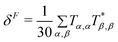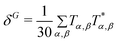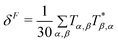DOI:
10.1039/C3RA40790J
(Paper)
RSC Adv., 2013,
3, 9247-9257
Influence of electronic effects on one- and two-photon absorption in porphyrin isomers†
Received
15th February 2013
, Accepted 3rd April 2013
First published on 4th April 2013
Abstract
The one- and two-photon absorption characteristics of the Porphyrin isomers Porphycene, Corrphycene, Hemiporphycene, N-confused Porphyrin, cis- and trans-Doubly confused Porphyrins, the newly synthesised Neo-confused Porphyrin, and a set of core-substituted Porphycenes are systematically investigated using linear and quadratic density functional response theory. It is shown that, despite the one-photon spectra displaying characteristic Soret- and Q-regions, the classic Gouterman four orbital theory has limitations for the analysis of the absorptions in this series of compounds. Generated spectra for the two-photon absorptions are shown to be very sensitive to the isoelectronic changes, contrary to the one-photon spectra. This accesses a potentially powerful tool by which the two-photon absorption can be fine-tuned without large structural changes to the nature of the macrocycle.
1 Introduction
The photochemistry of porphyrin (P in Fig. 1) systems is a vast area that involves chemistry and biology as well as materials and medicinal science. Research is mainly driven by the many attractive applications potentially attainable with a deeper understanding of these molecular systems, for example in the development of dye-sensitized solar cells for use in artificial photosynthesis, molecular semiconductors in photovoltaic cells, photo catalysts, biological imaging dyes and photosensitisers for use in photodynamic therapy, PDT.1–9
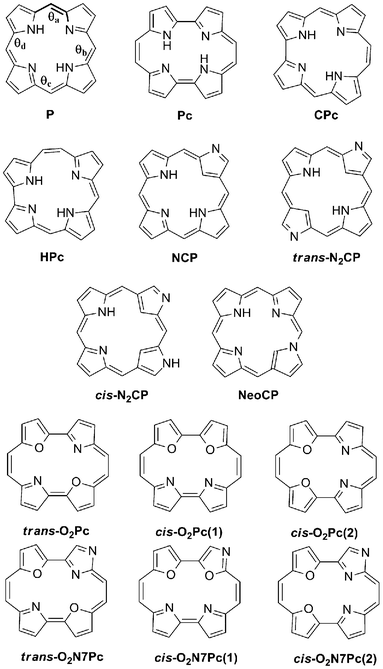 |
| | Fig. 1 The structural electronic isomers investigated herein. The dihedral angles referred to in geometry discussion is highlighted in P (spanning from C in the meso–position to N on the subsequent pyrrole unit, as per the bold bonds in θa). | |
Non-linear absorption properties, such as two-photon absorption (TPA), is a fairly recent focus of the photochemistry of P. For applications of non-linear materials, such as 3D optical storage, this is clearly a key property, but it is also of great interest in applications in a biological environment. This is due the fact that conventional linear absorption of P-type compounds rarely have their wavelengths of maximum absorption located inside the optical window of tissue penetration, a limiting factor for biological applications. The use of two longer wavelength photons to reach the same region of absorption as conventional one-photon absorption (OPA) processes, coupled with the accompanying change in parity selection rules for the transition, therefore provides a potential tuning mechanism whereby the wavelength of maximum absorption of chromophores can be moved into the more transparent region. TPA can therefore be a useful tool for deeper access into biological samples.
There are numerous investigations into porphyrin type systems and their OPA and TPA spectra.10–19 The probability of TPA in any molecular system is defined through its TPA cross section, which is proportional to its TPA transition strength, δTPA, which is controlled directly by the molecular electronic structure. General strategies to increase δTPA in porphyrin derivatives have previously mainly involved the addition of electron donor- and acceptor groups, extending conjugation path lengths and coordination of the macrocycle to a metal centre.11,20–26 We have recently investigated the TPA of the reduced porphyrin isomer porphycene (Pc in Fig. 1) and its dioxa-derivatives and found that the effect on the TPA can be more subtle than expected.27 Small changes in the electronic structure of the core, through the substitution of nitrogen atoms for oxygen atoms in the pyrrole-units of the macrocycle had a minor effect the OPA signature, whilst it afforded a very large change in the TPA spectra. Similar effects have also been seen in extended porphyrinic macrocycles.28,29 As this can provide a powerful mechanism, where the TPA can be improved without large structural additions, we set out to systematically investigate the P macrocycle and its reduced and isoelectronic isomers and their OPA and TPA properties. The first constitutional isomer of the core P to be presented was the Porphycene (Pc) macrocycle, synthesised in the late 1980's and since then related varieties such as Hemiporphycene (HPc) and Corrphycene (CPc) have also been developed (Fig. 1).30,31 These structural isomers have similar properties to the parent P in terms of structure and coordination, and have been shown to have porphyrin–like OPA spectra with variations in intensity profiles caused by change in the symmetries of the species.32 A slight change in the linear spectra can however afford large changes in the non-linear case (vide infra).27 As early as the 1940's it was realised that a structural isomer did not need all its Nitrogen atoms pointing into the centre of the core macrocycle, but could experience confusion of the pyrrole unit, and keep its stability.33 It was not until 1994 that the first of the confused porphyrin isomers were synthesised, with the N-confused porphyrin (NCP) being reported independently by two groups.34,35 Due to the change in conjugation pathway in the molecule, confused porphyrins have properties that varies from the parent P, in physical, structural and coordination properties. Examples are their multivalent character as a metal ligand, fusion properties in aggregates, and a red–shift seen in absorption measurements.36 Further work also showed the doubly-confused (N2CP) to be relatively stable.37 Research into new and more efficient synthetic routes continues to lead to the discoveries of novel P isomers, the latest being the Neo-confused porphyrin (NeoCP) with one pyrrole unit oriented so that its N atom is fused to a meso-carbon of the macrocycle (Fig. 1).38 The preliminary investigation into the photochemical properties of this latest structural isomer show a porphyrin–like linear absorption pattern.
This work focuses on the δTPA as calculated using Density Functional Theory (DFT). Density functional theory is essential for the optimisation of the geometries of P-type molecules as the model scales favourably with the size of the system, making accurate geometries possible for large structures. The basic Hartree–Fock method is also well known to fail to accurately describe the aromaticity pattern of these systems, unless coupled with computationally expensive correlation methods.39,40 These advantages of DFT are combined with photochemical properties through the use of time-dependent DFT, where the linear and quadratic response functions gives OPA and TPA information respectively.15,16,41–43 Here, the calculations were carried out for P, Pc, HPc, CPc, NCP, N2CP and NeoCP, to provide an overview of constitutional porphyrin isomers and their OPA and TPA properties. In order to get a deeper understanding of how δTPA is effected by the electronic structure of the core, we also present the series of dioxa-porphycenes mentioned previously, together with a set of dioxa-porphycenes with a Nitrogen atom in its confused position, according to Fig. 1. A change of the central atom to a group 16 atom has also been shown to change the compound ability to chelate to a metal, which has been suggested to improve the availability for bio-applications.44
Several experimental and theoretical investigations have been carried out on the TPA qualities of P as well as Pc, and the latter shows exceptional promise as a TPA PDT sensitiser due to its exceptionally large δTPA compared to the P core.32,45,46 The δTPA of NCP have also been investigated experimentally, and show similar properties to the P core.47 As far as we are aware neither HPc, CPc nor NeoCP have been reviewed for their TPA properties.
2 Computational methods
The ground state geometries of each molecular system were optimised using the coloumb-attenuated version of the hybrid B3LYP exchange correlation functional, CAM-B3LYP, with the 6-31G** Pople-type basis set on all atoms. Analytical Hessian calculations were preformed to confirm the nature of each stationary point on the potential energy surface.
At each confirmed ground state optimised geometry vertical one- and two-photon absorption spectra were computed using linear response and quadratic response DFT respectively. All geometry optimisations and OPA linear response calculations were computed using Gaussian 09,48 whilst TPA quadratic response theory calculations were performed using a local version of the Dalton 2.0 program.49 The linear and quadratic response calculations were carried out with CAM-B3LYP. As this functional gives a better description of the long range exchange potential than local hybrid functionals do, it has been shown to greatly improve the determination of various properties compared to B3LYP, without significant increase in computational cost. Crucially for this work, the CAM-B3LYP functional has become one of the standard DFT functionals for TPA due to the ability of this functional to describe excitations to and from the virtual state (vide infra) in a robust manner. This leads to results that are very reliable even when comparing DFT results with high level coupled-cluster calculations.50
In response theory calculations, the OPA spectra are obtained from the linear response function and the TPA spectra from the quadratic response function, which both have poles at the excitation energies of the system. The residues of the response functions give the electronic dipole transition moments to the excited states. The first residue of the quadratic response function gives the two-photon transition tensor, T. Its spatially dependent components is conveniently expressed in a sum-over-states equation:
| | 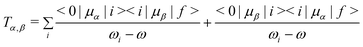 | (1) |
where
ω is the frequency of the irradiating light, and
ωi is the excitation frequency of the intermediate state
i. The
labels α and
β refers to the Cartesian components
x,
y and
z of the dipole operator. Thus the TPA proceeds through a so-called virtual state, which can be written as a linear combination of ail real eigenstates
i. This is an important concept in that it highlights the need for a robust functional such as CAM-B3LYP, which can describe such a state, for the correct calculation of TPA absorption qualities. It is worth to note here that the actual machinery of response theory allows for the recovery of the TPA tensor directly, without the need to sum over all states available to the molecule. As the TPA tensor is dependent on the accurate description of the virtual state, and hence the number of states used if employing a sum-over-states calculation, this fact highlights the advantage of the use of response theory for large molecules with a high density of states. The use of the sum-over-states-expression in
eqn (1) is however very convenient when interpreting the TPA spectra, as seen in the following Results and Discussion section.
The tensor T is related to δTPAviaeqn (2).
| | | δTPA = FδF + GδG + HδH | (2) |
The values F, G and H depend on the polarisation of the incident photons and the conditions applied in this work was linear polarised parallel photons, in which case F = G = H = 2.
For theoretical work on TPA an added difficulty is the relation of δTPA and the experimentally observed TPA cross section, reported in units of cm4 s photon−1, as it involves molecule–dependent line shape functions. However, when comparing and contrasting trends, δTPA provides the information needed for conclusions with regards to structure–TPA relationships, without the need for unit conversion.
The results achieved from a linear response calculation, apart from excitation energies, are a set of excitation and deexcitation amplitudes that describes the contribution of a particle-hole pair to the specific transition. In order to analyse the nature of the electronic transitions in detail Natural Transition Orbitals, NTOs were calculated for each transition.51 A NTO is a calculated orbital representation of the electronic transition where the transition amplitudes have been reduced to a set of particle-hole amplitudes, in terms of eigenvectors of the transition density matrix. The NTO representation of the excitation is complementary to the response eigenvectors using the Kohn–Sham orbitals but generally gives a compact, and somewhat more intuitive, representation of the state. NTO representations was calculated for each excited state of interest from the linear response calculations.
The calculation of properties of molecules of the size of porphyrins is a challenging task that has been under way for many years,52–56 and as the development of novel isomers in this important family of chromophores continues, it also continues to be important issue. With DFT and especially response theory maturing to the task of accurately modelling the non-linear optical properties of increasingly large molecules it constitutes an important tool for structure–property prediction.
3 Results and discussion
3.1 Reduced porphyrin isomers
All four N-sites in the P parent are possible binding sites of the two inner hydrogen atoms and even though the cis-isomers, with hydrogens on adjacent N-sites, in general are less stable they are known to take part in the step-wise N–H tautomerism observed in P.57–59 It was therefore considered pertinent to include all possible cis-isomers of the reduced isomers investigated here, and analyse their optical response. The optimization results of the reduced porphyrin isomers, with their pyrrolic hydrogen atoms in the cis as well as trans-position, all resulted in a planar series of systems, apart from cis-Pc-2 (Fig. 2 and Fig. 3). Cis-Pc-2 did not suggest itself to be stable as a free base, and theoretical and experimental work suggests that there is in fact a crossing point between the cis and trans surfaces along the tautomerisation path, rendering this isomer inaccessible for the unsubstituted free base.60 No optical response was therefore calculated for this isomer. The overall results confirm previous theoretical, as well as experimental, studies of these compounds.40,61,62Table 1 describes the relative energies and symmetries, which can be rationalised by the consideration of angle strain as well as hydrogen bonding stabilising factors.
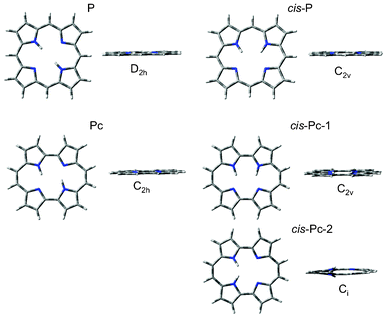 |
| | Fig. 2 The optimised geometries of P and Pc, and their respective cis and trans-isomers, from which the absorption qualities were calculated. | |
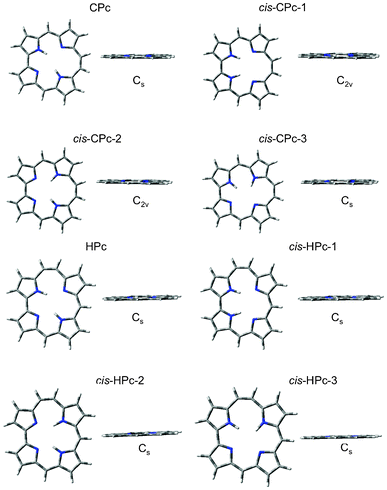 |
| | Fig. 3 Corrphycene and Hemiporphycene isomer optimised geometries. These were used to calculate vertical OPA and TPA transitions. | |
Table 1 Optimised parameters of the series of reduced porphyrin isomers. The relative energies (kJ mol−1) of the trans-isomers are with respect to the P parent macrocycle, whilst the relative energies of the cis-isomers are with respect to their respective stable trans-conformer
| Compound |
Point Group |
Energy (a. u.) |
ΔE (kJ mol−1) |
|
P
|
D
2h
|
−989.0240 |
0 |
|
cis-P |
D
2h
|
−989.0105 |
34.11 |
|
Pc
|
C
2h
|
−989.0254 |
−3.54 |
|
cis-Pc-1 |
C
2v
|
−989.0223 |
8.14 |
|
cis-Pc-2 |
C
i
|
−988.9784 |
123.40 |
|
CPc
|
C
s
|
−989.0062 |
46.74 |
|
cis-CPc-1 |
C
2v
|
−988.9977 |
21.47 |
|
cis-CPc-2 |
C
2v
|
−988.9966 |
24.25 |
|
cis-CPc-3 |
C
s
|
−988.9775 |
72.51 |
|
HPc
|
C
s
|
−989.0180 |
15.58 |
|
cis-HPc-1 |
C
s
|
−989.0108 |
18.19 |
|
cis-HPc-2 |
C
s
|
−989.0078 |
25.77 |
|
cis-HPc-3 |
C
s
|
−988.9854 |
64.17 |
The smaller rectangular cavity of Pc contributes to favourable hydrogen bonding interactions, making it the most stable isomer investigated. For the two isomers with trapezoidal cavity hydrogen bonding stabilises HPc to a larger extent than CPc, counteracting angle strain, rendering the latter the least stable of the reduced set of isomers. These steric strains are not enough to take the (4n + 2)π-electron structures out of planarity, and thereby break the aromaticity. The energies of the more symmetrical of the cis-isomers of CPc suggest that they are stable, and indeed rapid tautomerisation of the central hydrogen atoms has been shown experimentally.31
The simulated spectra of the set of reduced porphyrin isomers show two dominant regions of absorption for the series, one in the 200–350 nm region and one at 470–570 nm as seen in Fig. 4 and Table 2. These two regions have been characterised extensively for the porphyrin family over the years, and in fact no other compound have probably been analysed in terms of its UV–visible spectrum more than the P free base.
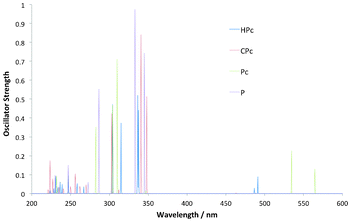 |
| | Fig. 4 Simulated OPA spectra for a selection of the reduced porphyrin isomers, as well as the parent porphyrin (P), all showing main absorption into the 300–350 nm Soret-region. The absorption into the longer wavelength (470–570 nm) Q-region is present for all but strongest for Pc and HPc. | |
Table 2 Overview of the wavelengths (λ) and oscillator strengths (f) for key transitions from the linear response of reduced P-isomers
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
f
|
λ (nm) |
f
|
|
P
|
559 |
0.0021 |
345 |
0.8416 |
|
|
507 |
0.0015 |
333 |
1.1671 |
|
cis-P |
552 |
0.0002 |
346 |
0.7953 |
|
|
516 |
0.0061 |
339 |
0.8892 |
|
Pc
|
564 |
0.1578 |
310 |
0.7557 |
|
|
534 |
0.2343 |
304 |
0.7721 |
|
cis-Pc-1 |
557 |
0.1835 |
309 |
0.6951 |
|
|
533 |
0.1979 |
300 |
0.9271 |
|
CPc
|
497 |
0.0003 |
348 |
0.5164 |
|
|
463 |
0.0028 |
340 |
1.1576 |
|
cis-CPc-1 |
494 |
0.0035 |
338 |
0.5945 |
|
|
470 |
0.0683 |
336 |
1.0433 |
|
cis-CPc-2 |
496 |
0.0001 |
349 |
0.4497 |
|
|
476 |
0.0325 |
343 |
0.9356 |
|
cis-CPc-3 |
548 |
0.0121 |
356 |
0.3191 |
|
|
503 |
0.1318 |
342 |
0.8802 |
|
HPc
|
491 |
0.0899 |
337 |
0.4450 |
|
|
486 |
0.0380 |
336 |
0.6407 |
|
cis-HPc-1 |
521 |
0.1432 |
334 |
0.2599 |
|
|
478 |
0.0383 |
330 |
0.5922 |
|
cis-HPc-2 |
538 |
0.0229 |
344 |
0.5115 |
|
|
521 |
0.0647 |
333 |
0.6249 |
|
cis-HPc-3 |
514 |
0.1454 |
332 |
0.4894 |
|
|
477 |
0.0619 |
318 |
0.5610 |
|
|
|
|
304 |
0.7344 |
The transitions of shorter wavelength occurs in a region called the Soret-region whilst the transitions into the longer wavelength region are termed Q-region transitions. The rationale behind these two regions for P was developed as early as in the 1960's by Martin Gouterman in the classic Gouterman Four Orbital (GFO) Model. According to this model the Soret and Q-regions are a result of absorption from the HOMO and HOMO-1 orbitals to the LUMO and LUMO + 1 pair. In the P macrocycle the latter pair is near-degenerate, which leads to a cancellation of transition moments into the Q-region and a complementary strengthening of transition moments into the Soret–region, which is reproduced here. There are plenty of theoretical results which support the use of the GFO model for the assignment of the transitions into these regions in P.16 When it comes to the reduced Pc and HPc derivatives however, there is a clear strengthening of the Q- relative to the Soret-region. This is a well known phenomenon and results from the two LUMO–orbitals no longer being near degenerate. This leads to the transition no longer being described in terms of cancellation of transition moments of as large a magnitude as for P. The relative location of the states in the Q-region will be important when discussing the TPA absorption pattern (vide infra).
The optical responses of the cis-isomers of the reduced isomers follow the same trends, with clear Soret- and Q-bands, as their trans-isomers, including the relative strengthenings of the Q-bands in Pc and HPc (Table 2). cis-CPc-3 experiences strengthening of the transition in the Q-region, contrary to what was found in the parent trans-isomer, similar to the case of Pc.
In DFT the Kohn–Sham orbitals generated are difficult to analyse in terms of the electron density. The NTO's however, present the density picture in terms of the transition densities, by accounting for where the electron density is located before and after the transition, and is a powerful tool to analyse linear response DFT spectra. In the Pc and HPc derivatives the NTOs that describe the transitions making up the Q-band show absorption from the HOMO and HOMO-1 orbitals exclusively into one of the LUMO pair, in accordance with the GFO model. In the Soret–region however, HPc show more than the two transitions that are expected above 300 nm, if the GFO model is used. Analysis of the NTOs confirms that the region consists of transitions amongst non-frontier-type orbitals (see at 315 nm in Fig. 5). For Pc, a significant contribution can also be seen at higher energy than the Soret–region from transitions from an orbital that mainly can be described as the HOMO-2 frontier orbital. These facts questions the applicability of using only four orbitals in the GFO model for analysis of the absorption spectra for reduced porphyrins, and the expansion of this model is an issue already opened for debate in the case of P and Pc.32 It is also crucial to keep the position of the states involved in the transitions in mind, and therefore it might be more pertinent to talk about these transitions in using a state–based approach, rather than an expanded orbital–based version. This is especially true when discussing TPA transitions, as the states available and their relative position is crucial (vide infra).
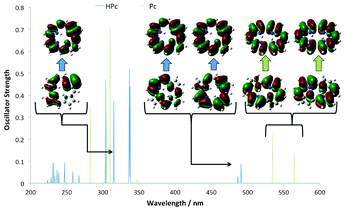 |
| | Fig. 5 NTOs calculated for Pc and HPc show GFO transitions into the Q-region, whilst the Soret–region displays non-GFO transitions, which highlights that this method needs to be expanded in order to fully describe relevant states for reduced isomers. | |
The TPA spectra all show a clear main absorption into the Soret-region, at 200–300 nm, and very small absorption into the Q-region (Fig. 6 and Table 3). The effect on the absorption wavelength of the reduced isomers in comparison to the parent P is a blue shift with the wavelength of maximum absorption going from 285 nm for P to 264 nm for Pc, 246 nm for HPc and 226 nm for CPc. It is worth to note that these corresponds to a wavelength of incoming light of twice that wavelength, in the area of 450–528 nm.
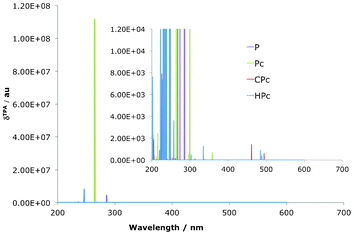 |
| | Fig. 6 Simulated TPA spectra for reduced porphyrin isomers. Note the change in scale between the two versions. All isomers show absorption into the 200–350 nm Soret-region. | |
Table 3 Key transitions from the quadratic response calculations of the reduced P-isomers
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
δ (a. u.) |
λ (nm) |
δ (a. u.) |
|
P
|
419 |
1.11 × 103 |
321 |
4.47 × 103 |
|
|
|
|
285 |
4.51 × 106 |
|
cis-P |
549 |
88.1 |
337 |
19.4 |
|
|
515 |
54.8 |
309 |
42.0 |
|
Pc
|
|
|
299 |
3.42 × 104 |
|
|
|
|
267 |
2.37 × 108 |
|
cis-Pc-1 |
554 |
3.11 × 102 |
279 |
3.55 × 106 |
|
|
530 |
1.51 × 102 |
260 |
1.28 × 107 |
|
CPc
|
494 |
6.12 × 102 |
338 |
5.75 × 101 |
|
|
461 |
2.03 × 103 |
261 |
1.43 × 102 |
|
cis-CPc-1 |
492 |
3.81 × 102 |
338 |
2.78 × 102 |
|
|
466 |
2.41 × 103 |
309 |
4.35 × 103 |
|
cis-CPc-2 |
547 |
3.82 × 102 |
276 |
1.14 × 105 |
|
|
502 |
1.45 × 103 |
255 |
3.55 × 105 |
|
cis-CPc-3 |
488 |
1.27 × 102 |
238 |
3.56 × 104 |
|
|
466 |
2.01 × 103 |
231 |
1.66 × 106 |
|
HPc
|
488 |
6.04 × 102 |
246 |
1.40 × 107 |
|
|
484 |
1.21 × 103 |
235 |
4.49 × 105 |
|
cis-HPc-1 |
519 |
6.40 × 102 |
260 |
3.49 × 107 |
|
|
575 |
5.68 × 102 |
239 |
3.15 × 107 |
|
cis-HPc-2 |
535 |
4.12 × 102 |
263 |
2.15 × 105 |
|
|
519 |
2.87 × 102 |
259 |
1.25 × 109 |
|
cis-HPc-3 |
513 |
1.27 × 103 |
259 |
4.34 × 106 |
|
|
475 |
4.49 × 102 |
237 |
2.68 × 108 |
The δTPA for all reduced isomers is also presented in Table 3 and Fig. 6 and the main feature is the exceptionally large value that can be seen for Pc, compared to the parent P, an increase that is also evident in the cis-HPc-2 isomer, as well as to some degree for cis-CPc-3. These three were the same set of compounds that were shown to have strong, non-cancelling, transition strengths in the Q-region of the OPA spectrum. The reason for the corresponding increases in δTPA is caused by so-called one photon resonance enhancement of the two photon signal.26 The phenomenon of resonance enhancement is best illustrated by consideration of the sum-over-states expression for the TPA transition tensor (eqn (1)). The magnitude of T depends on the difference of the i-th state and the frequency of the total excitation through the denominator in the expression, (ωi − ω). As this value is approaching zero if the frequency of the incident light approaches the frequency difference between the intermediate and the ground state, the δTPA is expected to be very large at these points. An allowed OPA Q-transition accidentally degenerate at half the Soret-transition energy will therefore greatly enhance the TPA transition into the Soret region of the TPA spectrum. The effect of resonance enhancement is clearly seen in the case of the free Pc macrocycle as well as in cis-HPc-2 and cis-CPc-3. The large difference in the TPA transition strength in compounds differing in only the position of the coordination of the core hydrogen shows how exceptionally subtle the effects can be, and how sensitive TPA properties are to the position of the various states and the possibility of resonance enhances transitions. Resonance enhancement also rationalises why there are only very weak TPA transitions to be seen in the Q-region for any of the investigated chromophores, as there is no allowed one-photon transition near half the wavelength value of this region. It is worth to note that the values of the δTPA at wavelengths that are near or at resonance can not be determined explicitly using the method described here, as they require the use of damped response functions.42,63,64 At resonance enhanced TPA transitions it is also expected that vibronic contributions plays a non-negligible role, which leads to so called intensity stealing by otherwise forbidden signals in the symmetric molecules and also potentially accesses purely vibrational channels which distorts and shifts the absorption peak.65–68 Nevertheless, it is important to note that non-dampened response theory is very useful in the interpreting of general trends in the TPA spectra upon varying the electronic structure.
3.2 Confused porphyrin isomers
The resulting geometries of the confused porphyrin isomers are displayed in Fig. 7 as well as Table 4.
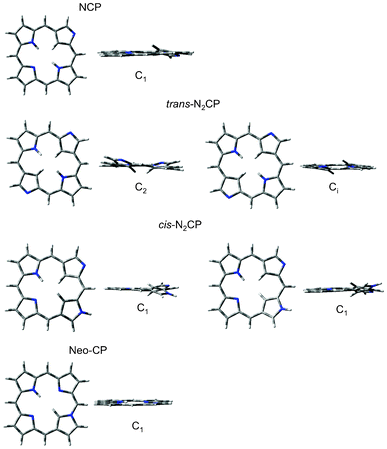 |
| | Fig. 7 The optimised geometries of the confused series of porphyrin isomers. | |
The planar NCP structure optimised to a second order saddle point, with two imaginary vibrational frequencies. Both of these vibrations were related to the relief of steric crowding in the macrocyclic centre, by the out-of-plane movement of the confused pyrrole ring, with the methylene H in the centre of the ring bending out of the plane made by the remaining pyrrolic rings (θa in Table 4). Optimisation of the planar trans-N2CP lead to a fourth order saddle point with the four vibrational modes also describing relief of the steric crowding in the centre. Optimisation along the four modes resulted in two minima, one belonging to the C2 point group (trans-N2CP(1) in Table 4) and one with 12.71 kJ mol−1 higher minimum energy and with Ci symmetry (trans-N2CP(2) in Table 4). The cis-N2CP optimisation also yielded two geometries, both of C1 symmetry (cis-N2CP(1) and (2) in Table 4). It is evident from their relative energies and dihedral angle data in Table 4 that these are each others inverts.
Table 4 Optimised parameters of the series of confused porphyrin isomers. The dihedral angle is described in Fig. 1
| Compound |
Optimised dihedral angles |
Point Group |
Energy (a. u.) |
ΔE (kJ mol−1) |
|
|
θ
a
|
θ
b
|
θ
c
|
θ
d
|
|
|
|
|
NCP
|
10.04 |
−1.78 |
0.00 |
−3.61 |
C
1
|
−988.9960 |
|
|
trans-N2CP(1) |
−11.57 |
5.77 |
−11.57 |
5.77 |
C
2
|
−988.9668 |
0 |
|
trans-N2CP(2) |
10.12 |
4.66 |
−10.12 |
−4.66 |
C
i
|
−988.9620 |
12.71 |
|
cis-N2CP(1) |
−8.34 |
14.88 |
1.91 |
0.62 |
C
1
|
−988.9682 |
0 |
|
cis-N2CP(2) |
8.31 |
−14.87 |
−1.91 |
−0.62 |
C
1
|
−988.9682 |
2.31 × −4 |
|
NeoCP
|
0.29 |
−2.64 |
0.14 |
0.52 |
C
1
|
−988.9835 |
|
The OPA spectra for the N-confused isomers are displayed in Fig. 8 and Table 5. The classic GFO-related transitions can be seen for these compounds as well as the reduced isomers, in a clear dominant region of absorption in the 200–370 nm Soret-region. The absorption into the Q-region is significantly weaker than in the classic case however, with only two clear transitions at 505 nm and 560 nm for NeoCP and cis-N2CP(2) respectively. An NTO analysis of these compounds show the classic absorption amongst the Gouterman orbitals for all compounds, with the expected transitions amongst the GFOs in the Soret and Q-region. These are visualised in the NeoCP example in Fig. 9 by transition 4 and 1. There are also, however large contributions into the Soret-region by non-GFO transitions. Transition 8 show an orbital that can mainly be described as HOMO-2 to a GFO LUMO whilst transition 3 show main electron density in a LUMO + 2–like porphyrin orbital after the transition. This yet again highlights the fact that an orbital–based model must be used with caution when analysing spectra of electronic isomers of porphyrins.
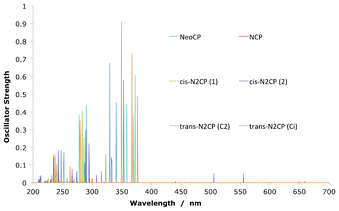 |
| | Fig. 8 One-photon absorption transitions for the set of confused porphyrins. All show absorption into the Soret- and the Q-band. | |
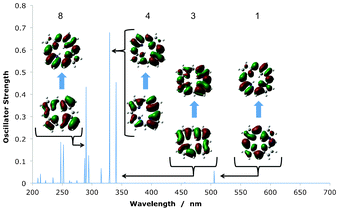 |
| | Fig. 9 Key transitions in the absorption spectra of NeoCP, as visualised using NTOs. Transition number 8 clearly show a transition amongst non-Gouterman frontier orbitals. | |
Table 5 Key transitions from the OPA calculations of the confused set of isomers, in terms of their wavelengths (λ) and oscillator strengths (f)
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
f
|
λ (nm) |
f
|
|
NCP
|
597 |
0.0023 |
369 |
0.5473 |
|
|
518 |
0.0014 |
349 |
0.9191 |
|
trans-N2CP(1) |
649 |
0.0176 |
372 |
0.6344 |
|
|
535 |
0.0015 |
358 |
0.8573 |
|
trans-N2CP(2) |
658 |
0.0151 |
376 |
0.6236 |
|
|
546 |
0.0027 |
366 |
0.7776 |
|
cis-N2CP(1) |
555 |
0.0583 |
353 |
0.6288 |
|
|
440 |
0.0088 |
332 |
0.2940 |
|
cis-N2CP(2) |
555 |
0.0582 |
353 |
0.6920 |
|
|
440 |
0.0088 |
332 |
0.2938 |
|
NeoCP
|
505 |
0.0548 |
340 |
0.4742 |
|
|
456 |
0.0037 |
329 |
0.6805 |
The TPA spectrum for the confused set of porphyrins are shown in Table 6 as well as Fig. 10, where the P signals have been added for comparison. As with the reduced isomers, one photon resonance arguments rationalises the fact that all compounds have little or no absorption into the Q-region, as there is no OPA signal near half of the TPA. Contrary to the reduced isomers, there is a negative effect on the δTPA in comparison with the parent P. Inspection of the OPA spectra would suggest that NeoCP and cis-N2CP(2) have potential for resonance enhanced TPA into the Soret-region, as they both exhibit relatively strong OPA into the Q-region. This turns out to influence the TPA of those two species, which do have clear TPA peaks. However, when comparing with P it becomes evident that confusion does not benefit the over-all TPA qualities of the compound. It is worth noting here though, that this does not exclude the use of a confused porphyrin derivative in a biological solvent. The results presented here, suggests that the newly discovered NeoCP and the cis-N2CP(2) are potential candidates. Indeed cis-N2CP, and especially its Cu and Ag derivatives, have already been shown to be promising in the development of photosensitizers for use in OPA PDT, and could therefore potentially be probed for TPA PDT development.36
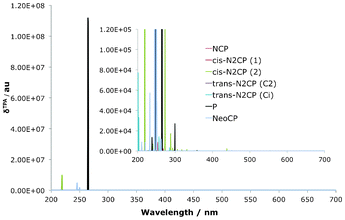 |
| | Fig. 10 TPA spectra for the confused isomers, with P added as a reference. Confusion have negative impact on the δTPA in comparison with the porphyrin parent. | |
Table 6 A selection of key transitions from the TPA calculations of the confused set of isomers. Wavelengths (λ) and δTPA.
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
δ (a. u.) |
λ (nm) |
δ (a. u.) |
|
NCP
|
594 |
1.47 × 101 |
268 |
4.02 × 103 |
|
|
517 |
3.61 × 101 |
253 |
1.27 × 102 |
|
trans-N2CP(1) |
|
|
285 |
1.57 × 103 |
|
|
|
|
2.57 |
1.39 × 104 |
|
trans-N2CP(2) |
|
|
284 |
4.03 × 103 |
|
|
|
|
256 |
1.41 × 104 |
|
cis-N2CP(1) |
554 |
1.89 × 102 |
288 |
2.77 × 104 |
|
|
438 |
2.49 × 103 |
272 |
1.33 × 105 |
|
cis-N2CP(2) |
554 |
1.89 × 102 |
288 |
2.75 × 104 |
|
|
438 |
2.49 × 103 |
272 |
1.38 × 105 |
|
NeoCP
|
500 |
2.32 × 102 |
249 |
3.51 × 106 |
|
|
451 |
1.32 × 103 |
245 |
7.49 × 106 |
3.3 Dioxa-porphycene isomers
Both the series of O2Pc and O2N7Pc optimised geometries are presented in Fig. 11 and Fig. 12, as well as in Table 7. The whole series optimised to planar geometries, with the trans-isomers being most stable (Note that trans here refers to the substitution site, as these do not have centre hydrogen atoms coordinated to the core).
 |
| | Fig. 11 The optimised geometries of the dioxa-porphycene series. | |
 |
| | Fig. 12 The optimised geometries of the dioxa-porphycene series, bearing a Nitrogen atom in the confused 7-position of the Pc macrocycle. | |
Table 7 Optimised parameters of the series of dioxa-porphycene isomers
| Compound |
Point Group |
Energy (a.u.) |
ΔE (kJ mol−1) |
|
trans-O2Pc |
C
2h
|
−1029.20223202 |
0 |
|
cis-O2Pc(1) |
C
2v
|
−1029.19462111 |
19.98 |
|
cis-O2Pc(2) |
C
2v
|
−1029.19689738 |
14.00 |
|
trans-O2N7Pc |
C
s
|
−1045.24981542 |
0 |
|
cis-O2N7Pc(1) |
C
1
|
−1045.24028475 |
25.02 |
|
cis-O2N7Pc(2) |
C
1
|
−1045.24367034 |
16.13 |
Table 8 Selected OPA calculation results for the core–substituted porphyrins, in terms of their wavelengths (λ) and oscillator strengths (f)
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
f
|
λ (nm) |
f
|
|
trans-O2Pc |
619 |
0.0900 |
317 |
0.9271 |
|
|
528 |
0.2414 |
303 |
1.3285 |
|
cis-O2Pc(1) |
570 |
0.0972 |
317 |
0.9901 |
|
|
514 |
0.1488 |
311 |
1.0832 |
|
cis-O2Pc(2) |
569 |
0.1006 |
321 |
1.0384 |
|
|
512 |
0.1525 |
605 |
1.1899 |
|
trans-O2N7Pc |
624 |
0.1087 |
316 |
0.8603 |
|
|
529 |
0.2353 |
294 |
1.0586 |
|
cis-O2N7Pc(1) |
558 |
0.1276 |
312 |
0.9819 |
|
|
500 |
0.1348 |
308 |
0.9810 |
|
cis-O2N7Pc(2) |
580 |
0.1100 |
321 |
0.9903 |
|
|
512 |
0.1647 |
297 |
0.8788 |
Table 9 A selection of key transitions from the TPA calculations on the core–substituted porphyrins. Wavelengths (λ) and transition strengths (δTPA)
| Compound |
Absorption |
|
|
Q-region |
Soret-region |
|
|
λ (nm) |
f
|
λ (nm) |
f
|
|
trans-O2Pc |
618 |
8.77 × 10−7 |
269 |
4.34 × 107 |
|
|
526 |
6.60 × 10−8 |
235 |
1.04 × 106 |
|
cis-O2Pc(1) |
569 |
1.72 × 103 |
265 |
3.48 × 105 |
|
|
513 |
4.06 × 102 |
249 |
2.32 × 105 |
|
cis-O2Pc(2) |
567 |
2.17 × 103 |
255 |
1.07 × 1011 |
|
|
510 |
4.59 × 102 |
254 |
6.28 × 102 |
|
trans-O2N7Pc |
621 |
3.19 × 100 |
268 |
1.50 × 108 |
|
|
528 |
3.26 × 101 |
246 |
1.25 × 105 |
|
cis-O2N7Pc(1) |
556 |
2.44 × 103 |
247 |
3.66 × 107 |
|
|
498 |
4.00 × 102 |
238 |
9.17 × 105 |
|
cis-O2N7Pc(2) |
577 |
2.07 × 103 |
297 |
2.27 × 106 |
|
|
510 |
5.73 × 102 |
252 |
5.81 × 108 |
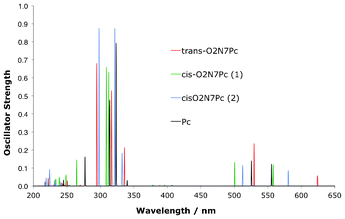
In Fig. 13 and Table 8 the OPA spectra of the O2Pc derivatives are seen to display the classic GFO transitions, as expected for Pc. The transitions overlap each other to a large degree across the dioxa-substitution pattern. There is however a slight but clear split of the main transitions into the Q-region with respect to the Pc macrocycle, as indicated in Fig. 13.
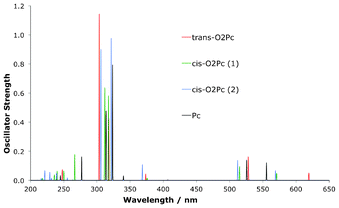 |
| | Fig. 13 Simulated spectra for OPA in dioxa-porphycene derivatives. | |
The OPA spectra for the O2N7Pc set of structures is presented in Fig. 14 and Table 8, and show a pattern analogous to the O2Pc derivatives. The absorption into both the Soret- and Q-transitions are reproduced, and again the main difference lies in the splitting pattern amongst the Q-region transitions in comparison to the Pc absorption signals.
The TPA spectra are presented in Fig. 15, Fig. 16 and Table 9. Clearly the very small difference in the Q-region splitting seen in the OPA spectrum has an very large impact on the TPA qualities of the molecules, as shown previously in the reduced set of isomers. A small shift moves the OPA Q-transition to a region where a TPA exists at twice the wavelength which means an effective strengthening of the signal through resonance enhancement. A similar TPA pattern can be seen for the O2N7Pc compounds, with a large δTPA for O2N7Pc(2) in comparison to the other derivatives (Fig. 16). Interestingly, the resonance effect is not nearly as pronounced for this series of compounds as for the O2Pc(2) set. In Fig. 17 the effect of resonance enhancement is presented in a purely state–based approach in which it can clearly be seen to be an effect of the position of a Q-region transition at near half the wavelength of the TPA transition.
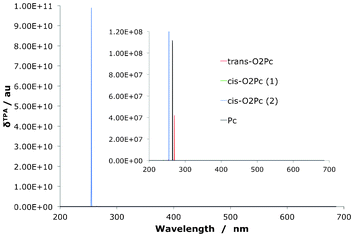 |
| | Fig. 15 The two-photon absorption spectrum for dioxa-porphycenes as well as the Pc core for comparison. The signal for the O2Pc(2) is several orders of magnitude larger than the related derivatives. | |
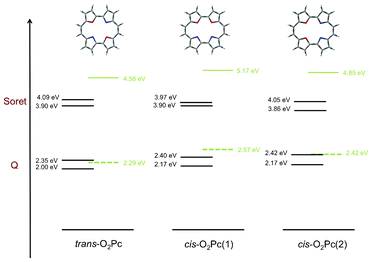 |
| | Fig. 17 This absorption scheme illustrates the concept of resonance enhancement in the series of O2Pc derivatives. Black horizontal lines are states available for OPA, clearly forming the Soret- and the Q-regions. Green lines are the dominant TPA transition, with the dashed line representing the virtual state. When the latter state can be represented in terms of a real state, resonance is expected to affect the TPA signal by greatly enhancing it. | |
4 Conclusion
In this work linear and quadratic density functional response theory was used to systematically study the effects of the core electronic structure of porphyrin isomers on their OPA and TPA characteristics. The OPA spectra reproduced the well known Gouterman pattern of absorption signals in the Soret- and Q-regions across the series of compounds. However, the use of natural transition orbitals to investigate the nature of the transition pointed to the fact that the Gouterman orbital picture might not be sufficient to describe these transitions fully. The GFO model was developed for the P macrocycle and was derived based on the degeneracy of the two LUMO orbitals. This degeneracy is successively being broken in derivative compounds and although the GFO model accurately describes some trends (such as the Q-transition strengthening in Pc relative to P) it also fails to account for some key transitions (such as the Soret–region contributions in NeoCP). There is therefore a need to also look specifically into the location of the states if one wants to rationalise the full absorption pattern in porphyrin isomers. Interestingly, it was shown that the TPA pattern was very much more sensitive to the changes in the electronic structure than previously expected. Confusion of the N-atoms in the porphyrin macrocycle did little to improve the TPA qualities whilst substitution of the pyrrolic Nitrogen atoms by Oxygens, as well as changing the Hydrogen coordination of the core, could lead to an incredible increase in the TPA transition strength. These large changes in the TPA spectra were not reproduced in the OPA spectra, where only small changes were seen. However, these small changes were sufficient to move the OPA signal to a region where it could contribute to one-photon resonance enhancement of the TPA signal, and thereby making the effect on the TPA more significant. This subtlety in the non-linear TPA means that a change in the core electronic structure of compounds is a potentially very powerful avenue, by which the TPA qualities of porphyrinic compounds can be fine-tuned, without the need for large structural changes to the extremities of the macrocycle. This also highlights the importance of computational techniques for use in structure-reactivity relationship studies, as a large number of compounds can be probed and unexpected absorption qualities such as the ones presented here can be identified.
Acknowledgements
This work was supported by the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013/ERC Grant agreement number: 258990).
References
- K. Szacilowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 105, 2647–2694 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS.
- H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032–4045 RSC.
- S. M. Costa, S. M. Andrade, D. M. Togashi, P. M. Paulo, C. A. Laia, M. Isabel Viseu and A. M. Gonalves da Silva, J. Porphyrins Phthalocyanines, 2009, 13, 509–517 CrossRef CAS.
- J. Barber, Nature, 1984, 307, 596 CrossRef.
- J. Arnbjerg, M. J. Paterson, C. B. Nielsen, M. Jrgensen, O. Christiansen and P. R. Ogilby, J. Phys. Chem. A, 2007, 111, 5756–5767 CrossRef CAS.
- P. C. Jha, B. Minaev and H. Agren, J. Chem. Phys., 2008, 128, 074302 CrossRef.
- E. Dahlstedt, H. A. Collins, M. Balaz, M. K. Kuimova, M. Khurana, B. C. Wilson, D. Phillips and H. L. Anderson, Org. Biomol. Chem., 2009, 7, 897–904 CAS.
- M. Drobizhev, F. Q. Meng, A. Rebane, Y. Stepanenko, E. Nickel and C. W. Spangler, J. Phys. Chem. B, 2006, 110, 9802–9814 CrossRef CAS.
- A. Karotki, M. Khurana, J. R. Lepock and B. C. Wilson, Photochem. Photobiol., 2006, 82, 443–452 CrossRef CAS.
- J. D. Baker and M. C. Zerner, Chem. Phys. Lett., 1990, 175, 192–196 CrossRef CAS.
- S. J. A. van Gisbergen, A. Rosa, G. Ricciardi and E. J. Baerends, J. Chem. Phys., 1999, 111, 2499–2506 CrossRef CAS.
- B. Minaev, Y.-H. Wang, C.-K. Wang, Y. Luo and H. Agren, Spectrochim. Acta, Part A, 2006, 65, 308–323 CrossRef.
- M. Drobizhev, Y. Stepanenko, Y. Dzenis, A. Karotki, A. Rebane, P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 2004, 126, 15352–15353 CrossRef CAS.
- B. Minaev and H. Agren, Chem. Phys., 2005, 315, 215–239 CrossRef CAS.
- D. Sundholm, Phys. Chem. Chem. Phys., 2000, 2, 2275–2281 RSC.
- O. Rubio-Pons, Y. Luo and H. Agren, J. Chem. Phys., 2006, 124, 094310 CrossRef.
- M. Albota, D. Beljonne, J.-L. Bredas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCorg-Maughon, J. W. Perry, H. Rockel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS.
- M. Drobizhev, Y. Stepanenko, Y. Dzenis, A. Karotki, A. Rebane, P. N. Taylor and H. L. Anderson, J. Phys. Chem. B, 2005, 109, 7223–7236 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS.
- P. C. Ray and Z. Sainudeen, J. Phys. Chem. A, 2006, 110, 12342–12347 CrossRef CAS.
- J. Shen, W.-D. Cheng, D.-S. Wu, Y.-Z. Lan, F.-F. Li, S.-P. Huang, H. Zhang and Y.-J. Gong, J. Phys. Chem. A, 2006, 110, 10330–10335 CrossRef CAS.
- M. Johnsen, M. J. Paterson, J. Arnbjerg, O. Christiansen, C. B. Nielsen, M. Jorgensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2008, 10, 1177–1191 RSC.
- L. T. Bergendahl and M. J. Paterson, Chem. Commun., 2012, 48, 1544–1546 RSC.
- G. Karthik, M. Sneha, V. P. Raja, J. M. Lim, D. Kim, A. Srinivasan and T. K. Chandrashekar, Chem. Eur. J., 2013 Search PubMed.
- H. Rath, J. Sankar, V. PrabhuRaja, T. K. Chandrashekar, A. Nag and D. Goswami, J. Am. Chem. Soc., 2005, 127, 11608–11609 CrossRef CAS.
- E. Vogel, M. Kocher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1986, 25, 257–259 CrossRef.
- J. L. Sessler, E. A. Brucker, S. J. Weghorn, M. Kisters, M. Schafer, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1994, 33, 2308–2312 CrossRef.
- J.-Y. Hasegawa, K. Takata, T. Miyahara, S. Neya, M. J. Frisch and H. Nakatsuji, J. Phys. Chem. A, 2005, 109, 3187–3200 CrossRef CAS.
- M. O. Senge, Angew. Chem., Int. Ed., 2011, 50, 4272–4277 CrossRef CAS.
- P. J. Chmielewski, L. Latos-Grazynski, K. Rachlewicz and T. Glowiak, Angew. Chem., Int. Ed. Engl., 1994, 33, 779–781 CrossRef.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS.
- A. Srinivasan and H. Furuta, Acc. Chem. Res., 2005, 38, 10–20 CrossRef CAS.
- H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2000, 122, 803–807 CrossRef CAS.
- T. D. Lash, A. D. Lammer and G. M. Ferrence, Angew. Chem., Int. Ed., 2011, 50, 9718–9721 CrossRef CAS.
- J. Almlof, T. H. Fischer, P. G. Gassman, A. Ghosh and M. Haeser, J. Phys. Chem., 1993, 97, 10964–10970 CrossRef CAS.
- A. Ghosh, Acc. Chem. Res., 1998, 31, 189–198 CrossRef CAS.
- R. Bast, U. Ekstrom, B. Gao, T. Helgaker, K. Ruud and A. J. Thorvaldsen, Phys. Chem. Chem. Phys., 2011, 13, 2627–2651 RSC.
- P. Norman, Phys. Chem. Chem. Phys., 2011, 13, 20519–20535 RSC.
- P. Salek, O. Vahtras, J. Guo, Y. Luo, T. Helgaker and H. Agren, Chem. Phys. Lett., 2003, 374, 446–452 CrossRef CAS.
- D. G. Hilmey, M. Abe, M. I. Nelen, C. E. Stilts, G. A. Baker, S. N. Baker, F. V. Bright, S. R. Davies, S. O. Gollnick, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2002, 45, 449–461 CrossRef CAS.
- J. Arnbjerg, A. Jimenez-Banzo, M. J. Paterson, S. Nonell, J. I. Borrell, O. Christiansen and P. R. Ogilby, J. Am. Chem. Soc., 2007, 129, 5188–5199 CrossRef CAS.
- L. T. Bergendahl and M. J. Paterson, J. Phys. Chem. B, 2012, 116, 11818–11828 CrossRef CAS.
- J. S. Lee, J. M. Lim, M. Toganoh, H. Furuta and D. Kim, Chem. Commun., 2010, 46, 285–287 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09 Revision A.2, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- DALTON, a molecular electronic structure program, Release 2.0 (2005), see http://daltonprogram.org/.
- M. J. Paterson, O. Christiansen, F. Pawlowski, P. Jorgensen, C. Hattig, T. Helgaker and P. Salek, J. Chem. Phys., 2006, 124, 054322 CrossRef.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- V. Sauri, L. Serrano-Andres, A. R. M. Shahi, L. Gagliardi, S. Vancoillie and K. Pierloot, J. Chem. Theory Comput., 2011, 7, 153–168 CrossRef CAS.
- L. Serrano-Andres, M. Merchan, M. Rubio and B. O. Roos, Chem. Phys. Lett., 1998, 295, 195–203 CrossRef CAS.
- Y. Tokita, J. Hasegawa and H. Nakatsuji, J. Phys. Chem. A, 1998, 102, 1843–1849 CrossRef CAS.
- S. R. Gwaltney and R.
J. Bartlett, J. Chem. Phys., 1998, 108, 6790–6798 CrossRef CAS.
- M. Gouterman, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef CAS.
- D. K. Maity, R. L. Bell and T. N. Truong, J. Am. Chem. Soc., 2000, 122, 897–906 CrossRef CAS.
- J. Baker, P. M. Kozlowski, A. A. Jarzecki and P. Pulay, Theor. Chem. Acc., 1997, 97, 59–66 CrossRef CAS.
- M. Schlabach, H. H. Limbach, E. Bunnenberg, A. Y. L. Shu, B. R. Tolf and C. Djerassi, J. Am. Chem. Soc., 1993, 115, 4554–4565 CrossRef CAS.
- A. L. Sobolewski, M. Gil, J. Dobkowski and J. Waluk, J. Phys. Chem. A, 2009, 113, 7714–7716 CrossRef CAS.
- Y.-D. Wu, K. W. K. Chan, C.-P. Yip, E. Vogel, D. A. Plattner and K. N. Houk, J. Org. Chem., 1997, 62, 9240–9250 CrossRef CAS.
- A. Ghosh and J. Almloef, J. Phys. Chem., 1995, 99, 1073–1075 CrossRef CAS.
- K. Kristensen, J. Kauczor, T. Kjaergaard and P. Jorgensen, J. Chem. Phys., 2009, 131, 044112 CrossRef.
- K. Kristensen, J. Kauczor, A. J. Thorvaldsen, P. Jorgensen, T. Kjaergaard and A. Rizzo, J. Chem. Phys., 2011, 134, 214104 CrossRef.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 CrossRef CAS.
- A. Painelli, L. D. Freo and F. Terenziani, Chem. Phys. Lett., 2001, 346, 470–478 CrossRef CAS.
- P. Macak, Y. Luo and H. Agren, Chem. Phys. Lett., 2000, 330, 447–456 CrossRef CAS.
- N. Lin, Y. Luo, K. Ruud, X. Zhao, F. Santoro and A. Rizzo, ChemPhysChem, 2011, 12, 3392–3403 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full cartesian coordinates of all optimised geometries. See DOI: 10.1039/c3ra40790j |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a