 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of surfactant alcohols with various chemical moieties at the hydrophilic–hydrophobic interface†
Pierre-Léonard
Zaffalon
a,
Vincenza
D'Anna
b,
Hans
Hagemann
*b and
Andreas
Zumbuehl
*ac
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest-Ansermet 30, 1211 Geneva, Switzerland
bDepartment of Physical Chemistry, University of Geneva, Quai Ernest-Ansermet 30, 1211 Geneva, Switzerland. E-mail: hans-rudolf.hagemann@unige.ch
cDepartment of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.zumbuehl@unifr.ch
First published on 21st March 2013
Abstract
The melting behavior, the solubility, and the influence of hydrogen bonds were analyzed for a series of single- and double-tailed surfactant alcohols. Various effects such as the presence of free amides or the intermolecular spacing were found to be important factors for increasing or decreasing the melting temperature of a surfactant. Furthermore, we present a model for the packing of diamido-lipids and study the temperature-dependence of the IR signals.
Introduction
Recently, we have reported on the synthesis of Pad–PC–Pad, a 1,3-diamidophospholipid with chemical modifications of the natural glycerol backbone: the ester linkages were replaced with amido groups and the natural 1,2-diacyl substitution pattern was changed to a 1,3-arrangement.1 The vesicles formulated from this lipid showed promising mechanosensitive properties: a dye-loaded vesicle did not release its cargo spontaneously but did so when it was exposed to shear-stress or simple shaking. Such vesicles might find interesting applications as drug delivery devices.2 During the synthesis of the phospholipid, we were confronted by the surprising near-insolubility of the free alcohol N,N′-(2-hydroxypropane-1,3-diyl)dipalmitamide (10). Intermolecular forces such as hydrogen bonding and van der Waals interactions between the hydrophobic chains are present in a balanced fashion and will be influenced by chemical modification of the hydrogen donor–acceptor moieties involved. The low solubility of this molecule might be induced by intermolecular hydrogen bonds and this hypothesis prompted us to have a closer look at various one- and two-tailed hydrocarbon surfactants with different end-group moieties.Here, we provide a concise list of single-tail and double-tail surfactants containing various chemical moieties at the hydrophilic–hydrophobic interface and study their melting behavior and their solubility in various solvents. In order to ease the comparison between molecules, we use the nomenclature that we proposed for artificial phospholipids.1 For single-tail surfactants the C16 or palmitic aliphatic chain is abbreviated by the capital letter P. This is followed by the functional group present, i.e., et for ether, es for ester, and ad for amide. For double-tail surfactants the positions of the glycerol backbone are numbered 1 through 3 following the Hirschmann notation and the aforementioned abbreviations for the tails and the chemical moieties are used.3 Therefore, N,N′-(2-hydroxypropane-1,3-diyl)dipalmitamide is now named Pad–OH–Pad (10), making the comparison between molecules straightforward.
The literature on single- or double-chain aliphatic ethers, esters, or amides at the hydrophobic–hydrophilic interface spans several decades and is therefore contradictory in itself. The situation is easiest for single-chain aliphatic amides where the amides are both hydrogen bond donors and acceptors: two molecules pack head-to-head and form a centrosymmetric amide pair, a hydrogen bonded dimer.4,5 The force attributed to these two palmitic amide hydrogen bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–NH) is estimated at 33 kJ mol−1.6 The additional free amide protons are expected to form hydrogen bonds to the neighboring pair of molecules.7 Each of these additional hydrogen bonds is contributing a stabilizing energy of approximately 19 kJ mol−1.7 It is interesting to note that the long-chain N-methyl amides “are peppery in taste, provoke an intense burning taste and numb the tongue”, making them candidates for local anaestetics.8
O⋯H–NH) is estimated at 33 kJ mol−1.6 The additional free amide protons are expected to form hydrogen bonds to the neighboring pair of molecules.7 Each of these additional hydrogen bonds is contributing a stabilizing energy of approximately 19 kJ mol−1.7 It is interesting to note that the long-chain N-methyl amides “are peppery in taste, provoke an intense burning taste and numb the tongue”, making them candidates for local anaestetics.8
1,3-Diglycerides have been suggested as drug delivery vectors.9 These lipids are symmetrical molecules with 2 aliphatic chains in the 1 and 3 positions and a free alcohol in position 2 of the glycerol backbone. Initially, the X-ray data were interpreted to show a head-to-head arrangement of the alcohols.10 In analogy to the crystal structure of the 1,3-diglyceride of 3-thiadodecanoic acid,11 the molecules are now thought to pack in an extended form.12 The 1,3-diamido lipid Pad–OH–Pad (10) has already been reported previously and its low solubility has been mentioned several times.9,13–15 However, a concise study of this phenomenon has been missing until now.
Experimental
General remarks
Starting compounds and solvents were purchased from Sigma-Aldrich/Fluka or Acros and were used without further purification. Column chromatographic separations were carried out using 230–400 mesh silica gel. TLC plates were developed with KMnO4. 1H and 13C NMR spectra were recorded (as indicated) on either a Bruker 300 MHz, 400 MHz or 500 MHz spectrometer and are reported as chemical shifts (δ) in ppm relative to TMS (δ = 0). Spin multiplicities are reported as a singlet (s), doublet (d), or triplet (t) with coupling constants (J) given in Hz, or multiplet (m). Broad peaks are marked as br. HRESI-MS were performed on a QSTAR Pulsar (AB/MDS Sciex) spectrometer and are reported as mass-per-charge ratio m/z. IR spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrometer (ATR, Golden Gate). Additional IR spectra were performed using a Biorad Excalibur Instrument equipped with high and low temperature Specac Golden Gate ATR setups. DSC traces were obtained with a Mettler Toledo DSC1 Star Systems differential scanning calorimeter from 3 to 5 mg samples (5 °C min−1, under N2).Synthesis of Pet–CH3 (4)
The synthesis was improved from an existing procedure.16 1-Bromohexadecane (1.00 g, 3.28 mmol) was dissolved in 18 mL of dry MeOH containing cesium carbonate (1.04 g, 3.28 mmol). The solution was put under microwave radiation at 150 °C for 1 h. 50 mL of CH2Cl2 was added and the mixture was filtered in order to remove any insoluble salts. Then the organic solvents were evaporated under reduced pressure and the compound was purified by column chromatography (pentane then pentane–CH2Cl2 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The product was obtained as a colorless oil (683 mg, 2.66 mmol, 81%). Rf = 0.5 (pentane–CH2Cl2 3:1). 1H NMR (300 MHz, CDCl3): δ 3.39–3.19 (m, 5H), 1.59–1.43 (m, 2H), 1.22 (s, 26H), 0.84 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 72.85, 58.29, 31.96, 29.75, 29.66, 29.55, 29.42, 26.18, 22.68, 13.99. Mp: 19–21 °C.
:1). The product was obtained as a colorless oil (683 mg, 2.66 mmol, 81%). Rf = 0.5 (pentane–CH2Cl2 3:1). 1H NMR (300 MHz, CDCl3): δ 3.39–3.19 (m, 5H), 1.59–1.43 (m, 2H), 1.22 (s, 26H), 0.84 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 72.85, 58.29, 31.96, 29.75, 29.66, 29.55, 29.42, 26.18, 22.68, 13.99. Mp: 19–21 °C.
Synthesis of Pes–OH–Pes (9)
2-(Benzyloxy)propane-1,3-diyl dipalmitate (275 mg, 0.417 mmol) was dissolved in 8 mL of THF and Pd/C 10% was added (180 mg, 0.152 mmol). Hydrogen was injected into the system with Parr apparatus and the solution was stirred at 50 psi overnight. The catalyst was filtered with celite and the product was purified by silica gel column chromatography (pentane–ethyl acetate 9:1) to give the product as a white solid (175 mg, 0.308 mmol, 74%). Rf = 0.18 (pentane–ethyl acetate 9:1). 1H NMR (400 MHz, CDCl3): δ 4.20–4.11 (m, 5 H), 2.41 (s, 1 H), 2.34 (t, J = 7.3 Hz, 2 H), 1.63–1.61 (m, 4 H), 1.25 (s, 48 H), 0.88 (t, J = 8.0 Hz, 6 H). HRMS (ESI) m/z calcd for C37H71O7 [M + MeCOO−]− 627.5205, found 627.5205.
Synthesis of Pad–OH–Pad (10)
The synthesis was performed similar to a literature procedure.13 1,3-Diamino-2-propanol (385 mg, 4.19 mmol) was dissolved in a mixture of CH2Cl2 (18 mL), toluene (7 mL) and NaOH (625 mg, 15.6 mmol) in H2O (13 mL). At 20 °C, palmitoyl chloride (2.62 mL, 8.57 mmol) was added in one step. After stirring for 3 h, the precipitate was washed with 100 mL of diethyl ether (100 mL), H2O (100 mL) and methanol (100 mL). 2.00 g, 3.53 mmol, 84%) of a white powder were obtained. 1H NMR (500 MHz, CDCl3): δ 6.13 (s, 2H), 3.82 (s, 1H), 3.76 (q, J = 5.0 Hz, 1H), 3.44–3.36 (m, 2H), 3.27 (dd, J = 20.1, 5.2 Hz, 2H), 2.43–1.86 (m, 4H), 1.63 (q, J = 7.1 Hz, 4H), 1.27 (s, 48H), 0.91–0.87 (m, 6H). IR (cm−1): 3294, 3100, 2916, 2849, 1640, 1565, 1466, 1439, 1376, 1268, 1250, 1230, 1116, 1086, 843, 721, 634. HRMS (ESI) m/z calcd for C35H71N2O3 [M + H]+ 567.5459, found 567.5456. Mp: 132–134 °C.Synthesis of cyclo-Pad–OH–Pad (11)
Hexahydropyrimidin-5-ol (18) (402 mg, 3.94 mmol) was dissolved in a mixture of CH2Cl2 (18 mL), toluene (7 mL), NaOH (693 mg, 17.3 mmol) in H2O (13 mL). At 20 °C, palmitoyl chloride (2.4 mL, 7.9 mmol) was added in one step. After stirring for 3 h, the solution was extracted with saturated NaHCO3 (50 mL) and washed twice with 50 mL of CH2Cl2 (50 mL). The organic phases were dried over MgSO4 and the solvents were removed under reduced pressure. The crude material was purified by silica gel column chromatography using a gradient of CH2Cl2 then CH2Cl2–ethyl acetate 1:1. Cyclo-Pad–OH–Pad (11) was isolated as a white powder (935 mg, 1.62 mmol, 41%). Rf = 0.5 (CH2Cl2–ethyl acetate 1:1). 1H NMR (500 MHz, CDCl3): δ 5.69 (d, J = 13.1 Hz, 1H), 4.54 (d, J = 13.1 Hz, 1H), 4.16 (dd, J = 13.6, 3.4 Hz, 1H), 3.95–3.85 (m, 1H), 3.73 (dd, J = 13.8, 4.1 Hz, 1H), 3.54 (d, J = 13.6 Hz, 1H), 3.46 (d, J = 13.5 Hz, 1H), 2.67–2.50 (m, 2H), 2.33–2.39 (m, 2H), 1.66 (d, J = 46.1 Hz, 9H), 1.26 (s, 49H), 0.89 (t, J = 6.8 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 174.35, 172.91, 64.20, 55.91, 51.01, 47.83, 33.16, 32.91, 31.94, 29.71, 29.38, 25.29, 24.95, 22.71, 14.21. HRMS (ESI) m/z calcd for C36H71N2O3 [M + H]+ 579.5459, found 579.5466. IR (cm−1): 3414, 2918, 2850, 1658, 1619, 1468, 1255, 1145, 885, 722. Mp: 84–86 °C.
Synthesis of Pad–OTIPS–Pad (12)
To Pad–OH–Pad (440 mg, 0.78 mmol) and NEt3 (430 μL, 3.10 mmol) in CH2Cl2 (12 mL) was added TIPS-triflate (470 μL, 1.75 mmol) and the mixture was stirred for 20 h at 20 °C. Then the mixture was extracted with saturated NaHCO3 (50 mL) and washed 2 times with CH2Cl2 (2 × 50 mL). The organic phases were dried over MgSO4 and removed under reduced pressure. The crude material was purified by silica gel column chromatography using a solvent gradient (pentane–EtOAc 9:1 then 4:1) to give the product as a white powder (480 mg, 0.66 mmol, 94%). Rf = 0.5 (pentane–ethyl acetate 4:1). 1H NMR (400 MHz, CDCl3): δ 6.23 (dd, J = 8.6, 3.8 Hz, 2H), 3.94 (tt, J = 6.6, 3.3 Hz, 1H), 3.88–3.77 (m, 1H), 2.71 (ddd, J = 13.7, 6.6, 4.2 Hz, 2H), 2.31–2.11 (m, 3H), 1.73–1.53 (m, 4H), 1.26 (d, J = 15.9 Hz, 43H), 1.07 (d, J = 4.7 Hz, 19H), 0.87 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 174.18, 69.03, 41.44, 36.88, 31.98, 29.72, 29.55, 29.40, 25.80, 22.74, 18.09, 14.17, 12.28. IR (cm−1): 3303, 2923, 2854, 1650, 1549, 1465, 1380, 1225, 1118, 1012, 883, 734, 679. HRMS (ESI) m/z calcd for C35H71N2O3 [M + H]+ 723.6793, found 723.6791. Mp: 51–53 °C.
Synthesis of hexahydropyrimidin-5-ol (17)
The synthesis was performed according to a literature procedure.17 A solution of 1,3-diaminopropan-2-ol (1.00 g, 11.1 mmol) and paraformaldehyde (0.31 g, 10.4 mmol) in methanol (17 mL) was heated under reflux for 48 h. Then the solvent was removed under reduced pressure. The resulting solid residue was washed with THF to afford a yellowish crystalline solid (1.05 g, 10.3 mmol, 93%). 1H NMR (300 MHz, D2O): δ 3.68 (d, J = 13.0 Hz, 1H), 3.61 (dd, J = 7.2, 3.5 Hz, 1H), 3.50 (d, J = 12.9 Hz, 1H), 3.11 (dd, J = 13.4, 3.2 Hz, 2H), 2.64 (dd, J = 13.3, 7.4 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 63.78, 58.94, 49.26.Synthesis of 2-(benzyloxy)propane-1,3-diyl diparmitate (19)
Palmitoyl chloride (3.74 mL, 12.0 mmol) was added dropwise to 2-(benzyloxy)propane-1,3-diol (18, 960 mg, 5.22 mmol) and pyridine (1.10 mL, 13.6 mmol) in 20 mL of CH2Cl2 at 0 °C. Then the solution was stirred for 20 h at rt under a N2 atmosphere. After addition of H2O (20 mL), the solution was stirred for 5 min. Extraction was achieved with the addition of aqueous HCl 1 M (40 mL). The aqueous phase was washed with CH2Cl2 (50 mL). The organic phases were washed with NaHCO3 10% (50 mL). The aqueous phase was washed again with CH2Cl2 (50 mL) and all the organic phases were dried with MgSO4. The crude material was purified by silica gel column chromatography (CH2Cl2) to give 2-(benzyloxy)propane-1,3-diyl dipalmitate as a white solid (3.42 g, 5.19 mmol, 99%). Rf = 0.5 (CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.66 (d, J = 11.5 Hz, 1H), 4.56 (d, J = 9.0 Hz, 1H), 4.27–4.03 (m, 1H), 3.90 (s, 1H), 3.73 (d, J = 8.5 Hz, 1H), 3.68–3.38 (m, 2H), 2.73–2.48 (m, 2H), 2.36 (d, J = 23.3 Hz, 3H), 1.62 (s, 5H), 1.26 (s, 48H), 1.02–0.76 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 173.56, 137.84, 128.44, 127.82, 74.50, 72.12, 63.02, 34.20, 31.96, 29.73, 29.50, 24.93, 22.73, 14.15. FTIR (cm−1): 2923, 2853, 1741, 1458, 1378, 1167, 1115, 1053, 735, 697. HRMS (ESI) m/z calcd for C42H78NO5 [M + NH4]+ 676.5874, found 676.5892.Results and discussion
Synthesis
Several C16-surfactants have already been reported in the literature. However, in order to complete the picture and to be able to draw meaningful conclusions, we have synthesized additional new compounds.Cyclo-Pad–OH–Pad (11) was available through a double amide formation between pyrimidin-5-ol (17) and the corresponding palmitoyl chloride (see Scheme 1). Pyrimidin-5-ol (17) itself was made beginning from the commercially available 1,3-diaminopropanol (16) via a condensation reaction using paraformaldehyde.
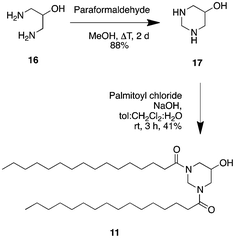 | ||
| Scheme 1 Synthesis of cyclo-Pad–OH–Pad (11). | ||
Again, starting from 1,3-diaminopropanol (16), Pad–OH–Pad (10) was made via a double amide formation using palmitoyl chloride (see Scheme 2). A protection with triisopropyl triflate led to Pad–OTIPS–Pad (12).
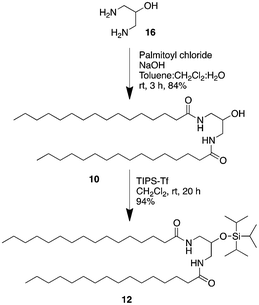 | ||
| Scheme 2 Synthesis of Pad–OTIPS–Pad (12). | ||
We have also included an improved synthesis of Pes–OH–Pes (9, see Scheme 3):18 starting from 2-(benzyloxy)propane-1,3-diol, a double esterification with two equivalents of palmitoyl chloride led to the benzyl-protected Pes–OBn–Pes (19) that was readily deprotected with H2 over Pd to give Pes–OH–Pes (9).
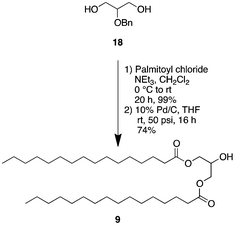 | ||
| Scheme 3 Synthesis of Pes–OH–Pes (9). | ||
Single-tail surfactants
First, we focused on a series of single-tail surfactants containing 16 carbons and various chemical motifs at the hydrophilic end of the molecule (see Table 1). A clear correlation between the melting point of the pure surfactant and its propensity to form hydrogen bonds was found: as expected, an amide functionality (capable of both accepting and donating H-bonds) would increase the melting temperature of a molecule compared to molecules containing chemical moieties which would form weaker H-bonds such as esters and ethers. Here, the amide Pad–H (3) melted 53 K higher than the ether Pet–H (1) and 42 K higher than the acid Pes–H (2).
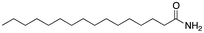

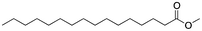
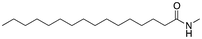
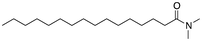
The introduction of a methyl end group on the hydrophilic side of the molecule would be expected to reduce a tight packing of the surfactants and thus a lower melting point was expected when going from Pet–H (1) to Pet–CH3 (4). Indeed, a reduction by 30 K was found. The same 30 K reduction was also found between Pes–H (2) and Pes–CH3 (5), where the inhibition of the hydrogen bond donation moiety apparently only had a minor effect on the melting temperature. The mono-methylation of Pad–H (3) to Pad–CH3 (6) only led to a reduction by 7 K meaning that neither the packing nor the hydrogen bonding was significantly reduced (see Fig. 1). On the other hand, the fully methylated Pad–(CH3)2 (7) melted at a temperature that was 58 K lower than Pad–H (3) and both the disruption of hydrogen bonding and molecular packing became evident.
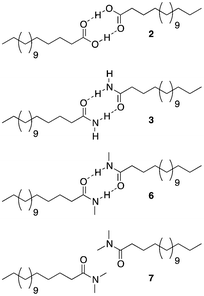 | ||
| Fig. 1 Increasing intermolecular hydrogen bonding interactions leads to higher melting temperatures (2 to 3). The effect is reversed by blocking the hydrogen bond donating protons on the amide nitrogens (3 to 6 to 7). | ||
Double-tail surfactants
We next analyzed a series of double-tail surfactants (see Table 2).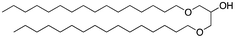
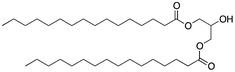
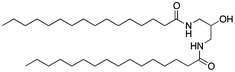
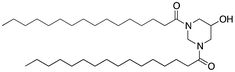
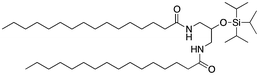
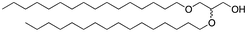
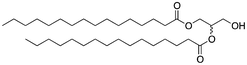
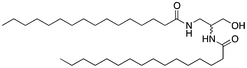
Here, an ether-based backbone moiety would be expected to lead to the lowest melting point of all the listed molecules due to the absence of hydrogen bonding between molecules. A slightly higher melting point would be reached for the more hydrogen bonding prone esters and the highest melting point would be expected for amides. Indeed, Pet–OH–Pet (8) does melt at 16.8 K lower than its ester analog Pes–OH–Pes (9) and a full 76.8 K lower than the amide (10).
If an amide such as Pad–OH–Pad (10) would form hydrogen bonds, the substitution of the amide proton by an alkyl group would significantly reduce this effect. We have therefore synthesized cyclo-Pad–OH–Pad (11), a molecule which has both amides bridged by a methylene group. Indeed this molecule (11) shows a large reduction of the melting point by 46 K compared to Pad–OH–Pad (10). We hypothesize that this effect is mainly due to the presence or absence of intermolecular hydrogen bonding interactions.
The introduction of a bulky head-group will create a large steric barrier and will force the surfactant molecules to increase their intermolecular distance (see Fig. 2). This will reduce the contribution of the hydrogen bonding and overrule this effect of the chemical group linking together the backbone and the alkyl chains. Indeed, Pad–OTIPS–Pad (12) shows a low melting point at the same level as Pet–OH–Pet (8), a surfactant that cannot form intermolecular hydrogen bonds.
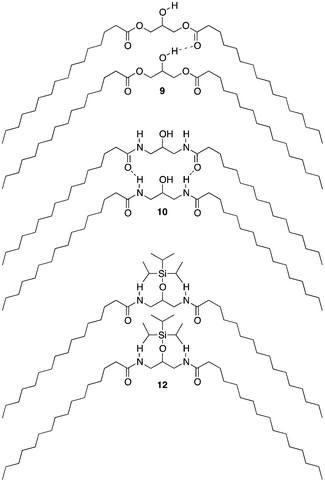 | ||
| Fig. 2 Increasing hydrogen bonding from esters (9) to amides (10) leads to increased melting temperatures. Weakening the intermolecular forces by spacing apart the molecules (12) leads to a drastic drop in melting temperature. | ||
A change from a 1,3-binding moiety (as found e.g. in Pet–OH–Pet (8)) to a more natural 1,2-binding moiety (e.g. in the 1,2-diether lipid Pet–Pet–OH (13)) will lead to a rearrangement of the hydrophilic portion of the crystal packing. However, this effect apparently has only a minor influence on the melting point (54 °C for Pet–OH–Pet (8) vs. 58 °C for Pet–Pet–OH (13)) and does not significantly change the overall stability of the crystal packing.
A second, more pronounced effect can be seen when hydrogen bonds are involved. The case is analogous to phospholipids: 1,2-phospholipids were found to have the glycerol backbone organized almost parallel to the membrane normal.26 The acyl chain at sn-1 extends the zig-zag conformation of the backbone and immerses right into the membrane. The acyl chain at sn-2 first organizes parallel to the membrane surface and only turns down into the membrane at C2. A 1,3-phospholipid is organized more symmetrically and will have both acyl chains extending out parallel to the membrane surface and bending down into the membrane at the C2 position.26 Analogously, this will organize the esters of Pes–OH–Pes (9) and the amides of Pad–OH–Pad (10) at the same vertical level and allow a good preorganization for the formation of hydrogen bonds. A 1,2-backbone moiety will place the esters of Pes–Pes–OH (13) and the amides of Pad–Pad–OH (14) on different vertical levels and the preorganization for hydrogen bonding is less optimal. Although Pes–Pes–OH (13) and Pad–Pad–OH (14) show a hydrogen bonding effect, it is less pronounced than in Pes–OH–Pes (9) or Pad–OH–Pad (10) as can be seen by the lowered melting temperatures (62 °C vs. 72 °C and 110 °C vs. 132 °C, respectively).
It is interesting to note that going from a single-tail to a double-tail surfactant, the melting temperature should significantly increase. Indeed, for all the reasonable comparisons (e.g.4 and 8; 5 and 9; 6 and 10; 7 and 11), an increase of >30 K in the melting temperatures can be found. The proximity of two H-bond forming moieties in the same molecule (e.g.10) does not lead to a larger increase of the melting temperature. Apparently, the proximity effect between the two tails in a double-tail surfactant is mainly influenced by van der Waals forces.
Solubility studies
The presence of hydrogen bonds might be at the origin of the high melting temperatures measured for the diamido phospholipids. In parallel, such an intermolecular hydrogen bonding network might also reduce the solubility of the molecule. Both Pad–OH–Pad (10) and Pes–OH–Pes (9) are expected to form intermolecular hydrogen bonds and both molecules show a reduced solubility in organic solvents (see Table 3). In the case of cyclo-Pad–OH–Pad (11) and Pad–OTIPS–Pad (12) the propensity to form intermolecular hydrogen bonds is reduced due to the methylation of the amide nitrogen (11) or due to the increased intermolecular spacing (12). Both molecules show a high solubility in organic solvents. The solubility of Pad–OH–Pad (10) in organic solvents is temperature dependent as can be seen in Table 4.| Pes–OH–Pes (9) | Pad–OH–Pad (10) | Cyclo-Pad–OH–Pad (11) | Pad–OTIPS–Pad (12) | ||
|---|---|---|---|---|---|
| Solubility [mg mL−1] | THF | 50 | <0.8 | >100 | >100 |
| CHCl3 | 50 | <0.8 | >100 | >100 | |
| CH2Cl2 | 35 | <0.8 | >100 | >100 | |
| Solubility [mM] | THF | 88 | <1.4 | >173 | >138 |
| CHCl3 | 88 | <1.4 | >173 | >138 | |
| CH2Cl2 | 62 | <1.4 | >173 | >138 | |
| Solubility [mg mL−1] | |||
|---|---|---|---|
| THF | CHCl3 | CH2Cl2 | |
| 5 °C | 0.40 | 0.66 | 0.49 |
| 23 °C | 0.53 | 0.85 | 0.73 |
Differential scanning calorimetry measurements
Fig. 3 shows the DSC trace of Pet–OH–Pet (8) and Pad–OH–Pad (10). The ether lipid shows a single peak at 60.5 °C corresponding to the melting of the chains at a high transition enthalpy of ΔH = 126.9 kJ mol−1 ± 2.13 kJ mol−1. However Pad–OH–Pad shows two peaks: one for the chain melting at 134.8 °C (ΔH = 14.05 kJ mol−1 ± 1.06 kJ mol−1) and a broader peak at 119.2 °C (ΔH = 71.45 kJ mol−1 ± 1.83 kJ mol−1). This finding is consistent with DSC studies performed for the asymmetric 1,2-diamido-phospholipid Mad–Mad–PC where a similar broad peak was attributed to the breaking of the hydrogen bond region.27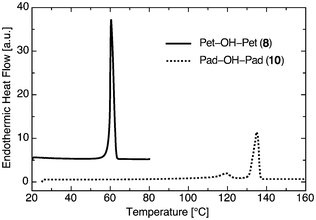 | ||
| Fig. 3 Differential scanning calorimetry scans of Pet–OH–Pet (8, solid line) and Pad–OH–Pad (10, dashed line). | ||
If Pad–OH–Pad (10) were equipped with a phospholipid head group such as phosphocholine, it would lead to the 1,3-diamido phospholipid Pad–PC–Pad. This substitution has a profound effect on the packing geometry and is expected to significantly reduce Tm, which is found with the Tm of 35 °C at a transition enthalpy of ΔH = 30.38 kJ mol−1 (in a vesicle bilayer and a U-shaped morphology).2
Infrared studies
The range of infrared signals between 3000 cm−1 and 3500 cm−1 can be used as a convenient means for detecting intramolecular hydrogen bonds. In order to study the influence of increasing temperature on hydrogen bonds, we analyzed a solid sample of Pad–OH–Pad (10, see Fig. 4). The spectra show two hydrogen bond related stretch vibrations at 3100 cm−1 (N–H) and at 3300 cm−1 (O–H). The position of the O–H band remains practically constant between 24 °C and 100 °C, then the position increases by about 10 cm−1 at 110–120 °C and increases further above 130 °C.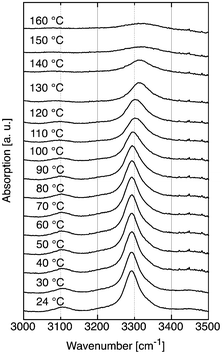 | ||
| Fig. 4 Temperature dependent IR study of the OH and NH stretch vibration region of Pad–OH–Pad (10). | ||
A significant line broadening is also observed above 130 °C. These changes correlate with the transitions observed by DSC (see Fig. 3). In the case of the IR data, the changes appear at a somewhat lower temperature, but in this case, the temperature was increased at a much slower rate (20 min for a 10 °C change) compared to the DSC experiments (5 °C min−1). This hints at the possibility that an intermolecular O⋯H–O hydrogen bond is present between the secondary alcohol and an amide carbonyl group of a neighboring molecule. This interaction seems to be a determining feature both of the structure of Pad–OH–Pad and its thermal behavior. The influence of the second hydrogen bond between an amide N–H and the carbonyl group of a second amide is clearly inferior to the first hydrogen bond as this stretching vibration is virtually absent at 130 °C .
Similarly, cyclo-Pad–OH–Pad (11) was analyzed (see Fig. 5). As in the case of Pad–OH–Pad (10), a strong hydrogen bond exists between the free secondary alcohol and the carbonyl group of an amide. This bond disappears above 80 °C, which is in perfect agreement with the melting temperature of cyclo-Pad–OH–Pad (84–86 °C). In contrast to Pad–OH–Pad, no amide N–H stretch vibration was found nor expected. This finding corroborates our analysis that hydrogen bonds are an important factor in determining the melting characteristics of the double-tail surfactants.
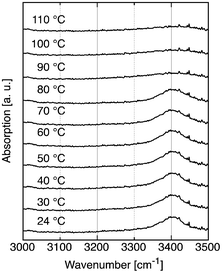 | ||
| Fig. 5 Temperature dependent IR study of the OH stretch vibration region of cyclo-Pad–OH–Pad (11). | ||
At low temperatures, the CH2 bending mode (at ca. 1460 cm−1) as well as the CH2 scissoring mode (at 720–730 cm−1) reveal a splitting which is characteristic for the chain packing found in the orthorhombic phase of the odd carbon containing n-alkanes such as C21H44.28,29 At 120 °C (above the first phase transition), this splitting disappears (see Fig. 6). This behavior is similar to the one observed in alkanes at the orthorhombic to hexagonal phase transition.28
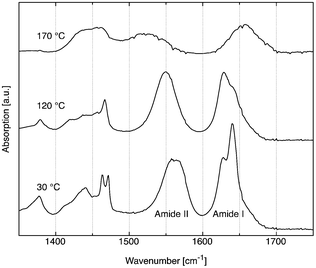 | ||
| Fig. 6 Comparison of the IR spectra of Pad–OH–Pad (10) at 30 °C, 120 °C, and 170 °C. | ||
Simultaneously, the amide I and, in a more pronounced way, the amide II bands [e.g.ref. 30] are changed. It has been shown31 that the relative intensity of the two components of the amide I band around 1640 cm−1 is related to the backbone conformation close to the amide groups. These observations show that at this phase transition, the packing is somewhat loosened not only from the inter-chain contacts, but also from some torsion of the peptide backbone.
Above the melting point, both amide bands are shifted and broadened significantly, and the CH2 bending mode has become much weaker and broader, hinting at conformational disorder of the alkane chain.29
Conclusions
In this article we have analyzed the melting and solubility properties of several single- and double-tailed surfactants containing various chemical functionalities between the hydrophobic and hydrophilic portions of the molecules. The presence of intermolecular hydrogen bonds between the free alcohol and the carbonyl oxygen of a neighboring molecule is suggested to have a major influence on the melting behaviour of the substances. Furthermore, we show that diamido-lipids are capable of forming an intermolecular hydrogen bonding network. The combination of alcohol–amide carbonyl as well as amide–amide hydrogen bonds is hypothesized to lead to the very low solubility of Pad–OH–Pad (10).Acknowledgements
The work was supported by the Swiss National Science Foundation (200020_132035), COST Action D43, and the NCCR in Chemical Biology. We thank the Mass Spectroscopy and NMR facilities at the University of Geneva for analytical services. We thank Jiri Mareda for fruitful discussions and helpful comments.Notes and references
- I. A. Fedotenko, P.-L. Zaffalon, F. Favarger and A. Zumbuehl, Tetrahedron Lett., 2010, 51, 5382 CrossRef CAS.
- M. Holme, I. A. Fedotenko, D. Abegg, J. Althaus, L. Babel, F. Favarger, R. Reiter, R. Tanasescu, P.-L. Zaffalon, A. Ziegler, B. Müller, T. Saxer and A. Zumbuehl, Nat. Nanotechnol., 2012, 7, 536 CrossRef CAS.
- H. Hirschmann, J. Biol. Chem., 1959, 235, 2762 Search PubMed.
- L. Leiserowitz and G. M. J. Schmidt, J. Chem. Soc. A, 1969, 2372 RSC.
- L. Abate, E. Badea, I. Blanco and G. D. Gatta, J. Chem. Eng. Data, 2008, 53, 959 CrossRef CAS.
- M. Davies and A. H. Jones, Trans. Faraday Soc., 1959, 55, 1329 RSC.
- M. Davies, A. H. Jones and G. H. Thomas, Trans. Faraday Soc., 1959, 55, 1100 RSC.
- G. F. D'Alelio and E. E. Reid, J. Am. Chem. Soc., 1937, 59, 109 CrossRef CAS.
- F. Mergen, D. M. Lambert, J. H. Poupaert, A. Bidaine and P. Dumont, Chem. Phys. Lipids, 1991, 59, 267 CrossRef CAS.
- T. Malkin, M. F. El Shurbagy and M. L. Meara, J. Chem. Soc., 1937, 1409 RSC.
- K. Larsson, Acta Crystallogr., 1963, 16, 741 CrossRef CAS.
- E. Mushayakarara and I. W. Levin, J. Phys. Chem., 1982, 86, 2324 CrossRef CAS.
- L. Elkihel, P. M. Loiseau, J. Bourass, P. Gayral and Y. Letourneux, Arzneim.-Forsch., 1994, 44, 1259 CAS.
- A. D. Morris, G. Atassi, N. Guilbaud and A. A. Cordi, Eur. J. Med. Chem., 1997, 32, 343 CrossRef CAS.
- H. Liu, Z. Zhu, H. Kang, Y. Wu, K. Sefan and W. Tan, Chem.–Eur. J., 2010, 16, 3791 CrossRef CAS.
- P. E. Sonnet, Synth. Commun., 1976, 6, 21 CrossRef CAS.
- T. Axenrod, J. Sun, K. K. Das, P. R. Dave, F. Forohar, M. Kaselj, N. J. Trivedi, R. D. Gilardi and J. L. Flippen-Anderson, J. Org. Chem., 2000, 65, 1200 CrossRef CAS.
- P.-L. Zaffalon and A. Zumbuehl, Synthesis, 2011, 778 CAS.
- S. Vandevoorde, K.-O. Jonsson, C. J. Fowler and D. M. Lambert, J. Med. Chem., 2003, 46, 1440 CrossRef CAS.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Chem. Commun., 2007, 624 Search PubMed.
- T. Li, J. Hamdi and M. F. Hawthorne, Bioconjugate Chem., 2006, 17, 15 CrossRef CAS.
- R. Darteil, K. C. Bertrand and J. Najib, FR 2850650 A1, 2004.
- J.-D. Lee, M. Ueno, Y. Miyajima and H. Nakamura, Org. Lett., 2007, 9, 323 CrossRef CAS.
- G. Guanti, L. Banfi, A. Basso, E. Bevilacqua, L. Bondanza and R. Riva, Tetrahedron: Asymmetry, 2004, 15, 2889 CrossRef CAS.
- C. Jia and A. H. Haines, J. Chem. Soc., Perkin Trans. 1, 1993, 2521 RSC.
- J. Seelig, R. Dijkman and G. H. De Haas, Biochemistry, 1980, 19, 2215 CrossRef CAS.
- J. Sunamoto, M. Goto, K. Iwamoto, H. Kondo and T. Sato, Biochim. Biophys. Acta, Biomembr., 1990, 1024, 209 CrossRef CAS.
- H. L. Casal, H. H. Mantsch, D. G. Cameron and R. G. Snyder, J. Chem. Phys., 1982, 77, 2825 CrossRef CAS.
- H. Hagemann, H. L. Strauss and R. G. Snyder, Macromolecules, 1987, 20, 2810 CrossRef CAS.
- P. Kupser, K. Pagel, J. Oomens, N. Polfer, B. Koksch, G. Meijer and G. von Helden, J. Am. Chem. Soc., 2010, 132, 2085 CrossRef CAS.
- J. Grdadolnik, S. G. Grdadolnik and F. Avbelj, J. Phys. Chem. B, 2008, 112, 2712 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra40704g |
| This journal is © The Royal Society of Chemistry 2013 |