The as-prepared aerogels were mortared and the X-ray diffraction data were recorded in order to determine the crystallographic phase. Afterwards the samples were heated up to 450 °C in a standard oven (Naber N 11) under air and the crystallographic data were measured again. The surface area, pore size and pore size distribution were measured by using a multiple point nitrogen gas adsorption BET surface area analyzer (Micromeritics Tristar). Before the aerogel samples were de-gassed at 120 °C for 30 min.Pore size distributions were calculated from the desorption isotherms using the BJH method.
B2 Processing
 | ||
| Scheme 1 Schematic presentation of the synthesis procedure. | ||
 | ||
| Fig. 1 (a) Metal shell with aerogels (HS_33) after supercritical drying. (b) DLR-made autoclave for supercritical drying. | ||
C Results and discussion
C1 Synthesis of the aerogels
Silicon-, titanium- and zirconium oxide aerogels are among the most common aerogels due to their sophisticated properties like thermal stability,41,79 photocatalytic properties,80–82 in general low density,1 high acoustical and thermal insulating properties.1 Therefore we started our investigations with finding alternative precursors and overall facile approaches allowing to access these aerogels as well as mixed metal oxide aerogels containing two of the elements Si, Ti, and Zr. As Tables TS1–TS4, ESI† show, provided that suitable reaction conditions (constitution and temperature of the reaction mixture, pH value, stirrer speed and time duration for adding the precursors) have been chosen, abrupt gel-formation takes place. All the gels presented in the tables could be transferred to aerogels via supercritical drying in CO2.We were successful in preparing silica aerogels starting from SiCl4 plus water or water THF mixtures, silica aerogels starting from SiCl4 plus water glass, titania aerogels starting from TiCl4 plus water or water/alcohol mixtures, zirconia aerogels starting from ZrCl4 plus water and zirconia–titania, silica–titania mixed metal oxide aerogels starting from the corresponding chlorides via reaction with water or water glass with or without propylene oxide as auxiliary agent. In most of the cases a sol gel transition occurs within seconds (see Fig. 2, 4, 5, 6). From alkoxy precursors it is known, that they, depending on reaction conditions and type of the precursors, require gelling times up to a tenth of an hour.24,28,29,31,35,96 Especially for silica aerogels long gelling times go along with low transmittance.83 Series syntheses show beyond that the gelling time is well reproducible. Supported not only by our findings it must be mentioned that the use of water glass up to some extent impairs the repeatability especially with respect to gelling time or overall gelation behaviour.84,85 Slight differences in the composition of water glass might be one reason. We obtained that e.g. the sodium content differs from supplier to supplier and hence commercially available sodium silicate solutions show Na2O concentrations with values between 8 and 14 wt.%. Even when purchased from one supplier the composition can vary and the Na2O content can reach values between 8.3 and 13 wt.%, respectively the SiO2 content can be in the range between 26.5 and 29 wt.%.86 This makes it difficult to design a reliable synthesis route. It turned out that in case that water glass was inserted in the reaction mixture abrupt gel-formation could be realized by adjusting the pH value in the range of 3. Depending on the ratio of alkaline compounds to acidic compounds i. e. the ratio of water glass to TiCl4, respectively the ratio of water glass to SiCl4, the pH value was controlled by adding either HCl or NaOH. From literature is known that for the generation of microporous silica aerogels starting from water glass a pH value in between 3 and 5 seems to be desirable.87–89
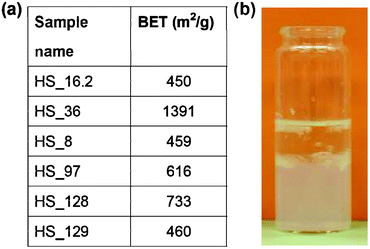 | ||
| Fig. 2 (a) BET surface area for silica aerogels. (b) Image of sol–gel transition of SiCl4 containing reaction mixture (sample HS_16.2). | ||
Supercritical drying with CO2 was found to be the method of choice for the replacement of liquid by gas in the pores of the generated gels in order to achieve aerogels of high quality. The elsewhere reported long diffusion time and hence, time consuming solvent exchange between solvent and CO290 could not be confirmed by us. The complete drying procedure using supercritical CO2 was completed within 48 h. In general the supercritical drying procedure was more and more optimized over a long time period by different groups91i.e. the statement that this drying technique is inefficient, expensive and not scaleable92 is more than questionable. CO2 technology is already commercially applied in a variety of fields – from pharmacy to food sciences to the textile industry. CO2 can be recycled easily, it is also non-flammable, non-toxic and inexpensive.93 In many cases the process costs are lower than comparable conventional processes.93 In comparison with other media used within the supercritical drying procedure like acetone94 or alcohols,80,95,96 CO2 benefits from its relatively mild supercritical conditions, especially the low supercritical temperature of 304.13 K. With the help of water glass, aerogels consisting of silica–titania mixed metal oxide aerogels were accessible even without additional auxiliary agent like propylene oxide (see Table TS4, ESI†). In all other cases propylene oxide acts, as a proton scavenger and raises the pH-value during the reaction. Reaction generated HCl is compensated, the hydrolysis and condensation can occur to form a three dimensional network.96 During the condensation reaction chloropropanol is formed and can be easily washed out with ethanol or acetone.
With this synthesis strategy the overall costs of the synthesis procedure could be kept within limits inasmuch as also the chosen auxiliary agent propylene oxide is with a FOB price of 1900 $ per ton97 a low cost compound. At the same time acceptable results could be obtained. Detailed information can be taken from Fig. 2–6, TS1–TS4, ESI† (supporting information) showing the constitution of the reaction mixtures, surface area, pore size distributions (BJH plots), the XRD powder pattern and SEM images.
C2 Properties of the aerogels
For silica aerogels gained from SiCl4, BET surface values within the range 450 to almost 1400 m2 g−1 were obtained (Fig. 2a). The hydrolysis reaction of SiCl4 with water led, under optimal reaction conditions, to an aerogel with a surface area of 1391 m2 g−1 (HS_36). The gel formation took place within less than 30 s. The corresponding white aerogel body was crack free but not transparent. The aerogel achieved by a similar synthesis route using 0.9 ml SiCl4 instead of 1 ml showed a drastic reduced surface area of 450 m2 g−1 (HS_16.2). The SEM image of the surface of this sample can be taken from Fig. 3b. It turned out that the hydrolysis of SiCl4 performed in a solvent mixture of water–THF led to transparent gel bodies (HS_8). However showing a BET value of 459 m2 g−1 the specific surface was much lower in comparison with our best sample. In addition the time required for gel formation was 2 days, which within this series of experiments represents the longest time. Some alkoxy precursor routes require duration times in the same dimension. Rao et al. reported on the synthesis and characterization of silica aerogels by hydrolysis reactions of TEOS, TMOS or polyethoxydisiloxane (PEDS).24 The hydrolysis of TEOS in alcohol–water mixtures delivered after 2.2 days a stable gel which could, via supercritical drying in CO2, transferred to an aerogel with a surface area of 800 m2 g−1.24 By using TMOS or PEDS instead of TEOS the gelation time could be decreased down to 30 min (TMOS), respectively 10 min for PEDS and at the same time the surface area was increased to 1000 m2 g−1 (TMOS), respectively 1100 m2 g−1 (PEDS). Anyway neither the use of TEOS, nor the use of TMOS is desirable most of all because of their toxicity.98 A silica aerogel with a BET value of 733 m2 g−1 from the reaction of SiCl4 with water glass followed by supercritical drying could be achieved (HS_128). Unfortunately the corresponding aerogel body, white in color, showed cracks. Tamon et al. investigated silica aerogels synthesized from TEOS and reported on the relation between visible light transmittance (wavelength: 600 nm) of silica aerogels and surface area.99 They found that the higher the surface area the higher the light transmittance. The possibility that the light transmittance of an aerogel body can be used as a criterion in order to predict the quality e.g. the BET surface seems to be valid only within a series of syntheses with similar characteristics with respect to precursors, reaction conditions and so on. While changing the reaction parameters within the synthesis series of our silica aerogels we could not prove this generally applicable rule. For the transparent sample HS_8 (see also SEM image Fig. 3d) a BET value of 459 m2 g−1 was determined whereas the turbid sample HS_36 showed a surface area of almost 1400 m2 g−1. The pore size distributions of selected silica aerogels can be taken from the BJH plots shown in Fig. 3a, 3c, S1–S3, ESI†.
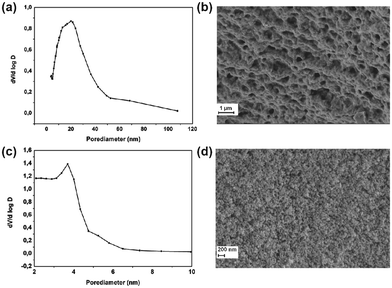 | ||
| Fig. 3 BJH plot from silica aerogel sample HS_16.2 (a) and sample HS_36 (c). SEM pictures of silica aerogel sample HS_16.2 (b) and sample HS_8 (d). | ||
The samples gained from the hydrolysis of SiCl4 with water showed, although the constitution of the reaction mixture only slightly differs, large deviations with respect to the pore size distribution. When 0.9 ml SiCl4 was used (HS_16.2) a broad size distribution was found, showing pore sizes in the range 5–50 nm (Fig. 3a). In an equal synthesis reaction 1 ml of the same precursor already led to very small pores below 6 nm and a narrow size distribution with pore sizes within the range of 2–6 nm (Sample HS_36; Fig. 3c). The reproducibility of both results was satisfying and it is actually not fully understood why small changes in the constitution of the reaction mixture has such a big influence on the results. Generally SiO2 exists in different crystalline modifications, crystalline phases like quartz, tridymite or christobalite. All as-prepared silica aerogels as well as all samples heated up to 450 °C consisted of X-ray amorphous SiO2. This can be taken from the powder diffraction pattern of selected samples (see Fig. S4–S7, ESI†). In case of HS_97 the diffraction pattern show peaks belonging to NaCl, JCPDS 01-077-2064 (see Fig. S7, ESI†). NaCl is a byproduct from the reaction of water glass and SiCl4. Before heat treatment the samples were washed with water to remove impurities coming from precursors or byproducts like NaCl. This is the reason that only traces of NaCl can be found in the heat treated sample (see Fig. S7, ESI†).
Hydrolysis of SiCl4 performed in H2O/THF mixtures followed by drying of the gels resulted in an aerogel with a pore size distribution of 3–23 nm (sample HS_8, Fig. S1, ESI†). A very broad range of pore sizes could be found for the aerogels derived from SiCl4 and water glass (samples HS_97, HS_128; Fig. S2, S3, ESI†).
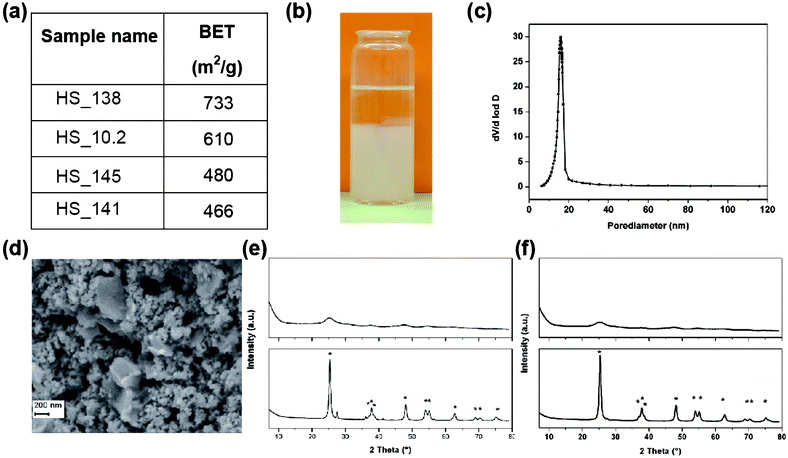 | ||
| Fig. 4 (a) BET surface areas for titania aerogels. (b) Image of sol–gel transition of TiCl4 containing reaction mixture (sample HS_10.2). (c) BJH plot from titania aerogel sample HS_10.2. (d) SEM pictures of silica aerogel sample HS_10.2. (e) Powder Diffraction pattern from titania aerogel HS_138 as-prepared (upper figure), respectively heat-treated (lower figure). Peaks corresponding to tetragonal TiO2,anatase modification (JCPDS 004-0477) are marked with an asterisk. (f) Powder diffraction pattern from titania aerogel HS_10.2 as-prepared (upper figure), respectively heat-treated (lower figure). Peaks corresponding to tetragonal TiO2 (anatase modification) (JCPDS 004-0477) are marked with an asterisk. | ||
Recently Anderson et al. reported on the synthesis of titania and mixed silica-/titania aerogels using autoclave technique in which both, gelation of the alkoxy precursors and supercritical drying was performed.27 The advantage of this method is that it is less time consuming (<6 h in total) than the classical sol–gel procedure. This synthesis procedure, developed by Gauthier et al.101 led to titania aerogels with surface areas in the range between 127 and 188 m2 g−127 so much lower than the ones we generated starting from inorganic chlorides. Small surface areas of around 220 m2 g−1 for titania aerogels were also reported by Ko et al.32 The aerogels were gained from classical synthesis procedures starting from titanium n-butoxide via sol–gel transition. Photocatalytic properties of titania based composite aerogels achieved from the alkoxy synthesis route were described in a recent publication written by Sunol et al.33 They compared their products with commercially available industry standard TiO2 compounds and found 5–10 times greater BET surface areas. Anyway the BET values obtained here were at least 50% lower in comparison with our findings.
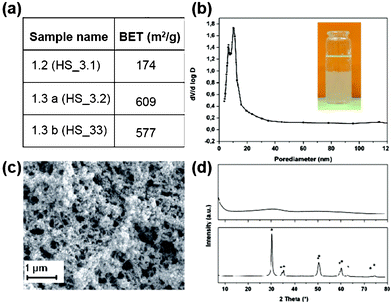 | ||
| Fig. 5 (a) BET surface area for zirconia aerogels. (b) BJH plot from zirconia aerogel sample HS_3.2. The inset showing the image of sol–gel transition of ZrCl4 containing reaction mixture (sample HS_3.2). (c) SEM pictures of silica aerogel sample HS_3.2. (d) Powder diffraction pattern from zirconia aerogel HS_3.1 as-prepared (upper figure), respectively heat-treated (lower figure). Peaks corresponding to tetragonal ZrO2 (JCPDS 01-080-0965) are marked with an asterisk. | ||
In comparison with synthesis procedures leading to titania aerogels which are actually all based on organic precursors, in case of zirconia aerogels some synthesis routes starting from inorganic precursors are described in the literature. Most of the synthesis procedures leading to zirconia aerogels reported so far are based on tetra alkoxy precursors like zirconium tetra-n-propoxide,28,29 zirconium tetra-i-propoxide,30 and zirconium tetra-n-butoxide.31
Ko et al. reported on the syntheses of zirconia aerogels starting from both, inorganic and organic Zr precursors28 and studied the effect of heat treatment on the physical properties of this material achieved from zirconium-n-propoxide. He obtained BET surface areas up to 134 m2 g−1 and mentioned in his report other groups who performed hydrolysis reactions of zirconyl chloride and zirconyl nitrate at last resulting in aerogels with surface areas in the same range. By using the same precursor (zirconium-n-propoxide) Suh et al. obtained slightly higher BET values and studied the effect of the reaction temperature on the gelation time as well as the influence of the aging conditions on the textural properties.29 Baiker published on the synthesis and characterization of zirconia aerogels starting from the hydrolysis of zirconium tetra butoxide in acid–alcohol mixtures.31 They obtained, depending on the nature of supercritical drying (CO2 low temperature or alcohol high temperature drying condition), BET values in the range 50–100 for CO2 dried species and 140–200 m2 g−1 for the high temperature dried aerogels.31 Surface areas in the range we found for zirconia aerogels were published by Hui et al. in a recent publication.102 Hydrolysis and condensation of zirconium nitrate with the help of propylene oxide followed by ambient pressure drying resulted in aerogels with nanoscale porous network showing pore sizes below 10 nm and surface areas in between 480 and 650 m2 g−1 whereas all procedures starting from organic zirconium precursors seem to result in aerogels with lower surface areas showing values in the range between 50 and 240 m2 g−1.28,29,31,103
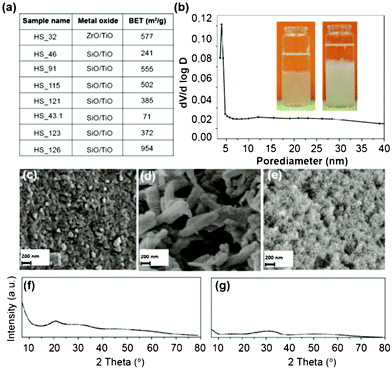 | ||
| Fig. 6 (a) BET surface area for mixed metal oxide aerogels. (b) BJH plot from zirconia-/titania aerogel sample HS_32. The inset showing image of sol–gel transition of ZrCl4 and TiCl4 containing reaction mixture (sample HS_32) (left) and of SiCl4 and water glass containing reaction mixture (sample HS_97) (right). (c–e) SEM pictures of silica aerogel sample HS_32 (c), HS_43.1 (d) and HS_126 (e). (f–g) Powder diffraction pattern of titania-zirconia aerogel HS_32 before (f) and after (g) annealing. | ||
Titania–zirconia composite aerogel materials have been prepared by sol–gel transition of the hydrolysis reaction of inorganic precursors Ti(SO4)2 and ZrOCl2 plus subsequent supercritical drying in CO2.79 This is indeed an interesting approach. The aerogels showed depending on the TiO2 to ZrO2 ratio and heat treatment BET-surface values up to 450 m2 g−1 and a quite broad pore size distribution with pore sizes in the range between 10 and 60 nm.79 However with respect to pore size and pore size distribution our experiment showed slightly better results.
We realized the synthesis of silica–titania aerogels by immersing TiCl4 into water glass solutions. The Ti–Si ratio was adjusted by the amount of TiCl4 respectively water glass (see also Table TS4, ESI†). The BET surface values vary, depending on the reaction conditions and Ti–Si ratios and are in the range 71 and 954 m2 g−1.
By far the best result was obtained in case of a Si–Ti ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 without additional amount of propylene oxide (954 m2 g−1; HS_126). Among the synthesized silica-titania aerogels this sample showed also the smoothest surface (see SEM image HS_126 Fig. 6e). Anyway, if in this procedure the alkaline compound NaOH is replaced by propylene oxide the surface area was reduced by more than 40% (HS_91, 555 m2 g−1). Poor results were obtained if the Si–Ti ratio was adjusted to 10 (HS_43.1). The aerogel showed a very low BET surface area (71 m2 g−1) and a very broad pore size distribution with pore sizes in the range 10–120 nm (see Fig. S9, ESI†), which is accompanied by a rough surface with pore sizes up to micrometer range (see SEM image in Fig. 6d). In contrary to the other mixed metal oxide aerogels, the aerogel network of this compound (HS_43.1) shows a more columnar structure. Interestingly within the series of mixed metal oxide aerogels only the titania–zirconia compound showed a narrow pore-size distribution (Fig. 6b). In case of the synthesized silica–titania aerogels the pore-size distribution was much broader with size intervals of 20–60 nm when propylene oxide as auxiliary agent was used (HS_91). The neutralization of an excess of acidic (HS_126) or alkaline (HS_43.1) precursors within the sol–gel reaction by NaOH (HS_126) or HCl (HS_43.1) obviously is responsible for wide pore size intervals ranging from 10–140 nm, respectively from 10–120 nm (HS_43.1). The crystallinity of all samples was checked by X-ray diffraction analyses – the results can be taken from Fig. 6f, g and 7, and S11–S14, ESI†. Although we performed many experiments no gel formation was observed starting from mixtures of TiCl4–SiCl4 and TiCl4–SiMeCl3.
:1 without additional amount of propylene oxide (954 m2 g−1; HS_126). Among the synthesized silica-titania aerogels this sample showed also the smoothest surface (see SEM image HS_126 Fig. 6e). Anyway, if in this procedure the alkaline compound NaOH is replaced by propylene oxide the surface area was reduced by more than 40% (HS_91, 555 m2 g−1). Poor results were obtained if the Si–Ti ratio was adjusted to 10 (HS_43.1). The aerogel showed a very low BET surface area (71 m2 g−1) and a very broad pore size distribution with pore sizes in the range 10–120 nm (see Fig. S9, ESI†), which is accompanied by a rough surface with pore sizes up to micrometer range (see SEM image in Fig. 6d). In contrary to the other mixed metal oxide aerogels, the aerogel network of this compound (HS_43.1) shows a more columnar structure. Interestingly within the series of mixed metal oxide aerogels only the titania–zirconia compound showed a narrow pore-size distribution (Fig. 6b). In case of the synthesized silica–titania aerogels the pore-size distribution was much broader with size intervals of 20–60 nm when propylene oxide as auxiliary agent was used (HS_91). The neutralization of an excess of acidic (HS_126) or alkaline (HS_43.1) precursors within the sol–gel reaction by NaOH (HS_126) or HCl (HS_43.1) obviously is responsible for wide pore size intervals ranging from 10–140 nm, respectively from 10–120 nm (HS_43.1). The crystallinity of all samples was checked by X-ray diffraction analyses – the results can be taken from Fig. 6f, g and 7, and S11–S14, ESI†. Although we performed many experiments no gel formation was observed starting from mixtures of TiCl4–SiCl4 and TiCl4–SiMeCl3.
Most of the synthesized silica–titania aerogels do not crystallize, neither at room temperature – nor at elevated temperature (450 °C). This finding coincides with the results from other groups. Schubert et al. obtained within the silica–titania system a very high onset temperature for the crystallization of anatase (800 °C) respectively cristobalite (1050 °C).75 Especially for the heat treated samples of HS_115 and HS_126 onset of crystallisation can be derived from peaks on the position typical for anatase type TiO2 (see Fig. 7). Why theses samples show some kind of crystallisation whereas similar performed syntheses do not lead to crystalline products remains, however unclear.
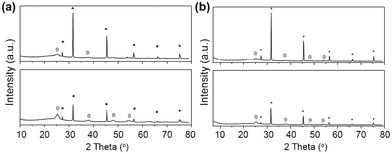 | ||
| Fig. 7 (a) Powder diffraction pattern from silica-titania aerogel HS_115 as prepared (upper figure), respectively heat-treated (lower figure). Peaks corresponding to NaCl (JCPDS 01-077-2064) are marked with an asterisk. Peaks corresponding to tetragonal TiO2, anatase modification (JCPDS 004-0477) are marked with a sphere. (b) Powder diffraction pattern from silica–titania aerogel HS_126 as prepared (upper figure), respectively heat-treated (lower figure). Peaks corresponding to NaCl (JCPDS 01-077-2064) are marked with an asterisk. Peaks corresponding to tetragonal TiO2, anatase modification (JCPDS 004-0477) are marked with a sphere. | ||
Hydrolysis of mixtures of Si- and Ti-alkoxides or single source alkoxy precursors were used by other groups in order to prepare silica-titania aerogels and xerogels. Organic based single source precursors for silica–titania aerogels and xerogels were developed.75 This procedure is hardly suitable for mass production of this material because of the expected high costs of the precursors. In addition the BET surface areas were, depending on the single source precursor used, 90 respectively 230 m2 g−1. Also the stepwise hydrolysis of separated Si and Ti precursors did not lead to convincing results in view of quite low BET surface areas and relatively high costs caused by the precursors.73 Yoda et al. generated silica–titania mixed aerogels by impregnation of organic titanium precursors into silica gels using different techniques.74,104,105 In terms of microstructure and porosity the results were brilliant. Large surface areas up to 900 m2 g−1 could be achieved74,104,105 and densities down to 0.15 g cm−13,105 were reached. On the other hand the procedures do not seem to be straightforward65 and in addition partially expensive and toxic precursors like tetramethoxysilane TMOS were used.105
D Conclusions
We have reported a rapid, straightforward synthesis of high quality, highly stable aerogels based on hydrolysis reactions of cheap precursors comprising titanium(IV) chloride, silicium(IV) chloride, zirconium(IV) chloride with or without water glass as additional network former. It should be noted that cheap propylene oxide can also be used as auxiliary agent. Apart from single metal oxide aerogels mixed-metal-oxide aerogels can be prepared by the hydrolysis of a mixture of chloride precursors. This technique provides a straightforward method for the preparation of homogeneous gels in aqueous solutions without the need for alkoxide precursors or complicate reaction procedures. Ethanol, required for solvent exchange was recycled and this step did not complicate the procedure. Supercritical drying of some of the gels led to crack-free aerogel bodies with nanoporous structure showing narrow pore size distribution with pore sizes in the nm range. X-ray powder diffraction analysis show that the as-prepared aerogels did not crystallize. After heat treatment at 450 °C titania and zirconia aerogels and some of the mixed oxide aerogels crystallize to form expected crystal structures. With nitrogen adsorption measurements, the BET surface areas of the aerogels up to 1400 m2 g−1 were determined, suggesting their potential use as an additives in foundry applications. For the use as aerogelic binders, aerogel granules, foundry sand and as thermal insulating compounds in vehicles, pipelines, packaging and home building the production costs have to be reduced drastically. The materials prepared by this method are currently being evaluated for these purposes and the results will be published elsewhere.Acknowledgements
The authors thank Markus Haases group for performing the XRD measurements and Martin Steinharts group for giving the possibility to perform a part of the experiments in his labs.References
- H. D. Gesseashr and P. C. Goswami, Chem. Rev., 1998, 89, 765 CrossRef.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22 CrossRef.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243 CrossRef CAS.
- K. Sinko, Materials, 2010, 3, 704 CrossRef CAS.
- J. L. Gurav, I.-K. Jung, H.-Ho. Park, E. S. Kang and D. Y. Nadargi, J. Nanomater., 2010 DOI:10.1155/2010/409310.
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- T. Sumiyoshi, I. Adachi, R. Enomoto, T. Iijima, R. Suda, N. Yokoyama and H. Yokogawa, Proceedings of the Fifth International Symposium on Aerogels (ISA 5), J. Non-Cryst. Solids, 1998, 225, 369 CrossRef CAS.
- T. Sumiyoshi, I. Adachi, R. Enomoto, T. Iijima, R. Suda, C. Leonidopoulos, D. R. Marlow, E. Prebys, R. Kawabata, H. Kawai, T. Ooba, M. Nanao, K. Suzuki, S. Ogawa, A. Murakami and M. H. R. Khan, Nucl. Instrum. Methods Phys. Res., Sect. A, 1999, 433, 385 CrossRef CAS.
- A. R. Buzykaev, A. F. Danilyuk, S. F. Ganzhur, E. A. Kravchenko and A. P. Onuchin, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 433, 396 CrossRef.
- A. K. Gougas, D. Ilie, S. Ilie and V. Pojidaev, Nucl. Instrum. Methods Phys. Res., Sect. A, 2000, 421, 249 CrossRef.
- T. M. Tillotson, W. E. Sunderland, I. M. Thomas and L. W. Hrubesh, J. Sol-Gel Sci. Technol., 1994, 1, 241 CrossRef CAS.
- C. M. Mo, Y. H. Li, Y. S. Liu, Y. Zhang and L. D. Zhang, J. Appl. Phys., 1998, 83, 4389 CrossRef CAS.
- B. B. Owens, S. Passerini and W. H. Smyrl, Electrochim. Acta, 1999, 45, 215 CrossRef CAS.
- J. W. Long, K. E. Swider, C. I. Merzbacher and D. R. Rolison, Langmuir, 1999, 15, 780 CrossRef CAS.
- G. M. Pajonk, Appl. Catal., 1991, 72, 217 CrossRef CAS.
- G. Matis, F. Juillet and S. Teichner, J. Bull. Soc. Chim. Fr., 1976, 1633 CAS.
- F. Blanchard, B. Pommier, J. P. Reymond and S. J. Teichner, J. Mol. Catal., 1982, 17, 171 CrossRef CAS.
- B. E. Yoldas, M. J. Annen and M. J. Bostaph, Chem. Mater., 2000, 12, 2475 CrossRef CAS.
- D. Quenard, B. Chevalier, H. Sallee, F. Olive and D. Giraud, Rev. Metall. Cah. Inf. Tech., 1998, 95, 1149 CAS.
- J. F. T. Conroy, B. Hosticka, S. C. Davis, A. N. Smith and P. M. Norris, Microscale Thermophys. Eng., 1999, 3, 199 CrossRef.
- M. Gronauer and J. Fricke, Acustica, 1986, 59, 177 CAS.
- L. C. Klein, Annu. Rev. Mater. Sci., 1985, 15, 227 CrossRef CAS.
- A. H. Boonstra and C. A. M. Mulder, J. Non-Cryst. Solids, 1988, 105, 201 CrossRef CAS.
- P. B. Wagh, R. Begag, G. M. Pajonk, A. V. Rao and D. Haranath, Mater. Chem. Phys., 1999, 57, 214 CrossRef CAS.
- S. Henning and L. Svensson, Phys. Scr., 1981, 23, 697 CrossRef CAS.
- M. Yamane, S. Inove and A. Yasumori, J. Non-Cryst. Solids, 1984, 63, 12 CrossRef.
- L. B. Brown, A. M. Anderson and M. K. Carrol, J. Sol-Gel Sci. Technol., 2012, 62, 404 CrossRef CAS.
- D. A. Ward and E. I. Ko, Chem. Mater., 1993, 5, 956 CrossRef CAS.
- D. J. Suh and T.-J. Park, Chem. Mater., 2002, 14, 1452 CrossRef CAS.
- S. J. Teichner, G. A. Nicolaon, M. A. Vicarini and G. E. E. Gardes, Adv. Colloid Interface Sci., 1976, 5, 245 CrossRef CAS.
- C. Stocker and A. Baiker, J. Non-Cryst. Solids, 1998, 223, 165 CrossRef CAS.
- L. K. Campbell, B. K. Na and E. I. Ko, Chem. Mater., 1992, 4, 1329 CrossRef CAS.
- H. Li, S. G. Sunol and A. K. Sunol, Nanotechnology, 2012, 23, 294012 CrossRef CAS.
- S. Kelly, F. H. Pollak and M. Tomkiewicz, J. Phys. Chem. B, 1997, 101, 2730 CrossRef CAS.
- B. Ksapabutr, E. Gulari and S. Wongkasemjit, Mater. Sci. Forum Vols., 2005, 549, 480 Search PubMed.
- C. N. Chervin, B. J. Clapsaddle, H. W. Chiu, A. E. Gash, J. H. Jr. Satcher and S. M. Kauzlarich, Chem. Mater., 2005, 17, 3345 CrossRef CAS.
- T. Okubo, T. Takahashi, M. Sadakata and H. Nagamoto, J. Membr. Sci., 1996, 118(2), 151 CrossRef CAS.
- C. Xia, H. Cao, H. Wang, P. Yang, G. Meng and D. Peng, J. Membr. Sci., 1999, 162, 181 CrossRef CAS.
- S. D. Bhagat, Y.-H. Kim, M.-J. Moon, Y.-S. Ahn and J.-G. Ye, Solid State Sci., 2007, 9(7), 628 CrossRef CAS.
- L. W. Hrubesh and R. W. Pekala, J. Mater. Res., 1994, 9(3), 731 CrossRef CAS.
- N. Olivi-Tran, A. Lecomte, P. Lenormand and A. Dauger, J. Phys.: Condens. Matter, 2000, 12, 7547 CrossRef CAS.
- Z. Zhao, D. Chen and X. Jiao, J. Phys. Chem. C, 2007, 111, 18738 CAS.
- A. E. Gash, T. M. Tillotson, J. H. Jr. Satcher, L. W. Hrubesh and R. L. Simpson, J. Non-Cryst. Solids, 2001, 285, 22 CrossRef CAS.
- A. E. Gash, J. H. Satcher and R. L. Simson, Chem. Mater., 2003, 15, 3268 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Jr. Satcher, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999 CrossRef CAS.
- B. J. Clapsaddle, A. E. Gash, J. H. Satcher and R. L. Simpson, J. Non-Cryst. Solids, 2003, 331(1–3), 190 CrossRef CAS.
- Y. P. Gao, C. N. Sisk and L. J. Hope-Weeks, Chem. Mater., 2007, 19, 6007 CrossRef CAS.
- S. K. Gill and L. J. Hope-Weeks, Chem. Commun., 2009, 4384–4386 RSC.
- Z. Deng, J. Wang, Y. Zhang, Z. Weng, Z. Zhang, B. Zhou, J. Shen and L. Cheng, Nanostruct. Mater., 1999, 11(8), 1313 CrossRef CAS.
- R. Takahashi, S. Sato, T. Sodesawa, K. Suzuki, M. Tafu, K. Nakanishi and N. Soga, J. Am. Ceram. Soc., 2001, 84(9), 1968 CrossRef CAS.
- R. Takahashi, K. Nakanishi and N. Soga, J. Sol–Gel Sci. Technol., 1997, 8, 71 CAS.
- R. Takahashi, K. Nakanishi and N. Soga, J. Ceram. Soc. Jpn., 1998, 106, 772 CrossRef CAS.
- H. Schäfer, H.-R. Stock and P. Mayr, Corros. Sci., 2005, 47, 953 CrossRef.
- C. C. Pavel, D. Vuono, L. Catanzaro, P. De Luca, N. Bilba, A. Nastro and J. B. Nagy, Microporous Mesoporous Mater., 2002, 56, 227 CrossRef CAS.
- D. S. van Vuuren, S. J. Oosthuizen and M. D. Heydenrych, The Journal of The Southern African Institute of Mining and Metallurgy, 2011, 111, 141 CAS.
- R. A. Gonzalez, C. D. Musick and J. N. Tilton, Process for controlling agglomeration in the manufacture of TiO2, U.S. Patent 5,508,015, April 16, 1996 Search PubMed.
- J. C. Deberry, M. Robinson, M. D. Pomponi, A. J. Beach, Y. Xiong and K. Akhtar, Controlled vapor phase oxidation of titanium tetrachloride to manufacture titanium dioxide, U.S. Patent 6,387,347, May 14, 2002 Search PubMed.
- Suplier: Hutong Global Co., Ltd, Chagugang town,wuqing district,Tianjin,China. Date of quotation: 09/17/2012.
- Suplier: Tomson Chemical Limited, Hangzhou, China, Price: 8400 $/ton; Date of quotation: 09/17/2012.
- T.-H. Wang, A. M. N. Lopez, S. Li, D. A. Dixon and J. L. Gole, J. Phys. Chem. A, 2010, 114, 7561 CrossRef CAS.
- W. Zheng, X. Liu, Z. Yan and L. Zhu, ACS Nano, 2009, 3(1), 115 CrossRef CAS.
- A. D. Paola, M. Bellardita, R. Ceccato, L. Palmisano and F. Parrino, J. Phys. Chem. C, 2009, 113, 15166 Search PubMed.
- D.-S. Lee and T.-K. Liu, J. Sol-Gel Sci. Technol., 2002, 25, 121 CrossRef CAS.
- N. Sasireka, B. Rajesh and Y.-W. Chen, Thin Solid Films, 2009, 518(1), 43 CrossRef.
- Producer: Shanghai Richem International Co., Ltd., Shanghai, China, Date of quotation: 09/17/2012.
- F. J. Broecker, W. Heckmann, F. Fischer, M. Mielke, J. Schroeder and A. Stange, Proceedings of the Second International Symposium on Aerogels (ISA2), Rev. Phys. Appl. Colloq., 1989, 10 Search PubMed.
- R. A. Boyse and E. I. Ko, Catal. Lett., 1996, 38, 225 CrossRef CAS.
- L. Chen, J. Hu and R. M. Richards, ChemPhysChem, 2008, 9, 1069 CrossRef CAS.
- H. W. Brinkmann, G. Z. Zhao and A. Burggraf, J. Solid State Ion, 1993, 37, 63 Search PubMed.
- A. Rahtu and M. Ritala, J. Mater. Chem., 2002, 12, 1484 RSC.
- A. K. Kivi, E.-L. Lakomaa, A. Root, H. Osterholm, J.-P. Jacobs and H. H. Brongersma, Langmuir, 1997, 13, 2717 CrossRef.
- J. C. Wang, S. H. Chiao, C. L. Lee, T. F. Lei and Y. M. Lin, J. Appl. Phys., 2002, 92, 3936 CrossRef CAS.
- M. A. Holland, D. M. Pickup, G. Mountjoy, E. S. C. Tsang, G. W. Wallidge, R. J. Newport and M. E. Smith, J. Mater. Chem., 2000, 10, 2495 RSC.
- S. Yoda, K. Otake, Y. Takebayashi, T. Sugeta and T. Sato, J. Non-Cryst. Solids, 2001, 285, 8 CrossRef CAS.
- W. Rüpp, N. Hüsing and U. Schubert, J. Mater. Chem., 2002, 12, 2594 RSC.
- K. W. Terry and T. D. Tilley, Chem. Mater., 1991, 3, 1001 CrossRef CAS.
- M. P. Coles, C. G. Lugmair, K. W. Terry and T. D. Tilley, Chem. Mater., 2000, 12, 122 CrossRef CAS.
- X. M. Wang and P. Xia, J. Mater. Res., 2006, 21(2), 355 CrossRef CAS.
- Y. Wan, J. X. Ma, W. Zhou, Y. J. Zhu, X. Y. Song and H. X. Li, Appl. Cat. A: General, 2004, 277(55), 1 Search PubMed.
- B. Malinowska, J. Walendziewski, D. Robert, J. V. Weber and M. Storalski, Int. J. Photoenergy, 2003, 5, 147 CrossRef CAS.
- B. Sen and A. Vannice, J. Catal., 1998, 113, 52 CrossRef.
- D. Robert and J. V. Weber, J. Clean. Prod., 1998, 6, 335 CrossRef.
- G. Poelz and R. Riethmuller, Cerenkov counters, Report 811055, DESY (Deutches Elektronen-Synchrotron), 1981 Search PubMed.
- A. V. Rao, G. M. Pajonk and N. N. Parvathy, J. Mater. Sci., 1994, 29, 1807 CrossRef CAS.
- S. Ki. Hong, Mi. Y. Yoon and H. Jin Hwang, J. Am. Ceram. Soc., 2011, 94(10), 3198 CrossRef CAS.
- Qingdao Sinoglory Chemical Co., Ltd, Qingdao, China.
- Y. C. Cha, C. E. Kim, S. Lee, H. J. Hwang, J. W. Moon, I. S. Han and S.-K. Woo, Sol. State Phenom. Vols., 2007, 671, 124 Search PubMed.
- A. V. Rao, A. P. Rao and M. M. Kulkarni, J. Non-Cryst. Solids, 2004, 350, 224 CrossRef CAS.
- C. J. Lee, G. S. Kim and S. H. Hyun, J. Mater. Sci., 2002, 37, 2237 CrossRef CAS.
- L. M. Hair, R. W. Pekala, R. E. Stone, C. Chen and S. L. Buckley, J. Vac. Sci. Technol., A, 1988, 6, 2559 CAS.
- C. A. Garcia-Gonzalez, M. C. Camino-Rey, M. Alnaief, C. Zetzl and I. Smirnova, J. Supercrit. Fluids, 2012, 66, 297 CrossRef CAS.
- F. Schwertfeger, D. Frank and M. Schmidt, J. Non-Cryst. Solids, 1998, 225, 24 CrossRef CAS.
- FeyeCon Development & Implementation B. V. 1382 GS Weesp, The Netherlands.
- C. Liang, G. Sha and S. Guo, J. Non-Cryst. Solids, 2000, 271, 167 CrossRef CAS.
- F. Kirkbir, H. Murata, D. Meyers and S. R. Chaudhuri, J. Non-Cryst. Solids, 1998, 225, 14 CrossRef CAS.
- T. M. Tillotson, W. E. Sunderland, I. M. Thomas and L. W. Hrubesh, J. Sol-Gel Sci. Technol., 1994, 1, 241 CrossRef CAS.
- Dongying Longxing Chemical Co., Ltd., Shandong, China, Date of quotation: 10/08/2012.
- G. B. Kolesar, W. H. Siddiqui, R. G. Geil, R. M. Malczewski and E. J. Hobbs, Fundam. Appl. Toxicol., 1989, 13, 285 CrossRef CAS.
- H. Tamon, T. Kitamura and M. Okazaki, J. Colloid Interface Sci., 1998, 197, 353 CrossRef CAS.
- B. Xia, H. Huang and Y. Xie, Mater. Sci. Eng., B, 1999, 57, 150 CrossRef.
- B. M. Gauthier, S. D. Bakrania, A. M. Anderson and M. K. Carroll, J. Non-Cryst. Solids, 2004, 350, 238 CrossRef CAS.
- X. Z. Guo, L. Q. Yan, H. Yang, J. Li, C. Y. Li and X. B. Cai, Acta. Phys. Chim. Sin., 2011, 27(10), 2478 CAS.
- C. Stöcker, M. Schneider and A. Baiker, J. Porous Mater., 1996, 2, 325 CrossRef.
- S. Yoda, Y. Tasaka, K. Uchida, A. Kawai, S. Ohshima and F. Ikazaki, J. Non-Cryst. Solids, 1998, 225, 105 CrossRef CAS.
- S. Yda, K. Ohtake, Y. Takebayashi, T. Sugeta, T. Sako and T. Sato, J. Mater. Chem., 2000, 10, 2151 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis procedures, additional results from nitrogen adsorption measurements as well as from powder diffraction analyses can be taken from the supporting information. This material is available free of charge via the internet. See DOI: 10.1039/c3ra41688g |
| This journal is © The Royal Society of Chemistry 2013 |