 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First principles calculations of oxygen vacancy formation in barium-strontium-cobalt-ferrite
C.
Zhang
and
P. D.
Bristowe
*
Department of Materials Science and Metallurgy, University of Cambridge, Cambridge, CB2 3QZ, UK. E-mail: pdb1000@cam.ac.uk
First published on 11th June 2013
Abstract
Perovskite-type barium-strontium-cobalt-ferrite (BSCF) is a potentially significant material in the development of electrodes for solid oxide fuel cells and electrolysis cells, primarily because of its large oxygen vacancy concentration and mobility. Using density functional theory (DFT) with the DFT + U approach, we perform first principles calculations of oxygen vacancy formation in bulk BSCF with the composition Ba0.5Sr0.5Co0.75Fe0.25O3−δ, in which a range of oxygen deficiencies (0.125 ≤ δ ≤ 0.875) is investigated. Contrary to previous DFT studies for δ = 0.125, DFT + U predicts that the non-stoichiometric structure is more stable than its stoichiometric counterpart with δ = 0, and that this originates from a significantly different atomic and electronic structure following the introduction of the Hubbard U term in the calculations. As more vacancies are created there is no tendency for ordering and the total vacancy formation energy becomes positive and increases as the oxygen deficiency increases. Using ab initio thermodynamics, the heat of formation of BSCF as a function of oxygen partial pressure and temperature is determined and the results are in broad agreement with available stoichiometry measurements.
1. Introduction
Solid-oxide fuel cells (SOFCs) are electrochemical devices that convert chemical energy into electrical energy, and offer significant prospects for efficient utilization of various fuels with low emissions. However, most electrode materials require a high operating temperature in SOFC applications and this results in high costs, material incompatibility and other issues. Barium-strontium-cobalt-ferrite (BSCF), which has a perovskite-type structure, has been shown to be a promising cathode material for reduced temperature SOFC operation due to its large concentration of oxygen vacancies, high oxygen ion mobility and significant electrocatalytic activity for the oxygen reduction reaction.1,2 More recently, it has been shown that BSCF could also be a potential candidate as an anode material for solid-state electrolysis cells (SOECs), which perform the reverse electrochemical process to SOFCs.3Understanding electrode materials at the atomic level can provide insight into improving the performance of SOFCs and SOECs. In particular, acquiring information on BSCF composition and structure is important in order to understand its ionic and electronic conductivity, its crystal stability and surface activity. Much effort has been made towards understanding BSCF with a particular emphasis on the composition Ba0.5Sr0.5Co0.8Fe0.2O3−δ. It has been shown experimentally4 that the ferrite can exhibit significant oxygen deficiency (δ = 0.66–0.81) between 873 and 1173 K and oxygen partial pressures of 1 × 10−3 to 1 atm. In first principles studies of BSCF using density functional theory (DFT) a slightly different composition, Ba0.5Sr0.5Co0.75Fe0.25O3, is often used5–9 for computational convenience. Formation of a single oxygen vacancy has been studied by removing a neutral O atom from the model BSCF structure, and with respect to the energy of an O2 molecule (i.e. in the O-rich limit) the vacancy formation energy has been calculated and found to be ∼1.3 eV. This is quite small compared to the vacancy formation energy of ∼5.0 eV, also calculated in the O-rich limit, for simpler perovskites such as SrTiO3 and LaMnO3.10,11 However, these DFT studies employed standard exchange and correlation functionals (e.g. the generalized gradient approximation, GGA), which are known to be an inadequate description of strongly correlated systems such as the one in the present study. Furthermore, most of the previous DFT studies have focused on single oxygen vacancy formation in the ideal model BSCF, and the composition with large oxygen deficiency reported experimentally has not been probed. In this paper, we perform DFT + U calculations on the formation of multiple oxygen vacancies in the model structure. The introduction of the Hubbard U term has been shown to improve the description of strongly correlated systems, and has now been used widely in studies of similar perovskites.12–15 Compositions with oxygen deficiency up to δ = 0.875 have been examined. We also use ab initio thermodynamics16 to determine how the heat of formation of the non-stoichiometric composition varies under environmental conditions close to those reported experimentally.
2. Computational details
We employ the plane-wave DFT approach as implemented in the computational package VASP.17,18 The generalized gradient approximation (GGA) parameterized by Perdew, Burke and Ernzerhof (PBE),19 along with the projector-augmented wave (PAW) method,20 are used to describe ionic cores. Specifically, we use PAW PBE potentials with electronic configurations O (2s2 2p4), Fe (3d7 4s1), Co (3d8 4s1), Sr (4s2 4p6 5s2) and Ba (5s2 5p6 6s2). To avoid the self-interaction errors that occur in the standard GGA functional for strongly correlated systems, the DFT + U approach, which accounts for the on-site Coulomb interaction in the relevant d orbitals, is adopted. Effective U values of 3.3 and 4.0 eV are used for Co and Fe respectively, which is guided by many DFT + U studies on similar systems.12,13,21–23 In our calculations a 4 × 4 × 4 k-point mesh is created by the Monkhorst–Pack scheme for the BSCF cell, and the cut-off energy for the plane-wave expansion is set to be 520 eV. With these choices, a convergence of 0.003 Å and 0.005 eV can be achieved for the geometry and energy respectively.Fig. 1a shows the model BSCF structure adopted in previous DFT studies and used to investigate oxygen vacancy formation.5–9 As can be seen, it is a cubic ABO3 perovskite-type supercell with composition Ba0.5Sr0.5Co0.75Fe0.25O3 containing 4 Ba, 4 Sr, 2 Fe, 6 Co and 24 O ions. It features alternating Ba and Sr ions on the A sites, and alternating Co and Fe ions on the B sites with the two Fe ions being well separated. The separation of Fe ions is based on observations that indicate that BSCF does not contain segregated regions.7 In order to compare with the previous studies, we have also performed DFT calculations with the standard PBE functional on the model structure with and without a single charge neutral O vacancy created by removing an oxygen atom. Our calculations reproduce most of the previously reported results,5–9e.g., the lattice constant of 3.91 Å for the defect-free model perovskite structure (i.e. 7.82 Å for the supercell constant), the high spin state (S = 2) for Fe, the intermediate spin state (S = 3/2) for Co, and the ferromagnetic spin arrangement as the most stable electronic configuration. The magnetic configurations are consistent with available experimental data24 and this result does not change using DFT + U. However, the vacancy formation energies, calculated in the standard way with respect to the model structure and an O2 molecule in its lowest energy triplet state, are somewhat different, despite the fact that the geometrical and electronic characteristics of the vacancies obtained in our calculations are similar to those reported previously. Specifically, we found that to create an O vacancy (VO) in between two Co ions (Co–VO–Co), shown schematically in Fig. 1b, or between a Co and a Fe ion (Co–VO–Fe), an energy of 0.82 eV and 0.91 eV is needed respectively, which differs from the reported values of 1.34 eV and 1.40 eV;6 both values are obtained in the O-rich limit. We believe that the discrepancy originates from the calculated energy of the gas-phase O2 molecule, for which a binding energy of 5.24 eV was reported in the previous calculation6 whereas we obtained 6.08 eV. Other relevant DFT calculations of the O2 binding energy using a similar GGA functional and O pseudopotential (5.99 eV25 and 6.04 eV22,25) are in closer agreement with our value. To further verify the results we have also used CASTEP26 to calculate the formation energy of an O vacancy between two Co ions, and found that the result obtained with this code (0.79 eV) is very close to that obtained with VASP (0.82 eV). It is important to note that DFT-GGA significantly overestimates the binding energy of an O2 molecule and a correction must be added to the calculated O2 energy. This issue has been addressed in detail by Lee et al.25 and Wang et al.22 where the correction is determined by fitting calculated and experimental formation enthalpies of metal oxides. Therefore, we will use below the reported corrected O2 total energy (−8.50 eV for the PBE functional and using the O PAW pseudopotential) in calculating the vacancy formation energy, unless otherwise stated.
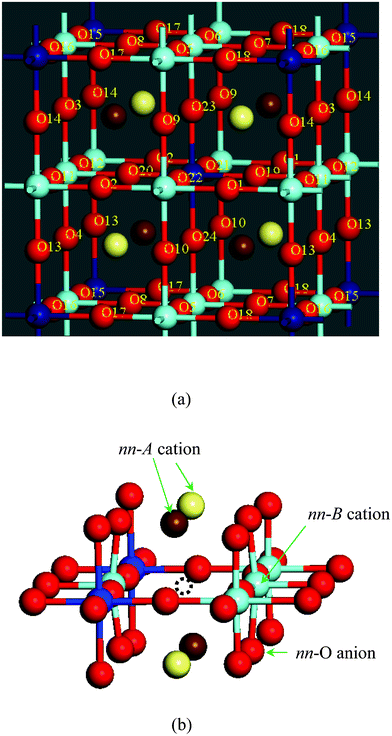 | ||
| Fig. 1 (a) Model structure of Ba0.5Sr0.5Co0.75Fe0.25O3 (BSCF). The light and dark brown circles represent Sr and Ba respectively, the light and dark blue circles represent Co and Fe respectively, and the red circles are O ions. All the O ions are labelled. (b) DFT-optimised local structure surrounding an oxygen vacancy, represented by a dashed circle, between two Co ions. Ions labelled nn are indicative of the nearest neighbours to the vacancy. | ||
In our DFT + U calculations of oxygen multi-vacancies, we started with a single VO in the model BSCF structure, i.e. one of the Co–VO–Co or Co–VO–Fe configurations, and investigated first all possible sites for an additional VO in order to find the most stable structure for two oxygen vacancies (the Fe–VO–Fe configuration was not considered since the separation between Fe ions is large in the model BSCF structure due to the low Fe concentration and the assumed homogeniety of the material). As more vacancies were created, however, many configurations with similar energies emerged. It became clear that there would be too many possible configurations to account for. Thus, to investigate the structure and energy of n oxygen vacancies (i.e. equivalent to an oxygen deficiency of δ = n/8), we only chose the several most stable (and most representative in some cases) configurations found for n − 1 vacancies and used them as the starting configurations for subsequent optimisation with another vacancy. This procedure was repeated until 7 vacancies were created, equivalent to an oxygen deficiency of δ = 0.875.
3. Results and discussion
3.1. Single oxygen vacancy: DFT versus DFT + U calculations
The DFT + U calculations are found to produce some significantly different geometrical and electronic structures compared to the standard DFT calculations on both the ideal model structure and the structure with a single VO. The calculated vacancy formation energy is quite different: without adding the O2 energy correction,22,25 values of −1.34 eV and −1.16 eV are obtained in the O-rich limit using the DFT + U method for the formation of Co–VO–Co and Co–VO–Fe vacancies respectively, which is in sharp contrast to the standard DFT values of around 0.8–0.9 eV, also obtained in the O-rich limit. To verify this difference, we have performed calculations with a hybrid HSE functional proposed by Heyd, Scuseria and Ernzerhof,27 which has recently been shown to give a good description of the physical properties of pervoskites.28 We found that the vacancy formation energy calculated with the HSE functional (−1.19 eV for Co–VO–Co and −1.03 eV for Co–VO–Fe in the O-rich limit) is in good agreement with the DFT + U results.We list in Table 1 some relevant bond lengths to illustrate the differences in atomic structure between the two calculations taking the Co–VO–Fe vacancy as an example. Since the lattice constant of the defect-free structure using DFT + U (3.93 Å) is slightly larger than that using standard DFT (3.91 Å), the Co–O and the Fe–O bonds from DFT + U are generally longer and thus weaker than those from standard DFT. After vacancy formation, the largest structural change occurs at the sites neighbouring the vacancy.6 For example, when a VO is created between a Co and a Fe ion, the two 5-coordinated metal ions move away from one another relative to their positions in the defect-free structure, resulting in a distortion of the ideal octahedral geometry. Interestingly, a larger distortion is found in the DFT + U calculations: e.g. with respect to its original position, the 5-coordinated Co ion moves away from the vacant site by 0.297 Å along the Co–VO–Fe bond axis (compared to 0.223 Å in the standard DFT). Furthermore, the metal-O bond lengths neighbouring the VO are in general shorter using DFT + U, suggesting that after vacancy formation the under-coordinated metal ions bind more strongly with their oxygen neighbours than they did in the DFT calculations.
| DFT | DFT + U | ||
|---|---|---|---|
| Co–O | Defect-free structure | 1.953 (4), 1.986 (2) | 1.966 (4), 1.955 (2) |
| Co–VO–Fe | 1.883 (4), 1.868 (1) | 1.817 (4), 2.188 (1) | |
| Fe–O | Defect-free structure | 1.920 (6) | 1.977 (6) |
| Co–VO–Fe | 1.868 (4), 1.799 (1) | 1.860 (4), 1.769 (1) | |
| ΔCo | 0.223 | 0.297 | |
| ΔFe | 0.255 | 0.262 | |
To illustrate the electronic structure differences between the two calculations, we again take the Co–VO–Fe vacancy as an example, and plot in Fig. 2 the total density of states (DOS) and the DOS projected onto the Co and the Fe ions that are nearest neighbours to the VO. For comparison, we also include the total DOS and the projected DOS for the defect-free structure. As can be seen from Fig. 2b and 2c, the addition of the Hubbard U in the calculations significantly affects the position of electronic states in both the defect-free and the vacancy system and, in particular, the metal d states shift toward higher energies whereas the p states appear at lower energies. Furthermore, after vacancy formation, more p states appear at lower energies (around −6.0 eV) in the DFT + U results, indicating a large contribution from these states to the stability of the metal-O bonds, whereas much smaller differences exist before and after the vacancy formation in the standard DFT results. These features are consistent with the larger geometrical changes induced by the vacancy in the DFT + U calculations compared to the standard DFT calculations. Another interesting result from the DFT + U calculations is the larger changes seen around the Fermi level in the total DOS after vacancy formation. The peak at 0.5 eV moves up to around 1.0 eV due to the upward shift of the d states of the metal ions neighbouring the vacancy. This leaves fewer states at the Fermi level implying a material with less metallic character after vacancy formation.
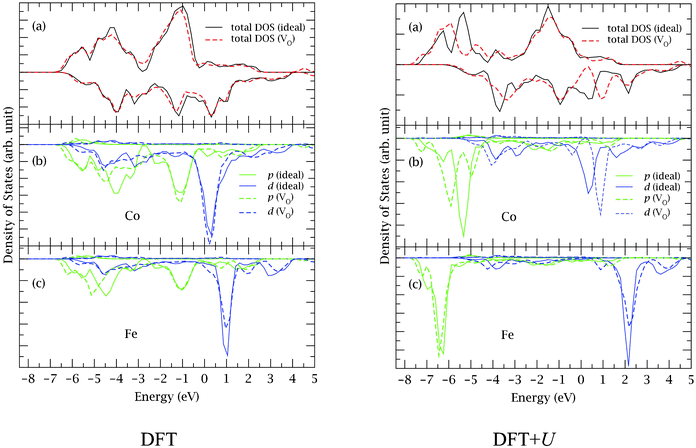 | ||
| Fig. 2 (a) Total densities of states (DOS) of the defect-free model BSCF structure (solid curves) and that containing an O vacancy (VO) between a Co and a Fe atom (dashed curves), from standard DFT (left) and DFT + U (right) calculations. (b) and (c) Projected DOS (PDOS) of a Co and a Fe atom in the model structure (solid curves) and PDOS of the Co and the Fe atom that are nearest neighbors to the VO (dashed curves). The Fermi levels are set at zero eV. For clarity, we neglect the s states in the PDOS. | ||
Oxygen vacancy formation leads to charge redistribution around the vacant site, for which the DFT + U calculations also show some significant differences from the standard DFT calculations. Using a Bader charge analysis,29 we summarise in Table 2 changes in ionic charge for ions adjacent to an oxygen atom following its removal from the model structure (again we use the Co–VO–Fe as an example). As can be seen, there are significantly smaller charge gains on the B cations and, to a lesser extent, on the O anions nearest to the VO using DFT + U, although the largest charge gain per ion still occurs at these B cations as found in standard DFT. On the other hand, more charge gains are found at the A cations and those O anions further away from the VO using DFT + U. This charge redistribution, which is found using DFT + U, helps stabilize the B-O bonds compared to the standard DFT result, since less extra charge is gained at those ions nearest to the vacancy, leading to much smaller Coulombic repulsion between them. It is not surprising that the charge gain at the B cations is different from the gain at the A cations since it is the B cations on to which the U term is explicitly applied. Usually DFT + U predicts more charge gain than standard DFT, i.e. more localised electronic configurations. For the oxygen vacancy in BSCF we find that the B cations gain less charge when compared to standard DFT as noted above. This may be due to the presence of the nearby A cations (on to which no U is applied) which suppresses this localisation effect.
| DFT | DFT + U | |
|---|---|---|
| a These refer to the rest of the A cations except the nn-A cations. b These nn-O anions directly bond to the two 5-fold B cations. c These refer to the O anions surrounding the VO except the nn-O anions. | ||
| nn-B cations | 1× Fe: −0.13 (−0.11) | 1× Fe: −0.05 |
| 1× Co: −0.23 (−0.26) | 1× Co: −0.15 | |
| nnn-B cations | 4× Co: −0.06 | 4× Co: −0.03 |
| nn-A cations | 2× Ba: −0.022 (−0.014) | 2× Ba: −0.043 |
| 2× Sr: −0.015 (−0.007) | 2× Sr: −0.023 | |
| other adjacent A cationsa | 2× Ba: −0.019 | 2× Ba: −0.037 |
| 2× Sr: −0.014 | 2× Sr: −0.010 | |
| nn-O anionsb | 10× O: −0.023 (−0.030) | 10× O: −0.016 |
| other adjacent O anionsc | 4× O: −0.004 | 4× O: −0.031 |
3.2. Multiple oxygen vacancies (0.25 ≤ δ ≤ 0.875)
Starting from the two distinct single oxygen vacancies defined above, i.e. Co–VO–Co and Co–VO–Fe, we examined all the possible configurations for another oxygen vacancy, corresponding to an oxygen deficiency in the crystal of δ = 0.25. Results of the most stable configurations are summarized in Table 3 where the specific vacancy sites are indicated (see also Fig. 1 for the vacancy labels). As can be seen, the two vacancies in these configurations are neighbours to each other, and in the most stable configuration (labelled 1 and 2 in bold) the vacancies lie between two Co ions. These features have been noted in a previous DFT investigation which studied 1 to 4 vacancies in the same size BSCF supercell as used here but with a somewhat different arrangement of B cation.24 Furthermore, we calculate the total vacancy formation energy in the O-deficient BSCF for the O-rich limit with respect to defect-free BSCF, which is defined as:| ΔEδV = EδV + 8δ(EO) − E0 | (1) |
| δ | V1 | V2 | V3 | V4 | V5 | V6 | V7 | ΔEδV | a |
|---|---|---|---|---|---|---|---|---|---|
| 0.125 | 1 | −0.79 | 3.970 | ||||||
| 22 | −0.54 | 3.967 | |||||||
| 0.25 | 1 | 2 | −0.39 | 3.991 | |||||
| 1 | 11 | −0.30 | 3.972 | ||||||
| 1 | 9 | −0.18 | 3.972 | ||||||
| 0.375 | 1 | 11 | 19 | +0.38 | 4.015 | ||||
| 1 | 11 | 22 | +0.38 | 4.015 | |||||
| 1 | 11 | 20 | +0.42 | 4.019 | |||||
| 1 | 11 | 21 | +0.42 | 4.019 | |||||
| 0.5 | 1 | 11 | 20 | 22 | +1.39 | 4.042 | |||
| 1 | 11 | 19 | 22 | +1.61 | 4.039 | ||||
| 1 | 11 | 21 | 22 | +1.64 | 4.006 | ||||
| 0.625 | 1 | 11 | 20 | 22 | 3 | +3.33 | 4.048 | ||
| 1 | 11 | 20 | 22 | 13 | +3.38 | 4.044 | |||
| 1 | 11 | 20 | 22 | 10 | +3.53 | 4.048 | |||
| 0.75 | 1 | 11 | 20 | 22 | 13 | 10 | +5.56 | 4.050 | |
| 1 | 11 | 20 | 22 | 13 | 18 | +5.66 | 4.057 | ||
| 1 | 11 | 20 | 22 | 13 | 23 | +5.71 | 4.048 | ||
| 0.875 | 1 | 11 | 20 | 22 | 13 | 10 | 6 | +8.39 | 4.074 |
| 1 | 11 | 20 | 22 | 13 | 10 | 19 | +8.40 | 4.064 | |
| 1 | 11 | 20 | 22 | 13 | 10 | 16 | +8.43 | 4.083 |
We note that energy differences between the many possible structures for δ = 0.25 are not large. Ideally, all structures should be taken into account when considering the introduction of further oxygen vacancies. However, this becomes impractical at higher O deficiencies as there would be too many possible vacancy arrangements to be explored. Thus, we only chose the most stable divacancy configuration shown in Table 3 to investigate further oxygen vacancy formation. At δ = 0.375, four low energy structures emerge with very small energy differences between them. As can be seen these are derived from the second most stable divacancy (labelled 1 and 11), and configurations resulting from the most stable divacancy are actually at least 0.2 eV higher in energy and thus not shown. For δ = 0.50, we list the three most stable structures which again are derived from the second most stable divacancy. Note that the previous standard DFT study which considered oxygen vacancy formation in BSCF found a vacancy tetramer with a square shape to be the most stable configuration,24 whereas we found this configuration (i.e. vacancies labelled 1, 11, 19 and 22) is ∼0.2 eV higher in energy than the most stable configuration (i.e. vacancies labelled 1, 11, 20 and 22) which exhibits a V-shape. Another salient feature at these vacancy concentrations is that the total vacancy formation energy becomes positive suggesting that in the O-rich limit vacancy formation is no longer favourable with respect to the defect-free crystal.
Considering even higher O deficiencies, we found that the most stable configurations are always derived from the lowest energy (or very close to the lowest energy) configurations at lower O deficiencies. For example, the most stable configuration at δ = 0.626 results from the most stable configuration with the V shape at δ = 0.50, whereas any configurations derived from the configuration with the square shape, which is about 0.2 eV higher in energy, are at least 0.38 eV less stable; the most stable configuration at δ = 0.75 results from the second most stable configuration at δ = 0.625 which is, however, only 0.04 eV less stable than the most stable configuration at the same vacancy concentration. These results suggest that although it is debatable whether or not our approach to search for vacancy structures can categorically produce the most stable ones, the structures obtained are certainly among the most stable as well as the most representative. Energetically, we found that the total vacancy formation energy continues to rise, and becomes significantly positive at high O deficiencies.
In optimising the geometries of the vacancy structures, we also relaxed the cell parameters. Although the cubic structure is largely preserved after vacancy formation, some deviations in the parameters ≤3% are typically found. We list in Table 3 the average of the three lattice constants calculated at each oxygen deficiency. As can be seen, the structure gradually expands with increasing δ. This result is consistent with the lattice expansion observed in experiments. In particular, the optimised lattice constant from our calculation at δ = 0.625–0.75 is around 4.05 Å, which is in a good agreement with the experimental measurement4 of 4.03–4.05 Å for the material having similar oxygen non-stoichiometry (δ = 0.66–0.81), excluding effects from experimental temperature and pressure.
In terms of electronic structure, we have also identified some interesting trends with increasing the O deficiency. These can be seen from a comparison between the DOS calculated for the most stable configurations, as shown in Fig. 3. As discussed earlier and seen in Fig. 3, for δ = 0.125 the single vacancy formation results in two distinct changes in the DOS, the downward shift of the peak around −6.0 eV and the appearance of a new peak at 1.0 eV above the Fermi level. Both changes originate from the B cations neighbouring the vacancy and the oxygen atoms associated with them. Furthermore, the downward shift of the states is directly related to the relative stability of the vacancy. As the O deficiency increases, there is a reduction of electronic states below −6.0 eV (particularly noticeable for δ ≥ 0.50), and more states appear at higher energies such as between −5.0 and −4.0 eV, and −2.0 and −1.0 eV which are mainly contributed from cations that are not immediate neighbours of the vacancies. These trends can be understood from the fact that as more vacancies are created, more electrons left by the removal of oxygen are distributed among all the cations in the system, leading to longer and thus weaker metal–O bonds. These electronic changes are also consistent with the trend in energetics which shows that the total vacancy formation energy increases with increasing O deficiency, and in particular it becomes significantly positive when δ ≥ 0.50. On the other hand, Fig. 3 also shows that as the O deficiency increases, the peak at 1.0 eV above the Fermi level moves upward substantially. Accompanying this shift, there is a significant reduction of states around the Fermi level, resulting in a small band gap-like feature at high O deficiencies.
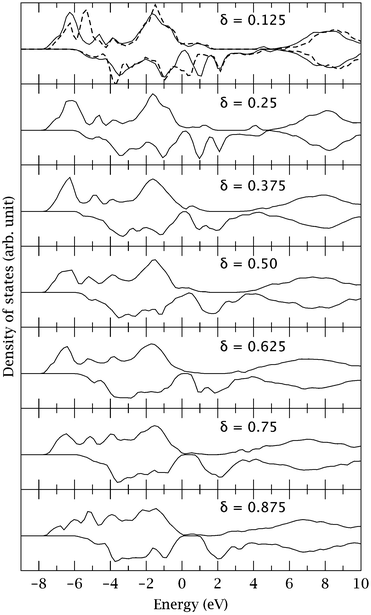 | ||
| Fig. 3 The uppermost panel: total density of states (DOS) for the defect-free BSCF structure (dashed curve) and the structure containing the Co–VO–Co vacancy (solid curve). Lower panels: DOS for the most stable configuration at different oxygen deficiencies (δ). The Fermi levels are set at zero eV. The DOS are also normlised with respect to the same number of electrons. | ||
3.3. Effect of oxygen atmosphere and temperature
So far we have only considered the O-rich limit, whereas in real applications the electrode material is always subjected to a particular oxygen partial pressure and temperature, which should be taken into account in assessing the stability of the vacancy structures. To this end, we have calculated the heat of formation (per formula unit) of the O-deficient BSCF, defined as:| ΔHδ = 0.125EδV − 0.5EBa − 0.5ESr − 0.75ECo − 0.25EFe − (3 − δ)(EO + μO(T, p)) | (2) |
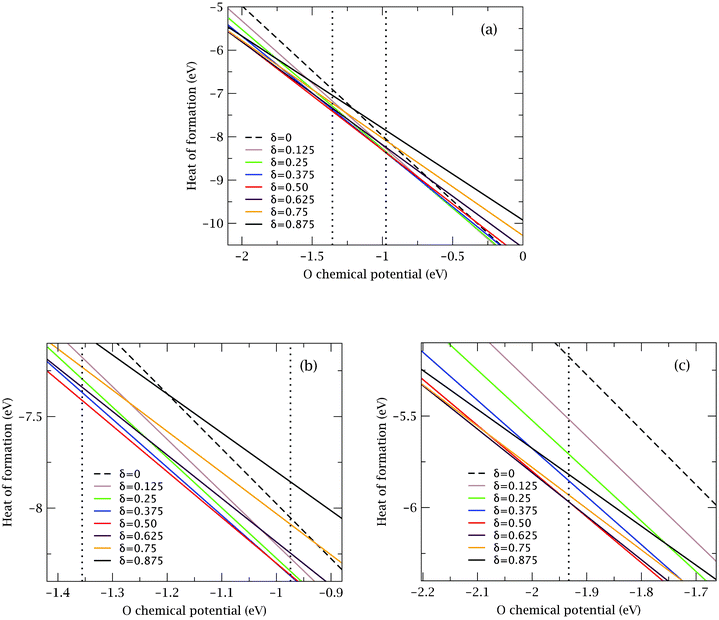 | ||
| Fig. 4 Heat of formation (per formula unit, f.u.) of BSCF (0 ≤ δ ≤ 0.875) as a function of oxygen chemical potential. The two vertical dotted lines in (a) represent relevant experimental conditions for which the O chemical potentials are −0.97 and −1.36 eV (see also text). For clarity, (b) and (c) show expanded views between these potentials and also near a potential of −1.93 eV. | ||
We note that in a neutron diffraction study4 of oxygen stoichiometry in BSCF, oxygen deficiencies were reported to have a minimum value of δ = 0.66 at 873 K and an oxygen partial pressure of 1 atm, and a maximum value of δ = 0.81 at 1173 K and an oxygen partial pressure of 10−3 atm. These experimental conditions correspond to an oxygen chemical potential range of −0.97 eV (873 K and 1 atm) to −1.36 eV (1173 K and 10−3 atm), which is indicated by two vertical dotted lines in Fig. 4a. As can be seen from the enlarged view in Fig. 4b, the most thermodynamically favorable stoichiometry in this range of μO is δ = 0.50, which is somewhat lower than the oxygen stoichiometry reported in the experiment (0.66–0.81). Furthermore, our results show that in the same range of μO many vacancy structures with other oxygen stoichiometries (e.g. δ = 0.250, 0.375 and 0.625) could still be viable since they are not significantly less favorable (<0.1 eV per f.u.) than the most stable structure with δ = 0.50. Although equilibrium kinetics may play a role in understanding this difference, other factors such as the arrangement of cations, which may be less ordered than assumed here, may subtly change the formation energies. Furthermore, we note from Fig. 4 that the relative stability of structures with different oxygen stoichiometries is quite sensitive to the thermodynamic conditions and a small variation can lead to different conclusions. Indeed Fig. 4c shows that when the O chemical potential is around −1.93 eV, which corresponds to a condition not far from the experimental ones (i.e. temperature of ∼1300 K and oxygen partial pressure of ∼10−4 atm), the most stable vacancy structure has an oxygen stoichiometry of 0.625, which is closer to the oxygen deficiency range reported experimentally. Despite these difficulties it is nevertheless clear from the calculations that extremes in stoichiometry (i.e. δ = 0 and δ = 0.875) are significantly less favorable than other stoichimetries and this agrees with all available observations.
4. Conclusions
BSCF, a promising electrode material for SOFC and SOEC applications, is non-stoichiometric in nature, yet the issue of oxygen vacancy formation has not been properly addressed in existing DFT simulations which used standard but often error-prone functionals in treating strongly correlated systems. As a consequence, we adopted a DFT + U approach with widely accepted U values for similar systems to study vacancy formation in BSCF. We first revisited the studies of isolated oxygen vacancy formation and our DFT + U calculations revealed that compared to the standard DFT results, the oxygen vacancy induces significantly larger geometry distortions as well as smaller ionic charge gains at those B cations neighbouring the vacant site, both effects increasing the stability of the vacancy. Consequently, the DFT + U vacancy formation energy is considerably smaller than found using standard DFT. We also probed the energy landscape for stable structures with high oxygen deficiencies corresponding to 0.25 ≤ δ ≤ 0.875. In particular, comparison has been made with the experimentally determined oxygen non-stoichiometry. Our results are in broad agreement with available data in that the material is significantly deficient in oxygen under the thermodynamic conditions studied and that this will need to be taken into account in further atomistic studies of the material, particularly of its surface since this is key to understanding its electrochemical performance.Acknowledgements
This work is supported by the Low Carbon Energy University Alliance (LCEUA). We are grateful to the UKCP Consortium for access to the UK National Supercomputing Service (HECToR) through EPSRC grant EP/F038356/1. Additional computing resources were provided by the University of Cambridge HPCS. Useful discussions with Prof. B. Yildiz and Prof. B. Yu are gratefully acknowledged.References
- Z. P. Shao and S. M. Haile, Nature, 2004, 431, 170 CrossRef CAS.
- F. S. Baumann, J. Fleig, H. U. Habermeier and J. Maier, Solid State Ionics, 2006, 177, 3187 CrossRef CAS.
- W. Zhang, B. Yu and J. Xu, Int. J. Hydrogen Energy, 2012, 37, 837 CrossRef CAS.
- S. McIntosh, J. F. Vente, W. G. Haije, D. H. A. Blank and H. J. M. Bouwmeester, Chem. Mater., 2006, 18, 2187 CrossRef CAS.
- E. A. Kotomin, Y. A. Mastrikov, M. M. Kuklja, R. Merkle, A. Roytburd and J. Maier, Solid State Ionics, 2011, 188, 1 CrossRef CAS.
- R. Merkle, Y. A. Mastrikov, E. A. Kotomin, M. M. Kuklja and J. Maier, J. Electrochem. Soc., 2012, 159, B219 CrossRef CAS.
- Y. A. Mastrikov, M. M. Kuklja, E. A. Kotomin and J. Maier, Energy Environ. Sci., 2010, 3, 1544 CAS.
- S. Gangopadhyay, A. E. Masunov, T. Inerbaev, D. Altilio, N. Orlovskaya, J. Mesit, R. Guha, A. Sleiti and J. Kapat, Abstracts of Papers of the American Chemical Society, 2009, 237, 1 Search PubMed.
- S. Gangopadhayay, A. E. Masunov, T. Inerbaev, J. Mesit, R. K. Guha, A. K. Sleiti and J. S. Kapat, Solid State Ionics, 2010, 181, 1067 CrossRef.
- Y. F. Zhukovskii, E. A. Kotomin, R. A. Evarestov and D. E. Ellis, Int. J. Quantum Chem., 2007, 107, 2956 CrossRef CAS.
- E. A. Kotomin, Y. A. Mastrikov, E. Heifetsa and J. Maier, Phys. Chem. Chem. Phys., 2008, 10, 4644 RSC.
- H. Ding, A. V. Virkar, M. Liu and F. Liu, Phys. Chem. Chem. Phys., 2013, 15, 489 RSC.
- Z. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 17696 CrossRef CAS.
- J. Hong, A. Stroppa, J. Iniguez, S. Picozzi and D. Vanderbilt, Physical Review B, 2012, 85 Search PubMed.
- H. Hsu, K. Umemoto, M. Cococcioni and R. M. Wentzcovitch, Phys. Earth Planet. Inter., 2011, 185, 13 CrossRef CAS.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter, 2001, 65, 035406 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Rollmann, A. Rohrbach, P. Entel and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 165107 CrossRef.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- J. W. Han and B. Yildiz, J. Mater. Chem., 2011, 21, 18983 RSC.
- S. Gangopadhayay, T. Inerbaev, A. E. Masunov, D. Altilio and N. Orlovskaya, ACS Appl. Mater. Interfaces, 2009, 1, 1512 CAS.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- J. He and C. Franchini, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 235117 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS.
- D. R. Stull and H. Probhet, JANAF Thermochemical Tables, U.S. National Bureau of Standards, Washington, D. C, 1971 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |