 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-headed nucleotides in DNA-zipper structures; base–base interactions and UV-induced cross-coupling in the minor groove†
Charlotte S.
Madsen
,
Lise J.
Nielsen
,
Nikolai S.
Pedersen
,
Anne
Lauritsen
and
Poul
Nielsen
*
Nucleic Acid Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark. E-mail: Pouln@sdu.dk; Fax: +45 6615 8780; Tel: +45 6550 2565
First published on 4th April 2013
Abstract
Nucleic acids containing double-headed nucleotides with additional nucleobases attached to the 5′(S)-position of thymidine through a methylene linker are studied. The additional bases are oriented towards base-base interactions in the minor groove of the DNA-double helix. Two new examples with adenine or cytosine as the additional bases as well as an analogue with a 4N-methylpiperazine in the same position are introduced, and in a combined study with the original double-headed nucleotide containing two thymines, interactions between the additional nucleobases across the minor groove are detected. Finally, a duplex with two thymines in the minor groove is cross-linked using UV irradiation.
Introduction
The nucleic acid duplex constitutes an excellent scaffold for supramolecular chemistry.1–3 By the introduction of modified nucleotides, the duplex has been used to organize different moieties, for instance chromophores, in proximity on the duplex surface.1–3 Recently, we and others have focused on interactions either at the duplex surface or in the duplex core by the introduction of double-headed or double-functionalized nucleotides into the duplex.4–13 As an example, a double-headed nucleotide with an additional thymine nucleobase positioned in the 5′(S)-position trough a methylene linker has been investigated.5 Each incorporation of this modification (termed TT in Fig. 1) into duplexes was found to cause a rather uniform decrease in the duplex thermal stability of around 5 °C. However, in duplexes where two incorporations of TT were performed in the opposite strands with exactly two interspacing unmodified base-pairs in what is defined as a (−3) zipper, a relative increase in stability was seen corresponding to a compensation of 6–7 °C. This indicates a strong thymine–thymine contact in the minor groove and modelling suggested this to be a stacking interaction.5 The interaction was lost by the extension of the methylene linkers to ethylene linkers, i.e. by the introduction of TetT (Fig. 1).6 On the other hand, a thermal compensation as high as 8–9 °C corresponding to a duplex with nearly the same stability as the unmodified duplex was found when one of the two thymines in the (−3) zipper was replaced by a phenyl group by the introduction of nucleotide TPh (Fig. 1).8 A range of double-functionalized nucleotides where the additional thymine of TT has been replaced by substituted or unsubstituted triazoles have also been investigated (Fig. 1, TtPh/TtU/TtT and T4t/T1t, respectively).8,9 However, no zipper interactions with similar specificity were found all though TtU demonstrated a strong tendency to form binding to the floor of the minor groove.8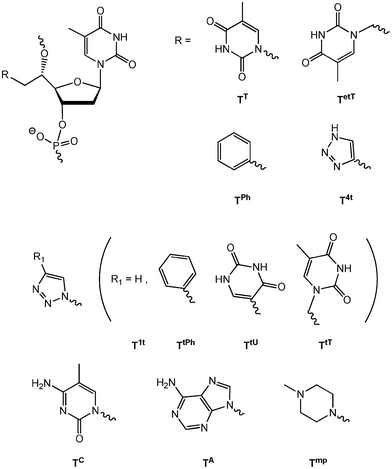 | ||
| Fig. 1 5′(S)-C-functionalised and double-headed nucleotides. | ||
Herein, we investigate the replacement of the additional thymine of nucleotide TT with the other nucleobases cytosine and adenine (nucleotide monomers TC and TA), in order to explore the possibility of various base–base interactions in the minor groove and to compare with the strong thymine-thymine and thymine-phenyl stacking interactions previously found in the (−3) zipper constructs. Furthermore, we decided to explore also a hydrophobic and basic moiety at the 5′-position, envisioning the possibility of π-cation interaction with the phenyl of TPh as well as charge neutralization of the phosphates. A 4-methylpiperazine group, which has been formerly studied in the 4′-position of oligonucleotides through a similar methylene linker,14 was chosen giving monomer Tmp when incorporated into oligonucleotides. Finally, we envisioned that the base–base interactions in the minor groove, especially between two thymines of TT could be used for UV-induced cross-linkages as T–T cross-linking is known as typical photolessions in DNA.15
Results and discussion
Chemical synthesis
The preparation of the three new double-headed nucleotides, TC, TA and Tmp (Fig. 1) is based on the ring opening of the epoxide 1 using the additional base as the nucleophile (Scheme 1). Epoxide 1 was obtained in five steps from thymidine16 and has been similarly used also in the preparation of TT.5 The reaction of 6N-benzoyladenine, 4N-benzoylcytosine and 4N-methylpiperazine with 1 afforded the double-headed nucleosides 2, 7 and 11, respectively, in moderate to good reaction yields. However, the use of NaHMDS as a strong base to deprotonate 6N-benzoyladenine gave rise to a TMS-ether in compound 2, and removal of the TMS group was subsequently performed using citric acid in methanol17 to give 3. A 2D NMR spectrum (HMBC) of 3 was used to confirm the N9-alkylation of the adenine, as a 3JHC coupling could be seen between the 6′-methylene protons and both C8 and C4 of the adenine. The pixyl group has been preferred over the more standard DMT-group for 5′-OH-protection of all our 5′-modified nucleoside analogues (Fig. 1),5,6,8 as it is more conveniently attached to sterically hindered secondary alcohols and works with similar efficiency in the automated solid phase DNA-synthesis.18 However, pixylation of the 5′-hydroxy group of 3 proved very problematic and several conditions were tested. It was found imperative to activate the pixyl chloride prior to use following a literature protocol.19 Among several attempted solvent systems, only a mixture of dioxane and pyridine proved successful giving the desired O5′-pixylated compound 4 in 53% yield. Deprotection of the 3′-hydroxy group to give 5, and subsequent phosphitylation gave the desired phosphoramidite 6 in 11% overall yield from epoxide 1. Phosphoramidite 6 was then used as a building block for oligonucleotide synthesis.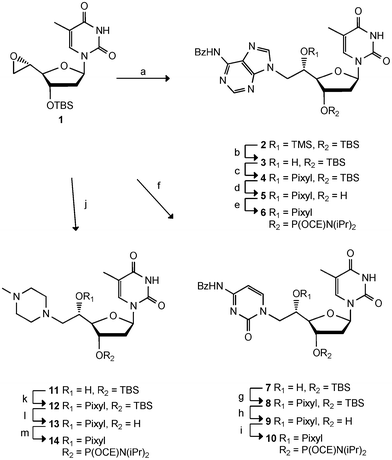 | ||
| Scheme 1 Reagents and conditions: a) 6N-benzoyl-adenine, NaHMDS, THF, 71%, b) citric acid, MeOH, 64%, c) PixylCl, pyridine, dioxane, 53%, d) TBAF, THF, 70%, e) NC(CH2)2OP(Cl)N(iPr)2, DIPEA, DCE, 63%, f) 4N-benzoyl-cytosine, NaHMDS, THF, 43%, g) PixylCl, pyridine, 53%, h) TBAF, THF, 63%, i) NC(CH2)2OP(Cl)N(iPr)2, DIPEA, DCE, 47%, j) 4N-methylpiperazine, THF, 76%, k) PixylCl, pyridine, 60%, l) TBAF, THF, 80%, m) NC(CH2)2OP(Cl)N(iPr)2, DIPEA, DCE, 96%. TBS = tert-butyldimethylsilyl. Pixyl = 9-phenylxanthen-9-yl. CE = cyanoethyl. | ||
Following a similar strategy, ring opening of 1 with 4N-benzoylcytosine and NaHMDS gave compound 7 in 43% yield as the only compound. Again a 2D NMR (HMBC) confirmed the desired configuration of the product as the H6′ coupled with C2 and C6 of the cytosine. Protection of the 5′-hydroxy group was achieved using reactivated pixyl chloride and pyridine, giving compound 8 in 53% yield. Subsequent cleavage of the TBS group to give 9 and phosphitylation gave phosphoramidite 10 in 7% overall yield from epoxide 1. The last building block was synthesised via ring opening of epoxide 1 with 4N-methylpiperazine to give 11 in 76% yield. Pixylation was accomplished using reactivated pixyl chloride in pyridine giving 12 in 60% yield. The TBS group was removed giving compound 13 in 80% yield, and finally, phosphitylation gave the desired phosphoramidite 14 in 96% yield (35% overall yield from 1).
Hybridisation studies
With the three phosphoramidites 6, 10, and 14 in hand, incorporations into DNA sequences were accomplished allowing for the evaluation of the effects of the 5′-modified nucleotides on duplex stability. Incorporation into DNA sequences was accomplished using standard automated solid phase DNA-synthesis with pyridinium chloride (pyrHCl)7,20,21 as activator and prolonged coupling time (20 min) for the modified monomers. Subsequently, standard acidic treatment after coupling also removed the pixyl group. Taking the sterical demands of the 5′-modified monomers into account, the coupling time for the following unmodified phosphoramidite was also prolonged (20 min). This allowed incorporation of monomers TC, TA, and Tmp and the following nucleotides in satisfactory >90% yield (over two coupling steps). The constitution and purity of the oligonucleotides were verified by MALDI-MS and RP-HPLC, respectively. Evaluation was performed by hybridization studies, and the synthesised oligonucleotides were mixed to form duplexes, which are shown in Table 1 in connection to their thermal stability data. The monomers TC, TA, and Tmp were examined for possible minor groove base–base interaction in a DNA zipper motif using the same design as described for previous studies. The results are shown in Table 1 as well as the corresponding results for monomer TT for direct comparison.5,8| ΔTm/°Ca [ΔΔTm]/°Cb | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Zipper | ON | Duplex | X = | TT | TC | TA | Tmp |
| a Differences in melting temperatures as compared to the unmodified duplex. ΔTm = Tm(x:y) − Tm(T1:T2), Tm(T1:T2)= 46.2 °C. Melting temperatures were obtained from the maxima of the first derivatives of the melting curves (A260vs. temperature) recorded in a medium salt buffer (Na2HPO4 (2.5 mM), NaH2PO4 (5 mM), NaCl (100 mM), EDTA (0.1 mM), pH 7.0) using 1.0 μM concentrations of each strand. b Differences in melting temperatures as compared to singly modified duplexes; ΔΔTm = ΔTm(x:y) − (ΔTm(x:T2) + ΔTm(T1:y)). c Data taken from ref. 5. | ||||||||
| 1 | T1 | 5′-d(CGC ATA TTC GC) | −5.4c | −4.2 | −5.9 | −1.0 | ||
| X1 | 3′-d(GCG XAT AAG CG) | |||||||
| 2 | T1 | 5′-d(CGC ATA TTC GC) | −4.7c | −5.6 | −5.9 | −0.5 | ||
| X2 | 3′-d(GCG TAX AAG CG) | |||||||
| 3 | T1 | 5′-d(CGC ATA TTC GC) | −10.9c | −10.4 | −14.5 | −1.5 | ||
| X3 | 3′-d(GCG XAX AAG CG) | |||||||
| 4 | X4 | 5′-d(CGC ATA TXC GC) | −4.4c | −3.2 | −6.0 | −1.5 | ||
| T2 | 3′-d(GCG TAT AAG CG) | |||||||
| 5 | X5 | 5′-d(CGC ATA XTC GC) | −4.9c | −5.0 | −5.6 | −2.0 | ||
| T2 | 3′-d(GCG TAT AAG CG) | |||||||
| 6 | X6 | 5′-d(CGC AXA XTC GC) | −12.0c | −11.0 | −15.0 | −3.2 | ||
| T2 | 3′-d(GCG TAT AAG CG) | |||||||
| 7 | (−1) | X5 | 5′-d(CGC ATA XTC GC) | −10.4 [−0.8]c | −9.4 [+1.2] | −12.1 [−0.6] | −3.0 [−0.5] | |
| X2 | 3′-d(GCG TAX AAG CG) | |||||||
| 8 | (−2) | X4 | 5′-d(CGC ATA TXC GC) | −10.0 [−0.9]c | −8.5 [+0.3] | −9.5 [+2.4] | −1.9 [+0.1] | |
| X2 | 3′-d(GCG TAX AAG CG) | |||||||
| 9 | (−3) | X5 | 5′-d(CGC ATA XTC GC) | −3.8 [+6.5]c | −7.3 [+1.9] | −7.9 [+3.6] | −4.0 [−1.0] | |
| X1 | 3′-d(GCG XAT AAG CG) | |||||||
| 10 | (−4) | X4 | 5′-d(CGC ATA TXC GC) | −9.8 [±0.0]c | −8.4 [−1.0] | −14.6 [−2.7] | −3.1 [−0.6] | |
| X1 | 3′-d(GCG XAT AAG CG) | |||||||
| 11 | (−2)/(−4) | X4 | 5′-d(CGC ATA TXC GC) | −17.0 [−1.7]c | −14.2 [−0.6] | −20.1 [+0.4] | −3.2 [−0.2] | |
| X3 | 3′-d(GCG XAXAAG CG) | |||||||
| 12 | (−1)/(−3) | X5 | 5′-d(CGC ATA XTC GC) | −9.0 [+6.8]c | −12.0 [+3.4] | −15.9 [+4.2] | −4.1 [−0.6] | |
| X3 | 3′-d(GCG XAX AAG CG) | |||||||
| 13 | (−3)/(−1) | X6 | 5′-d(CGC AXA XTC GC) | −10.4 [+7.0]c | −14.3 [+0.9] | −15.9 [+5.0] | −5.1 [−0.9] | |
| X1 | 3′-d(GCG XAT AAG CG) | |||||||
| 14 | (−1)/(+1) | X6 | 5′-d(CGC AXA XTC GC) | −18.2 [−1.5]c | −18.6 [−2.0] | −19.0 [+1.9] | −3.1 [+0.6] | |
| X2 | 3′-d(GCG TAX AAG CG) | |||||||
| 15 | (−1)/(−3)/(+1)/(−1) | X6 | 5′-d(CGC AXA XTC GC) | −16.8 [+6.1]c | −18.5 [+2.9] | −22.2 [+7.3] | −5.5 [−0.8] | |
| X3 | 3′-d(GCG XAX AAG CG) | |||||||
In all the duplexes studied, the introduction of one or more double-headed nucleotides led to various degrees of destabilization of the duplexes. Hence, Table 1 presents a range of negative ΔTm-values corresponding to the difference in Tm for each duplex as compared to the unmodified duplex (Tm = 46.2 °C). Single incorporations of monomer TC gave decreases in duplex stability ranging from 3.2 to 5.6 °C (entries 1, 2, 4 and 5), indicating that the monomer is tolerated in the duplex but that no favourable interactions are occurring. Two incorporations in the same strand (entries 3 and 6) gave similar decreases (−10.4 and −11.0 °C, respectively, corresponding to −5.2 and −5.5 °C for each modification). Overall, these results correspond very well to the hybridization data for monomer TT. The decrease in thermal stability for incorporation of monomer TA is slightly larger in all cases (ΔTm = −5.6 to −7.5 °C, the latter for each of two incorporations, entry 6) than for the additional pyrimidines, all though the results indicate that also monomer TA is accepted in the duplex. Possibly, the different sterical demands for the pyrimidines and the purine could explain why incorporation of the larger purine is slightly less favorable. Monomer Tmp, on the other hand, shows only slight destabilization of the duplex (ΔTm = −0.5 to −2.0 °C, covering all entries 1–6) for incorporation of each monomer. These results might be due to the increased flexibility of the aliphatic 4-methylpiperazine compared to the aromatic moieties, and to the possible charge neutralization of the phosphate groups. Previously, a range of different moieties have been introduced into oligonucleotides based on the 5′-C-position,22,23 and in general these 5′-C-branched nucleotides show a small destabilization of the duplexes with approximately 1 °C per modification,24–27 comparable to the observed destabilization caused by monomer Tmp. However, a 5′-C-aminoalkylated nucleotide has shown a small stabilization,28 and significant stabilization has been obtained with 5′-C-modified nucleotides in connection to conformationally restricted systems.29–32 In contrary to Tmp, the introduction of 4′-C-(N-methylpiperazino)methyl substituted nucleosides into DNA also increases the thermal stability of a DNA duplex.14
Hereafter, various zipper motifs were studied, and the results are depicted in Table 1 (entries 7–15). The zipper motifs position the additional bases in each strand with distances of 0–3 interspacing base-pairs (corresponding to (−1) to (−4) zipper motifs). As previously described, the (−3) zipper motif is ideal for base–base interaction in the minor groove when monomer TT is incorporated.5 This is evident, as a relative increase in Tm (ΔΔTm of 6–7 °C) is observed only when (−3) zipper contacts are possible (Table 1, entries 9, 12, 13 and 15). (These ΔΔTm-values are obtained by comparing the actual ΔTm's to expected ΔTm's, the latter based on the assumption that the two modifications are independent and the ΔTm's therefore additive.) In all other zippers with TT (entries 7, 8, 10, 11 and 14) the decreases in Tm are indeed additive, corresponding to the number of incorporations (i.e. ΔTm ∼−4–6 °C per modification and thereby very small ΔΔTm's).
For monomer TC the picture is less clear. Only small relative stabilizations (ΔΔTm's) are observed for (−3), and to some extent (−1) zippers, and the very specific contact seen in the (−3) zipper motif with monomer TT, is not observed with monomer TC. Monomer TA gives rise to some relative stabilizations in the (−3) and (−2) zipper motifs. Nevertheless, neither the interactions between two adenines are as specific as between the two additional thymines seen with monomer TT. Furthermore, all duplexes with monomer TA demonstrate lower Tm values than the corresponding duplexes with monomer TT.
The last monomer in the study, monomer Tmp, does not furnish any relative stabilization of the duplex by incorporation of two to four monomers in any zipper motif. The degree of destabilization of the duplex is significantly smaller compared with the three monomers TT, TC, and TA, and remarkably constant with ΔTm's for each modification of between −1 to −2 °C. Leumann and coworkers have previously investigated 5′(S)-C-alkyl modified nucleotides in various possible hydrophobic zipper motifs and observed similar results.26 In all cases the decrease in thermal duplex stability was remarkably unchanged, indicating that the main effect is distortion of the hydration pattern rather than any kind of intersubstituent communication.
Finally, the series of 5′ double-headed nucleosides was also investigated in mixed (−3) zipper constructs. Table 2 shows all possible combinations of the five building blocks TT, TC, TA, Tmp, and TPh. As described before, a significant relative stabilization is observed when monomer TPh and TT are incorporated in the (−3) zipper motif (ΔΔTm = +8.5 °C).8 However, none of the new results for the mixed (−3) zipper motifs can match this interaction, all though some of the combinations give relative stabilizations of the duplexes. The most significant are seen for the mixed zipper motifs containing monomers TT/TC and TPh/TC with ΔΔTm's of 3.7–5.8 °C. Presumably, these compensations are due to similar stacking interactions between the aromatic moieties, as it was seen for zipper motifs containing monomers TT/TT, and TPh/TT, however with slightly smaller stability. The TPh/TA mixed (−3) zipper constructs also displays some relative stabilization (ΔΔTm = +3.9 and 4.8 °C) at the same level as the TA/TA duplex. The combinations of TA and TT were theoretically interesting due to the potential of base pairing. However the duplexes are significantly destabilized and indicate that all observed interactions are stacking interactions and in this case less efficient than between the thymine and the phenyl group. This is similar to other studies of double-headed nucleotides, by which the possibility of T-A interactions were explored with additional bases placed on either the 2′-C-position, the 4′-C-position of on 2′-amino LNA, however, with no indication of basepairing.9,12,13 Finally, the present study also demonstrates that Tmp does not have any noteworthy effect on the mixed zipper motifs as no relative stabilizations were observed. Hence, the envisioned π-cation interaction was not indicated from these results.
5′-CGC ATA YTC GC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3′-GCG XAT AAG CG :3′-GCG XAT AAG CG |
||||||
|---|---|---|---|---|---|---|
| ΔTm/°Ca [ΔΔTm]/°Cb | ||||||
| X/Y | T | TT | TC | TA | Tmp | TPh |
| a See Table 1. The values in bold correspond to entry 9, Table 1. b See Table 1. The values in bold correspond to entry 9, Table 1. c Data taken from ref. 8. | ||||||
| T | 0 | −5.4 | −5.0 | −5.6 | −2.0 | −4.5 |
| TT | −5.3 | −3.8 [+6.5]c | −4.5 [+5.8] | −6.9 [+4.3] | −7.0 [+0.2] | −1.2 [+8.6]c |
| TC | −4.2 | −5.9 [+3.7] | −7.3 [+1.9] | −10.3 [−0.2] | −5.9 [+0.2] | −3.5[+5.2] |
| TA | −5.9 | −7.6 [+3.7] | −10.0 [+0.6] | −7.9 [+3.6] | −9.1 [−1.2] | −6.5 [+3.9] |
| Tmp | −1.0 | −6.1 [+0.3] | −5.8 [−1.7] | −8.1 [−1.2] | −4.0 [−1.0] | −5.6 [−0.2] |
| TPh | −3.2 | −0.1 [+8.5]c | −2.4[+5.8] | −4.0 [+4.8] | −5.4 [−0.3] | −6.3 [+1.4]c |
UV-induced formation of a T–T cross-link in the minor groove
Molecular modeling studies have indicated the reason for the observed specific interaction between the two TT monomers in the (−3) zipper duplex to be a stacking between the two additional thymine moieties in the minor groove.5,8 To shed further light on the interaction we decided to use this specific construct in a UV experiment. Thus, it has been demonstrated that adjacent thymines in a DNA duplex can form dimer complexes when exposed to UV irradiation in the range of 200–280 nm.15 Likewise, we envisioned that the two exohelical thymine moieties could potentially form a T–T dimer upon UV irradiation. The formation of such a product would demonstrate the close proximity of the two additional thymines. It is known that UV irradiation of cells induces the formation of DNA lesions in the form of covalent bonds between pyrimidines. Various lesions are formed, though the main lesion formed on irradiation of cells is the cis–syn cyclobutane pyrimidine dimer (CPD), which is formed between adjacent pyrimidine bases.15,33 The T–T dimer is formed in a photochemically induced [2π + 2π] cycloaddition reaction.15 Studies have shown that photodimers significantly destabilize B-type DNA but only decreases the stability of an A-type duplex to a small extent.34 CD-experiments of the (−3) TT/TT zipper construct previously described by us have shown that the duplex exhibit a slightly altered B-type DNA duplex.5 In the applied sequence context (Table 1, entry 9) there are in principle three possible sites for formation of photodimers between pyrimidines, namely the two additional thymines on monomer TT across the minor groove, and between two adjacent thymines, as well as between a thymine and a cytosine in one of the strands, i.e. the X5 strand (Fig. 2b).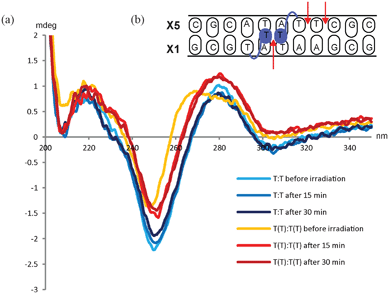 | ||
| Fig. 2 (a) CD-spectra of unmodified DNA duplex before and after irradiation at 254 nm (blue lines) and of the (−3) TT/TT zipper duplex before and after irradiation (orange and red lines). (b): Schematic illustration of the (−3) TT/TT zipper duplex. The arrows indicate the possible sites for dimer formation. | ||
In the experiment the sample was irradiated by a UV lamp (254 nm, 5 W) for 15 min. Recording CD-spectra of the (−3) TT/TT zipper duplex and the unmodified reference duplex allowed direct comparison of the two duplexes. The experiment demonstrated that irradiating the (−3) TT/TT zipper duplex for 15 min changed the overall duplex structure and that irradiating for additional 15 min did not give any further changes. When irradiating the unmodified duplex no changes in the overall structure were observed even after 30 min (see Fig. 2).
The tertiary structure of duplexes containing the cis–syn CPD has previously been analyzed by NMR spectroscopy, and only small distortions from the canonical B-type were reported.35,36 Therefore, the fact that no changes in the CD-spectra are observed for the unmodified duplex does not exclude an intrastrand dimer formation in the X5-strand (Fig. 2b). The CD-spectra of the (−3) TT/TT zipper duplex, on the other hand, demonstrated that irradiation changes the overall duplex structure. To verify the formation of the T–T dimer, the masses of the macromolecules were determined. The zipper duplex before irradiation displayed two mass peaks (m/z 3432.5 and 3522.1), corresponding to the two complementary strands (calcd. 3430.4 and 3520.7, respectively). However, after irradiating the duplex, three mass peaks were detected, namely, the two masses corresponding to the two strands (m/z 3427.1 and 3517.1) as well as a peak being equivalent to the covalently linked duplex (m/z 6943.4, calcd. 6949.1). Ion chromatography HPLC was used to determine the ratio between the covalently linked duplex containing the T–T dimer and the zipper duplex. To distinguish the species, the HPLC was run at 60 °C thereby ensuring that non-covalently bonded strands were separated during analysis. A peak eluting at 18.1 min arises from the two single stranded sequences. Only one peak is observed as the two strands have equal amount of negative charges. The covalently linked duplex containing the T–T dimer, appears after 19.4 min as a result of the increased amount of negative charges (see SI). The ratio between the two species after 15 min irradiation is approximately 2:3 of T–T dimer vs. non-covalently linked single stranded DNA. This is in agreement with previous experiments by Carell and coworkers, where an up to 40% conversion to dimers were demonstrated, when various hairpin structures were irradiated.37
This experiment demonstrates that the two additional thymines are prone to formation of the T–T dimer, and from the CD-spectra it seems that the reaction reaches equilibrium after 15 min, though more time points would be needed to elucidate this aspect. To clarify whether the dimer formation also changed the stability of the duplex, thermal denaturation studies were performed (Fig. 3). To allow for direct comparison, the measurements were performed on the same sample before and after irradiation. The unmodified duplex display very similar denaturation curves before and after irradiation giving Tm ∼46 °C in both cases indicating no major differences. Together with the unchanged CD-spectrum and the HPLC-results, this indicated that no significant intrastrand dimerization has taken place. On the other hand, the (−3) TT/TT zipper duplex exhibit a significant change. The hyperchromicity, being the increase in absorbance when a duplex is denatured to the single stranded structures, is significantly decreased for the (−3) TT/TT zipper duplex after irradiation, and the thermal stability for the (−3) TT/TT zipper duplex is decreased with ∼2.5 °C (Fig. 3). However, due to the 40% cross-linking these measurements were actually made on a mixture of the T–T dimer and the (−3) zipper duplex, and the melting transition does not necessarily represent a two state process. Separation of the two species is not possible due to the photoinduced reversibility of the cycloaddition reaction.37
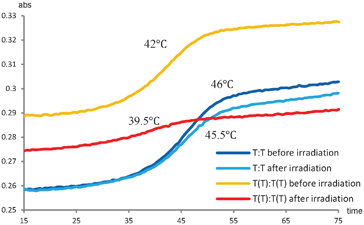 | ||
| Fig. 3 T m measurements before and after UV irradiation (254 nm) of the unmodified duplex (blue lines) and the (−3) TT/TT zipper duplex (orange and red lines). | ||
Discussion
In this study, the synthesis of three new analogues of double-headed nucleosides has been presented. The synthetic strategy was the same for the three analogues, and the introduction of 6N-benzoyladenine, 4N-benzoylcytosine and 4N-methylpiperazine to the 5′-C-epoxide 1 was achieved in moderate to good yields. With reactivated pixyl chloride, also the phosphoramidite building blocks were obtained in good yields. The three monomers TA, TC, and Tmp were hereby introduced in oligonucleotides and evaluation of the duplex stabilities revealed that the monomers all gave rise to destabilization of the duplexes. Monomer Tmp was found to be the best accepted in the duplex (also counting all other analogues shown in Fig. 1)‡ only causing small decrease in the thermal stability. This indicates that introducing charged hydrophobic moieties are preferred over heteroaromatic moieties, probably due to the increased flexibility of the system as well as some degree of charge neutralization of the backbone. However, monomer Tmp is not able to make significant favorable zipper interactions across the minor groove in line with similar hydrophobic zippers.26 Incorporation of monomer TC and TA destabilized the duplexes to a larger extent than monomer Tmp indicating some distortions in the minor groove. When evaluated in the zipper study none of the monomers were found to indicate a specific interaction in minor groove as seen for monomer TT in the (−3) zipper. In a mixed (−3) zipper study with sequences each containing one of the monomers TT, TC, TA, Tmp, and TPh, none of the new monomers were found to give better relative stabilization of the duplex than the interaction seen between phenyl and thymine in the TT/TPh zipper. However, some interactions were observed between the additional nucleobases thymine and cytosine, and between phenyl and cytosine or adenine in duplexes containing these monomer combinations, i.e.TT/TC, TPh/TC, TC/TPh and TPh/TA. In all cases, however, this indicates rather weak stacking interactions and in no cases the presence of hydrogen-bonding interactions. It might have been expected that the adenine of TA due to its larger surface should lead to stronger stacking interactions than the pyrimidines. However, any such effect seems lost due to the more unfavourable interactions by the adenine moieties in the minor groove. No indication of any π-cation interactions between Tmp and any of the aromatic moieties was detected.The easy UV-induced formation of a T–T dimer proves the close proximity of the two additional thymines in TT/TT, due to π–π-stacking. Overall, this is in line with the Tm analysis demonstrating that the interaction between the two thymines is the most selective in the series only surpassed by the mixed TT/TPh interaction, and with a molecular modeling analysis that demonstrated this to be a matter of proximity in the π–π-stacking.8
Conclusion
In this study, the series of 5′-C-modified thymidines has been extended with three new members, the two being other base-combinations of the original TT-monomer. The study of various zipper motifs has shown that the base–base interactions in the minor groove as organized in a (−3) zipper interaction is the only specific interaction and only based on stacking interactions. This is proved by the fact that no improved interactions were detected by combining different nucleobases and that two thymines in this zipper is preorganized for a UV-induced dimer formation. This knowledge adds to the understanding of the DNA duplex as a scaffold for nanoconstructs and enlighten the 5′-C-position for nucleic acid decoration.Experimental details
General
Reactions were performed under an atmosphere of nitrogen when anhydrous solvents were used. Microwave heated reactions were performed on an EmrysTM Creator. Column chromatography was carried out on glass columns using silica gel 60 (0.040–0.063 mm). Flash column chromatography was performed on a Biotage SP4TM Purification System. NMR spectra were recorded at 400 MHz for 1H NMR, 101 MHz for 13C NMR and 162 MHz for 31P NMR. The δ values are reported in ppm relative to tetramethylsilane as internal standard (for 1H and 13C NMR) and relative to 85% H3PO4 as external standard (for 31P NMR). Assignments of NMR spectra are based on 2D correlation spectroscopy (COSY, HSQC, and HMBC spectra) and follow standard carbohydrate and nucleoside style; i.e. the carbon atom next to a nucleobase is assigned C1′, etc. HR ESI mass spectra were recorded in positive-ion mode.5′(S)-C-(N6-Benzoyladenine-1-yl)methyl-3′-O-(tert-butyldimethylsilyl)-5′-O-(trimethylsilyl)thymidine (2)
A suspension of N6-benzoyladenine (950 mg, 3.95 mmol) in anhydrous THF (7 mL) was stirred at 55 °C and added a 1 M solution of NaHMDS in THF (3.95 mL). The mixture was stirred for 3 h at 55 °C, and then added to a stirred solution of epoxide 1 (0.973 mg, 2.64 mmol) in THF (10 mL). The reaction mixture was stirred at 55 °C for 44 h and then diluted with CH2Cl2 (50 mL) and washed with a saturated aqueous solution of NaHCO3 (50 mL). The aqueous phase was extracted with CH2Cl2 (3 × 50 mL) and the combined organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–5% MeOH in CHCl3) to give the product 1 (1.16 g, 71%) as a white foam. Rf 0.6 (EtOAc); 1H NMR (400 MHz, DMSO) δ 11.36 (s, 1H, NH(T)), 11.13 (s, 1H, NH(A)), 8.74 (s, 1H, H2(A)), 8.48 (s, 1H, H8(A)), 8.04 (d, J = 7.4 Hz, 2H, Bz), 7.78 (d, J = 1.0 Hz, 1H, H6(T)), 7.63 (t, J = 7.4 Hz, 1H, Bz), 7.54 (t, J = 7.4 Hz, 2H, Bz), 6.27 (t, J = 7.1 Hz, 1H, H1′), 4.52 (m, 1H, H3′), 4.48–4.41 (m, 2H, H6′), 4.37 (m, 1H, H5′), 3.89 (t, J = 2.0 Hz, 1H, H4′), 2.22–2.05 (m, 2H, H2′), 1.83 (d, J = 1.0 Hz, 3H, CH3), 0.87 (s, 9H, C(CH3)3), 0.10 (s, 6H, Si(CH3)2), −0.19 (s, 9H, Si(CH3)3); 13C NMR (101 MHz, DMSO) δ 165.79 (Bz), 163.97 (C4(T)), 153.21 (C4(A)), 151.76 (C2(A)), 150.70 (C2(T)), 150.51 (C6(A)), 145.45 (C8(A)), 135.92 (C6(T)), 133.75, 132.65, 128.72 (Bz), 125.63 (C5(A)), 110.13 (C5(T)), 87.65 (C4′), 84.31 (C1′), 73.91 (C3′), 71.08 (C5′), 46.60 (C6′), 40.47 (C2′), 25.93 (C(CH3)3), 17.95 (C(CH3)3), 12.58 (CH3(T)), 0.39 (Si(CH3)3), −4.35, −4.56 (Si(CH3)2); HR-ESI MS m/z 702.2832 ([M + Na]+, C32H45N7O6Si2Na+ calc. 702.2862).5′(S)-C-(N6-Benzoyladenine-1-yl)methyl-3′-O-(tert-butyldimethylsilyl)thymidine (3)
A solution of compound 2 (1.16 g, 1.7 mmol) in anhydrous MeOH (100 mL) was added citric acid (1.5 g, 7.8 mmol) and the mixture was stirred at room temperature for 24 h. The reaction mixture was cooled to 0 °C and a saturated aqueous solution of NaHCO3 (70 mL) was added slowly. The mixture was extracted with CHCl3 (3 × 100 mL) and the combined organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–10% MeOH in CHCl3) to give the product 3 (661 mg, 64%) as a white foam. Rf 0.3 (10% MeOH in CHCl3); 1H NMR (400 MHz, DMSO) δ 11.34 (s, 1H, NH(T)), 11.13 (s, 1H, NH(A)), 8.72 (s, 1H, H2(A)), 8.44 (s, 1H, H8(A)), 8.05 (d, J = 7.5 Hz, 2H, Bz), 7.93 (s, 1H, H6(T)), 7.65 (t, J = 7.5 Hz, 1H, Bz), 7.56 (t, J = 7.5 Hz, 2H, Bz), 6.26 (dd, J = 6.5, 7.5 Hz, 1H, H1′), 5.70 (d, J = 4.3 Hz, 1H, OH), 4.52 (m, 1H, H3′), 4.47–4.31 (m, 2H, H6′), 4.27 (m, 1H, H5′), 3.84 (s, 1H, H4′), 2.20 (m, 1H, H2′), 2.09 (ddd, J = 13.1, 5.9, 2.4 Hz, 1H, H2′), 1.83 (s, 3H, CH3), 0.87 (s, 9H, C(CH3)3), 0.02 (s, 6H, Si(CH3)2); 13C NMR (101 MHz, DMSO) δ 165.50 (Bz), 163.70 (C4(T)), 152.80 (C4(A)), 151.24 (C2(A)), 150.46 (C2(T)), 149.97 (C6(A)), 145.26 (C8(A)), 136.13 (C6(T)), 133.47, 132.32, 128.71, 128.41 (Bz), 125.44 (C5(A)), 109.64 (C5(T)), 87.19 (C4′), 83.99 (C1′), 73.20 (C3′), 67.85 (C5′), 46.59 (C6′), 40.14 (C2′), 25.64 (C(CH3)3), 17.64 (C(CH3)3), 12.39 (CH3(T)), −4.77, −4.90 (Si(CH3)2); HR-ESI MS m/z 630.2483 ([M + Na]+, C29H37N7O6SiNa+ calc. 630.2467).5′(S)-C-(N6-Benzoyladenine-1-yl)methyl-3′-O-(tert-butyldimethylsilyl)-5′-O-pixylthymidine (4)
Compound 3 (349 mg, 0.53 mmol) was co-evaporated with anhydrous pyridine (5 mL) and dissolved in a mixture of anhydrous 1,4-dioxane (18 mL) and anhydrous pyridine (3.5 mL). Pixyl chloride (350 mg, 1.19 mmol) was added and the reaction mixture was stirred in the dark at room temperature for 20 h, and then added a saturated aqueous solution of NaHCO3 (20 mL). The mixture was extracted with CH2Cl2 (3 × 50 mL) and the combined organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 0–5% MeOH in CHCl3) to give the product 4 (244 mg, 53%) as a white foam as well as unreacted starting material 3 (134 mg, 39%). Rf 0.4 (10% MeOH in CHCl3); 1H NMR (400 MHz, DMSO) δ 11.38 (s, 1H, NH(T)), 11.07 (s, 1H, NH(A)), 8.50 (s, 1H, H2(A)), 8.28 (s, 1H, H8(A)), 8.05 (d, J = 7.4 Hz, 2H, Bz), 7.64 (t, J = 7.4, 1H, Bz), 7.58 (d, J = 0.7 Hz, 1H, H6(T)), 7.55 (t, J = 7.4 Hz, 2H, Bz), 7.47–7.25 (m, 7H, pixyl), 7.12–6.90 (m, 4H, pixyl), 6.53 (t, J = 7.6 Hz, 1H, pixyl), 6.26 (d, J = 7.6 Hz, 1H, pixyl), 5.81 (dd, J = 9.5, 5.2 Hz, 1H, H1′), 4.24 (dd, J = 4.6, 14.0 Hz, 1H, H6′), 4.19 (m, 1H, H3′), 4.07 (dd, J = 8.4, 13.7 Hz, H6′), 3.96 (m, 1H, H5′), 2.95 (m, 1H, H4′), 2.15 (m, 1H, H2′), 1.87 (dd, J = 13.0, 5.2 Hz, 1H, H2′), 1.79 (d, J = 0.7 Hz, 3H, CH3), 0.74 (s, 9H, C(CH3)3), 0.02 (s, 6H, Si(CH3)2); 13C NMR (101 MHz, DMSO) δ 165.44 (Bz), 163.53 (C4(T)), 151.90, 151.05, 150.89, 150.26 (C2(T), C2(A), C6(A), pixyl), 146.91 (pixyl), 144.80 (C8(A), Bz), 135.63 (C6(T), 133.38 (Bz), 132.24, 131.00, 129.11 (pixyl), 128.33 (Bz), 127.56, 127.26, 126.95 (pixyl), 125.60 (C5(A)), 123.44, 123.0, 121.45, 116.33 (pixyl), 109.60 (C5(T)), 85.75 (C4′), 83.72 (C1′), 76.89 (pixyl), 71.24 (C3′), 69.98 (C5′), 44.12 (C6′), 40.08 (C2′), 25.41 (C(CH3)3), 17.27 (C(CH3)3), 12.06 (CH3), −4.84, −5.07 (Si(CH3)2); HR-ESI MS m/z 886.3315 ([M + Na]+, C48H49N7O7SiNa+ calc. 886.3355).5′(S)-C-(N6-Benzoyladenine-1-yl)methyl-5′-O-pixylthymidine (5)
To a solution of compound 4 (328 mg, 0.38 mmol) in anhydrous THF (7 mL) was added a 1 M solution of TBAF in THF (380 μL) and the mixture was stirred at room temperature for 20 h. A saturated aqueous solution of NaHCO3 (15 mL) was added and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 0–7% MeOH in CHCl3) to give the product 5 (215 mg, 70% containing 7% TBA) as a white foam. Rf 0.2 (10% MeOH in CHCl3); 1H NMR (400 MHz, DMSO) δ 11.37 (s, 1H, NH(T)), 11.10 (s, 1H, NH(A)), 8.53 (s, 1H, H2(A)), 8.32 (s, 1H, H8(A)), 8.06 (d, J = 7.5 Hz, 2H, Bz), 7.64 (t, J = 7.5 Hz, 1H, Bz), 7.58–7.50 (m, 3H, H6, Bz), 7.44–7.22 (m, 7H, pixyl), 7.14–6.99 (m, 4H, pixyl), 6.50 (t, J = 7.5 Hz, 1H, pixyl), 6.18 (d, J = 7.5 Hz, 1H, pixyl), 5.84 (dd, J = 8.7, 5.6 Hz, 1H, H1′), 5.09 (d, J = 4.7 Hz, 1H, OH), 4.26 (dd, J = 14.2, 4.5 Hz, 1H, H6′), 4.16–4.05 (m, 2H, H3′, H6′), 3.91 (m, 1H, H5′), 3.01 (m, 1H, H4′), 2.04 (m, 1H, H2′), 1.92 (dd, J = 12.1, 5.4 Hz, 1H, H2′), 1.78 (d, J = 0.7 Hz, 3H, CH3); 13C NMR (101 MHz, DMSO) δ 165.28 (Bz), 163.57 (C4(T)), 152.10 (C4(A)), 150.83, 150.67, 150.26, 149.75 (C2(T), C2(A), C6(A), pixyl), 147.64 (pixyl), 144.94 (C8(A), Bz), 135.63 (C6(T)), 132.25 (Bz), 131.27, 129.85, 129.15 (pixyl), 128.34 (Bz), 127.59, 127.06, 126.77 (pixyl), 125.62 (C5(A)), 123.45, 123.14, 123.00, 121.60, 116.00, 115.76 (pixyl), 109.53 (C5(T)), 84.99 (C4′), 83.48 (C1′), 76.53 (pixyl), 70.41 (C5′), 69.04 (C3′), 44.03 (C6′), 40.06 (C2′), 12.09 (CH3); HR-ESI MS m/z 772.2467 ([M + Na]+, C42H35N7O7Na+ calc. 772.2490).5′(S)-C-(N6-Benzoyladenine-1-yl)methyl-3′-O-(P-2-cyanoethoxy-N,N-diisopropylaminophosphinyl)-5′-O-pixyl-thymidine (6)
Compound 5 (108 mg, 0.14 mmol) was coevaporated with anhydrous DCE (2 × 4 mL) and redissolved in the same solvent (2 mL). DIPEA (502 μL, 2.9 mmol) and N,N-diisopropylamino-2-cyanoethylchlorophosphite (96 μL, 0.43 mmol) were added and the reaction mixture was stirred at room temperature for 4 h. 99.9% EtOH (3 mL) was added and the mixture was concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 0–5% MeOH in CH2Cl2) to give the product 6 (86 mg, 63%) as a white foam. Rf 0.4 (10% MeOH in CHCl3); 31P NMR (162 MHz, CDCl3) δ 150.38, 150.26; HR-ESI MS m/z 972.3540 ([M + Na]+, C51H52N9O8PNa+ calc. 972.3569).5′(S)-C-(N4-Benzoylcytosin-1-yl)methyl-3′-O-(tert-butyldimethylsilyl) thymidine (7)
To a suspension of N4-benzoylcytosine (2.6 g, 12.2 mmol) in anhydrous THF (15 mL) was added a 1 M solution of NaHMDS in THF (12.2 mL). The mixture was stirred at 55 °C for 1 h. A solution of epoxide 1 (1.52 g, 4.11 mmol) in anhydrous THF (15 mL) was added and the mixture was stirred at 55 °C for 24 h and then concentrated under reduced pressure. The residue was redissolved in CH2Cl2 (50 mL) and washed with a saturated aqueous solution of NaHCO3. The aqueous phase was extracted with CH2Cl2 (3 × 100 mL) and the combined organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–100% EtOAc in hexane) to give the product 7 (1.04 g, 43%) as a white foam as well as unreacted starting material 1 (303 mg, 20%). (Rf 0.5 EtOAc); 1H NMR (400 MHz, DMSO) δ 11.32 (s, 1H, NH(T)), 11.17 (s, 1H, NH(C)), 8.03–7.99 (m, 3H, H6(C), Bz), 7.86 (s, 1H, H6(T)), 7.64 (m, 1H, Bz), 7.50 (m, 2H, Bz), 7.31 (d, J = 7.1 Hz, 1H, H5(C)), 6.22 (dd, J = 7.7, 6.2 Hz, 1H, H1′), 5.67 (d, J = 5.8 Hz, 1H, OH), 4.48 (m, 1H, H3′), 4.08–4.01 (m, 2H, H5′, H6′), 3.81–3.73 (m, 2H, H4′, H6′) 2.19 (m, 1H, H2′), 2.09 (m, 1H, H2′), 1.81 (d, 3H, CH3(T)), 0.87 (s, 9H, (CH3)3), 0.09 (s, 6H, 2CH3). 13C NMR (101 MHz, DMSO) δ 167.08 (Bz), 163.61 (C4(T)), 163.02 (C4(C)), 155.40 (C2(C)), 151.23 (C6(C)), 150.36 (C2(T)), 136.01 (C6(T)), 133.09 (C4), 132.54, 128.79, 128.32, 128.09 (Bz), 109.51 (C5(T)), 95.33 (C5(C)), 87.34 (C4′), 83.83 (C1′), 73.00 (C3′), 66.92 (C5′), 53.25 (C6′), 40.08 (C2′), 25.60 (C(CH3)3), 17.57 (C(CH3)3), 12.33 (CH3), −4.82, −4.93 (SiCH3); HR-ESI MS m/z 606.2332 ([M + Na]+, C44H47N5O6SiNa+ calc. 606.2355).5′(S)-C-(N4-Benzoylcytosin-1-yl)methyl-3′-O-(tert-butyldimethylsilyl)-5′-O-pixylthymidine (8)
Compound 7 (373 mg, 0.64 mmol) was coevaporated with pyridine (2 × 5 mL) and redissolved in the same solvent (6.5 mL). Pixyl chloride (373 mg, 1.28 mmol) was added and the reaction mixture was stirred at room temperature for 48 h and then concentrated under reduced pressure. The residue was redissolved in CH2Cl2 (40 mL) and washed with a saturated aqueous solution of NaHCO3 (5 mL). The aqueous phase was extracted with CH2Cl2 (2 × 10 mL) and the combined organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 20–100% EtOAc in petrol ether) to give the product 8 (284 mg, 53%) as a white foam. Rf 0.7 (EtOAc); 1H NMR (400 MHz, DMSO) δ 11.39 (s, 1H, NH(T)), 11.17 (s, 1H, NH(C)), 8.04 (d, J = 7.4 Hz, 2H, Bz), 7.97 (d, J = 7.2 Hz, 1H, H6(C)), 7.70 (s, 1H, H6(T)), 7.68–7.64 (m, 2H, Bz), 7.57–7.53 (m, 2H, Bz), 7.50–7.23 (m, 10H, pixyl, H5(C)), 7.11 (m, 1H, pixyl), 7.00 (dd, J = 7.8, 1.5 Hz, 1H, pixyl), 6.86 (t, J = 7.3 Hz, 1H, pixyl), 6.71 (d, J = 7.8 Hz, 1H, pixyl), 5.84 (dd, J = 8.4, 6.0 Hz, 1H, H1′), 4.17 (s, 1H, H3′), 3.88 (m, 1H, H5′), 3.80 (d, J = 13.2 Hz, 1H, H6′), 3.55 (dd, J = 13.2, 8.2 Hz, 1H, H6′), 3.10 (s, 1H, H4′), 2.04–1.92 (m, 2H, H2′), 1.85 (s, 3H, CH3), 0.82 (s, 9H, C(CH3)3), 0.05 (s, 6H, Si(CH3)2); 13C NMR (101 MHz, DMSO) δ 167.08 (Bz), 163.61 (C4(T)), 163.02 (C4(C)), 154.63 (C2(C)), 151.14, 150.98 (pixyl, C6(C)), 150.18 (C2(T)), 147.18 (pixyl), 135.42 (C6(T)), 133.06, 132.55 (Bz), 130.91, 130.25, 130.13, 129.86 (pixyl), 128.32 (Bz), 127.59, 127.23, 126.93, 125.45, 123.41, 123.32, 122.88, 121.91, 116.40, 116.29 (pixyl), 109.49 (C5(T), 95.83 (C5(C)), 86.23 (C4′), 83.78 (C1′), 76.76 (pixyl), 71.55 (C3′), 69.77 (C5′), 51.04 (C6′), 40.07 (C2′), 25.44 (C(CH3)3), 17.30 (C(CH3)3), 12.08 (CH3), −4.84, −5.00 (Si(CH3)2); HR-ESI MS m/z 862.3271 ([M + Na]+, C47H49N5O8SiNa+ calc. 862.3243).5′(S)-C-(N4-Benzoylcytosin-1-yl)methyl-5′-O-pixylthymidine (9)
A solution of compound 8 (150 mg, 0.178 mmol) in anhydrous THF (2 mL) was added a 1 M solution of TBAF in THF (178 μL) and stirred at room temperature for 24 h. The mixture was concentrated under reduced pressure, and the residue was purified by column chromatography (1% pyridine and 0–5% MeOH in CH2Cl2) to give the product 9 (81 mg, 63%) as a white foam. Rf 0.3 (10% MeOH in CH2Cl2); 1H NMR (400 MHz, DMSO) δ 11.34 (s, 1H, NH(T)), 11.14 (s, 1H, NH(C)), 8.00 (d, J = 7.5 Hz, 2H, Bz), 7.96 (d, 7.0 Hz, 1H, H6(C)), 7.65–7.57 (m, 2H, Bz, H6(T)), 7.51 (t, J = 7.5 Hz, 2H, Bz), 7.44–7.00 (m, 12H, pixyl, H5(C)), 6.87 (t, J = 7.3 Hz, 1H, pixyl), 6.68 (d, J = 7.3 Hz, 1H, pixyl), 5.84 (t, J = 7.1 Hz, 1H, H1′), 5.08 (d, J = 4.6 Hz, 1H, OH), 4.03 (s, 1H, H3′), 3.84 (m, 1H, H5′), 3.75 (dd, J = 13.1, 4.8 Hz, 1H, H6′), 3.49 (dd, J = 13.1, 7.8 Hz, 1H, H6′), 3.12 (m, 1H, H4′), 2.02–1.89 (m, 2H, H2′), 1.80 (s, 3H, CH3); 13C NMR (101 MHz, DMSO) δ 167.36 (Bz), 163.62 (C4(T)), 162.96 (C4(C)), 154.70 (C2(C)), 150.84, 150.78, 150.16 (pixyl, C2(T), C6(C)), 147.93 (pixyl), 135.45 (C6(T)), 132.53 (Bz), 131.01, 130.03, 129.86, 129.77, 129.37, 128.83, 128.30, 127.86, 127.64, 126.96, 126.76, 125.76, 123.68, 123.42, 123.32, 122.82, 122.51, 122.23, 116.34, 116.11 (pixyl, Bz), 109.37 (C5(T)), 95.68 (C5(C)), 85.35 (C4′), 83.68 (C1′), 76.45 (pixyl), 69.98 (C5′), 69.40 (C3′), 50.59 (C6′), 40.07 (C2′), 12.11 (CH3); HR-ESI MS m/z 748.2936 ([M + Na]+, C41H35N5O8Na+ calc. 748.2383).5′(S)-C-(N4-Benzoylcytosin-1-yl)methyl-3′-O-(P-2-cyanoethoxy-N,N-diisopropylaminophosphinyl)-5′-O-pixyl-thymidine (10)
Compound 9 (310 mg, 0.43 mmol) was coevaporated with anhydrous DCE (2 × 5 mL) and redissolved in the same solvent (5 mL). DIPEA (1.48 mL, 8.5 mmol) and N,N-diisopropylamino-2-cyanoethylchlorophosphite (286 μL, 1.3 mmol) were added and the reaction mixture was stirred at room temperature for 40 min. 99.9% EtOH (6 mL) was added and the mixture was concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 0–100% EtOAc in petrol ether) and then dissolved in CH2Cl2 (4 mL) and precipitated in petrol ether (200 mL) to give the product 10 (190 mg, 47%) as a white foam. Rf 0.7 (0.05% pyridine in EtOAc); 31P NMR (162 MHz, CDCl3) δ 151.47, 149.54; HR-ESI MS m/z 926.4325 ([M + Na]+, C50H52N7O9PNa+ calc. 926.3644).3′-O-(tert-Butyldimethylsilyl)-5′(S)-C-(4-methylpiperazin-1-yl)methylthymidine (11)
Epoxide 1 (577 mg, 1.56 mmol) was coevaporated with anhydrous DCE (7 mL) and redissolved in anhydrous THF (15 mL). 4N-methylpiperazine (260 μL, 2.35 mmol) was added and the mixture was stirred at 66 °C for 20 h. The reaction mixture was added CH2Cl2 (15 mL) and washed with a saturated aqueous solution of NaHCO3 (25 mL). The aqueous phase was extracted with CH2Cl2 (3 × 30 mL) and the combined organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–10% MeOH in CH2Cl2) to give the product 11 (558 mg, 76%) as a white foam. Rf 0.2 (10% MeOH in CH2Cl2); 1H NMR (400 MHz, DMSO) δ 11.28 (s, 1H, NH), 7.88 (d, J = 0.9 Hz, 1H, H6), 6.16 (dd, J = 7.7, 6.0 Hz, 1H, H1′), 5.04 (s, 1H, OH), 4.44 (m, 1H, H3′), 3.84 (m, 1H, H4′), 3.75 (t, J = 6.4 Hz, 1H, H5′), 2.48–2.19 (m, 10H, H6′, NCH2CH2N), 2.16–2.08 (m, 4H, NCH3, H2′), 2.03 (ddd, J = 13.0, 6.0, 2.8 Hz, 1H, H2′), 1.75 (d, J = 0.9 Hz, 3H, CH3), 0.87 (s, 9H, C(CH3)3), 0.06 (s, 6H, Si(CH3)2). 13C NMR (101 MHz, DMSO) δ 163.59 (C4(T)), 150.33 (C2(T)), 136.09 (C6(T)), 109.17 (C5(T)), 87.32 (C4′), 83.67 (C1′), 73.19 (C3′), 66.65 (C5′), 60.57 (C6′), 54.66, 53.12 (NCH2CH2N), 45.62 (NCH3), 40.07 (C2′), 25.60 (C(CH3)3), 17.60 (C(CH3)3), 12.29 (CH3), −4.81, −4.92 (Si(CH3)2); HR-ESI MS m/z 469.2824 ([M + Na]+, C22H40N4O5SiNa+ calc. 469.2841).3′-O-(tert-Butyldimethylsilyl)-5′(S)-C-(4-methylpiperazin-1-yl)methyl-5′-O-pixylthymidine (12)
Compound 11 (895 mg, 1.9 mmol) was coevaporated with anhydrous pyridine (10 mL) and redissolved in the same solvent (20 mL). Pixyl chloride (753 mg, 2.6 mmol) was added, and the reaction mixture was stirred at room temperature for 24 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography (1% pyridine and 0–10% MeOH in CH2Cl2) to give the product 12 (824 mg, 60%) as a white foam. Rf 0.3 (10% MeOH in CH2Cl2); 1H NMR (400 MHz, DMSO) δ 11.43 (s, 1H, NH), 7.68 (d, J = 1.1 Hz, 1H, H6), 7.53–7.44 (m, 2H, pixyl), 7.42–7.27 (m, 7H, pixyl), 7.20–7.08 (m, 2H, pixyl), 7.01–6.92 (m, 2H, pixyl), 6.03 (dd, J = 9.4, 5.1 Hz, 1H, H1′), 4.00 (s, 1H, H4′), 3.42–3.19 (m, 2H, H3′, H5′), 2.33 (m, 1H, H6′), 2.27–1.97 (m, 7H, NCH2, NCH3), 1.97–1.68 (m, 9H, NCH2, CH3(T)), H2′), 1.38 (dd, J = 12.0, 3.5 Hz, 1H, H6′), 0.76 (s, 9H, C(CH3)3), −0.12 (s, 3H, Si(CH3)), −0.16 (s, 3H, Si(CH3)); 13C NMR (101 MHz, DMSO) δ 163.58 (C4), 151.16 (pixyl), 150.19 (C2), 146.88 (pixyl), 134.88 (C6), 131.53, 130.58, 130.22, 127.72, 127.14, 123.80, 123.56, 123.17, 122.61, 116.44 (pixyl), 109.50 (C5), 86.78 (C4′), 83.74 (C1′), 76.75 (pixyl), 73.74 (C3′), 70.29 (C5′), 57.33 (C6′), 54.50, 52.46 (NCH2CH2N), 45.43 (NCH3), 40.01 (C2′), 25.47 (C(CH3)3), 17.47 (C(CH3)3), 12.37 (CH3(T)), −4.88, −5.17 (Si(CH3)2); HR-ESI MS m/z 725.3721 ([M + Na]+, C41H52N4O6SiNa+ calc. 725.3729).5′(S)-C-(4-Methylpiperazin-1-yl)methyl-5′-O-pixylthymidine (13)
A solution of compound 12 (805 mg, 1.1 mmol) in anhydrous THF (11 mL) was added a 1 M solution of TBAF in THF (1.1 mL), and the mixture was stirred at room temperature for 20 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography (1% pyridine and 0–10% MeOH in CH2Cl2) to give the product 13 (616 mg, 80% containing 10% TBA) as a white foam. Rf 0.1 (10% MeOH in CH2Cl2); 1H NMR (400 MHz, DMSO) δ 11.37 (s, 1H, NH), 7.57 (s, 1H, H6), 7.48–7.23 (m, 9H, pixyl), 7.18–7.03 (m, 2H, pixyl), 7.02–6.92 (m, 2H, pixyl), 6.02 (m, 1H, H1′), 5.08 (d, J = 3.8 Hz, 1H, OH), 3.84 (t, J = 2.4 Hz, 1H, H4′), 3.40–3.31 (m, 2H, H3′, H5′), 2.30–2.23 (m, 2H, H6′), 2.18–2.01 (m, 4H, NCH2), 2.05 (s, 3H, NCH3), 2.01–1.83 (m, 6H, H2′, NCH2), 1.80 (s, 3H, CH3), 1.49 (m, 1H, H6′); 13C NMR (101 MHz, DMSO) δ 163.62 (C4), 150.99, 150.96 (pixyl), 150.15 (C2), 147.61 (pixyl), 135.19 (C6), 131.28, 130.68, 129.92, 129.82, 127.69, 126.99, 126.88, 123.41, 123.33, 123.26, 122.71, 116.32, 116.17 (pixyl), 109.37 (C5), 86.52 (C4′), 83.41 (C1′), 76.35 (pixyl), 70.55 (C3′, C5′), 57.95 (C6′), 54.30 (NCH2CH2N), 45.51 (NCH3), 40.06 (C2′), 12.30 (CH3); HR-ESI MS m/z 633.2699 ([M + Na]+, C35H38N4O6Na+ calc. 633.2684).3′-O-(P-2-Cyanoethoxy-N,N-diisopropylaminophosphinyl) 5′(S)-C-(4-methylpiperazin-1-yl)methyl-5′-O-pixyl-thymidine (14)
Compound 13 (133 mg, 0.22 mmol) was coevaporated with anhydrous DCE (4 mL) and redissolved in the same solvent (3 mL). DIPEA (1.14 mL, 6.5 mmol) and N,N-diisopropylamino-2-cyanoethylchlorophosphite (219 μL, 0.98 mmol) were added, and the reaction mixture was stirred at room temperature for 3 h. EtOH (99.9%, 1 mL) was added and the mixture was concentrated under reduced pressure. The residue was purified by column chromatography (1% pyridine and 0–6% MeOH in CH2Cl2) to give the product 14 (167 mg, 96%) as a white foam. Rf 0.4 (20% MeOH in CH2Cl2); 31P NMR (162 MHz, CDCl3) δ 149.93, 149.87; HR-ESI MS m/z 811.3930 ([M + Na]+, C44H55N6O7PH+ calc. 811.3943.Oligonucleotide synthesis and hydridization experiments
The oligodeoxynucleotides were synthesised using an automated system following the phosphoramidite approach. The syntheses were accomplished in a 0.2 μmol scale by using 2-cyanoethyl phosphoramidites of standard 2′-deoxynucleosides in combination with the modified phosphoramidites, 6, 10 and 14. The synthesis followed the regular protocol employing standard CPG supports and pyridinium chloride as the activator. The modified amidites were manually coupled using 0.02 M amidite and 0.25 M pyridinium chloride as activator in CH3CN for 20 min. The coupling time for the next standard amidite was extended to 20 min ensuring efficient coupling yields. The coupling yields for the modified phosphoramidites in combination with the following unmodified amidite were in the range of 50–100%. The 5′-O-DMT-ON oligonucleotides were removed from the solid support by treatment with concentrated aqueous ammonia at 55 °C for 16–24 h, which also removed the protecting groups. The oligonucleotides were purified by reversed-phase HPLC on a Waters 600 system using a Xterra prep MS C18; 10 μm; 7.8 × 150 mm column; Buffer A: 0.05 M triethylammonium acetate pH 7.4; buffer B: 75% CH3CN in buffer A. Program: 2 min 100% A, 100–30% A over 38 min., 30–0% A over 7 min, 10 min 100% B. All fractions containing 5′-O-DMT protected oligonucleotide were collected and concentrated. The products were detritylated by treatment with 80% aqueous acetic acid (100 μL) for 20 min at room temperature and subsequently quenched with water (100 μL). Sodium perchlorate (5 M, 15 μL), sodium acetate (3 M, 15 μL) and abs. ethanol (1 mL) were then added and allowed to precipitate at −18 °C over night. Finally, the ONs were washed with cold abs, ethanol (2 × 300 μL), solvent residues were removed by heating (55 °C) under a flow of nitrogen, and the ONs were redissolved in water. the purity and constitution of the synthesised ONs were verified by ion-exchange chromatography and MALDI-MS analysis recorded in positive ion mode on a PerSeptive Voyager STR spectrometer with 3-hydroxypicolinic acid as matrix. The concentrations were determined spectrometrically at 260 nm in medium salt buffer (0.1 mM EDTA, 100 mM NaCl adjusted to pH 7.0 by 10 mM NaH2PO4/5 mM Na2HPO4) assuming the extinction coefficients for the double headed nucleotide monomers to equal the sum of the individual nucleobases.The UV melting experiments were thereafter carried out on a UV spectrometer. Samples were dissolved in medium salt buffer with 1.0 μM concentrations of the two complementary sequences. The increase in absorbance at 260 nm as a function of time was recorded while the temperature was increased linearly from 10 to 80 °C at a rate of 0.5 °C min−1 by means of a Peltier temperature programmer. The melting temperature was determined as the local maximum of the first derivatives of the absorbance vs. temperature curve. The melting curves were found to be reversible. All determinations are averages of at least duplicates within ±0.5 °C.
Acknowledgements
The project was supported by The Danish Research Agency's Programme for Young Researchers, Nucleic Acid Center and The Danish National Research Foundation and The Danish Councils for Independent Research | Technology and Production Sciences (FTP) and Natural Science (FNU).Notes and references
- R. Varghese and H.-A. Wagenknecht, Chem. Commun., 2009, 2615–2524 RSC.
- V. L. Malinovskii, D. Wenger and R. Häner, Chem. Soc. Rev., 2010, 39, 410–422 RSC.
- T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138–148 RSC.
- S. L. Pedersen and P. Nielsen, Org. Biomol. Chem., 2005, 3, 3570–3575 CAS.
- M. S. Christensen, C. M. Madsen and P. Nielsen, Org. Biomol. Chem., 2007, 5, 1586–1594 CAS.
- C. Andersen, P. K. Sharma, M. S. Christensen, S. I. Steffansen, C. M. Madsen and P. Nielsen, Org. Biomol. Chem., 2008, 6, 3983–3988 CAS.
- M. S. Christensen, A. D. Bond and P. Nielsen, Org. Biomol. Chem., 2008, 6, 81–91 CAS.
- K. I. Shaikh, C. S. Madsen, L. J. Nielsen, A. S. Jørgensen, H. Nielsen, M. Pedersen and P. Nielsen, Chem.–Eur. J., 2010, 16, 12904–12919 CrossRef CAS.
- A. S. Jørgensen, K. I. Shaikh, G. Enderlin, E. Ivarsen, S. Kumar and P. Nielsen, Org. Biomol. Chem., 2011, 9, 1381–1388 Search PubMed.
- C. S. Madsen, S. Witzke, P. Kumar, K. Negi, P. K. Sharma, M. Petersen and P. Nielsen, Chem.–Eur. J., 2012, 18, 7434–7442 CrossRef CAS.
- T. F. Wu, M. Froeyen, G. Schepers, K. Mullens, J. Rozenski, R. Busson, A. Van Aershot and P. Herdewijn, Org. Lett., 2004, 6, 51–54 CrossRef CAS.
- T. F. Wu, K. Nauwelaerts, A. Van Aerschot, M. Froeyen, E. Lescrinier and P. Herdewijn, J. Org. Chem., 2006, 71, 5423–5431 CrossRef CAS.
- T. Umemoto, J. Wengel and A. S. Madsen, Org. Biomol. Chem., 2009, 7, 1793–1797 CAS.
- M. Raunkjær, T. Bryld and J. Wengel, Chem. Commun., 2003, 1604–1605 RSC.
- K. Heil, D. Pearson and T. Carell, Chem. Soc. Rev., 2011, 40, 4271–4278 RSC.
- G. Wang and P. J. Middleton, Tetrahedron Lett., 1996, 37, 2739–2742 CrossRef CAS.
- G. L. Bundy and D. C. Peterson, Tetrahedron Lett., 1978, 41–44 CrossRef CAS.
- H. Trafelet, E. Stulz and C. J. Leumann, Helv. Chim. Acta, 2001, 84, 87–97 CrossRef CAS.
- R. T. Day, D. Williams, P. Soriano and Y. S. Sanghvi, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 1135–1138 CAS.
- M. Beier and W. Pfleiderer, Helv. Chim. Acta, 1999, 82, 879–887 CrossRef CAS.
- S. Kumar, M. H. Hansen, N. Albæk, S. I. Steffansen, M. Petersen and P. Nielsen, J. Org. Chem., 2009, 74, 6756–6769 CrossRef CAS.
- J.-M. Escudier, C. Dupouy, M. A. Fountain, I. M. A. deæl Mundo, E. M. Jacklin and J. R. Morrow, Org. Biomol. Chem., 2009, 7, 3251–3257 CAS.
- D. James, J.-M. Escudier, E. Amigues, J. Schulz, C. Vitry, T. Bordenave, M. Szlosek-Pinaud and E. Fouquet, Tetrahedron Lett., 2010, 51, 1230–1232 CrossRef CAS.
- A. K. Saha, T. J. Caulfield, C. Hobbs, D. A. Upson, C. Waychunas and A. M. Yawman, J. Org. Chem., 1995, 60, 788–789 CrossRef CAS.
- G. Y. Wang, P. J. Middleton, C. Lin and Z. Pietrzkowski, Bioorg. Med. Chem. Lett., 1999, 9, 885–890 CrossRef CAS.
- H. Trafelet, S. P. Parel and C. J. Leumann, Helv. Chim. Acta, 2003, 86, 3671–3687 CrossRef CAS.
- X. Wu and S. Pitsch, Nucleic Acids Res., 1998, 26, 4315–4323 CrossRef CAS.
- V. Banuls, J. M. Escudier, C. Zedde, C. Claparols, B. Donnadieu and H. Plaisancie, Eur. J. Org. Chem., 2001, 4693–4700 CrossRef CAS.
- C. Dupouy, N. Iché-Tarrat, M.-P. Durrieu, A. Vigroux and J.-M. Escudier, Org. Biomol. Chem., 2008, 6, 2849–2851 CAS.
- C. Dupouy, P. Millard, A. Boissonnet and J.-M. Escudier, Chem. Commun., 2010, 46, 5142–5144 RSC.
- A. Boissonnet, C. Dupouy, P. Millard, M.-P. Durrieu, N. Tarrat and J.-M. Escudier, New J. Chem., 2011, 35, 1528–1533 RSC.
- P. P. Seth, C. R. Allerson, A. Siwkowski, G. Vasquez, A. Berdeja, M. T. Migawa, H. Gaus, T. P. Prakash, B. Bhat and E. E. Swayze, J. Med. Chem., 2010, 53, 8309–8318 CrossRef CAS.
- T. Lindahl, Nature, 1993, 362, 709–715 CrossRef CAS.
- J. Butenandt, L. T. Burgdorf and T. Carell, Angew. Chem., Int. Ed., 1999, 38, 708–711 CrossRef CAS.
- J. K. Kim, D. Patel and B. S. Choi, Photochem. Photobiol., 1995, 62, 44–50 CrossRef CAS.
- K. McAteer, Y. Jing, J. Kao, J. S. Taylor and M. A. Kennedy, J. Mol. Biol., 1998, 282, 1013–1032 CrossRef CAS.
- L. M. Kundu, U. Linne, M. Marahiel and T. Carell, Chem.–Eur. J., 2004, 10, 5697–5705 CrossRef CAS.
- P. Kumar, K. I. Shaikh, A. S. Jørgensen, S. Kumar and P. Nielsen, J. Org. Chem., 2012, 77, 9562–9573 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: MALDI data for all modified oligonucleotides, full Tm-data, HPLC chromatograms related to the T–T-dimer as well as NMR-spectra of all compounds. |
| ‡ Recently, a pyrene-triazole moiety in the 5′-C-position has led to some increase in thermal stability of a duplex but probably due to intercalation of the pyrene into the duplex.38 |
| This journal is © The Royal Society of Chemistry 2013 |