 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycogen and gold nanoparticle bioconjugates: controlled plasmon resonance via glycogen-induced nanoparticle aggregation†
Dušan K.
Božanić
a,
Adriaan S.
Luyt
b,
Lidija V.
Trandafilović
a and
Vladimir
Djoković
*a
aVinča Institute of Nuclear Sciences, University of Belgrade, P.O. Box 522, 11001 Belgrade, Serbia. E-mail: djokovic@vinca.rs
bDepartment of Chemistry, University of the Free State (Qwaqwa campus), Private Bag X13, Phuthaditjhaba 9866, South Africa
First published on 11th April 2013
Abstract
Hybrid nanosystems composed of glycogen biopolymers and gold nanoparticles were prepared by an in situ non-toxic synthetic procedure. Transmission electron microscopy (TEM) and various spectroscopic methods (optical absorption, X-ray photoelectron and photoluminescence spectroscopy) were used for the characterization of the obtained bioconjugates. The gold nanoparticles formed in the presence of glycogen were spherical in shape and approximately 15 nm in diameter. The TEM micrographs show that the nanoparticles aggregated on the surface of the glycogen biomolecules. This effect induced a shift of the surface plasmon resonance band towards higher wavelengths on an increase in gold ion concentration. Effective medium theory calculations were carried out in order to explain the observed redshift of the plasmon band in the absorption spectra of the Au–glycogen colloids. The interactions of the gold nanoparticles with glycogen strongly affected the photoluminescence of the glycogenin protein incorporated into its structure. The Au–glycogen hydrocolloids exhibited good chemical stability in the presence of saline, phosphate buffered saline, alanine, histidine and D-glucose.
1 Introduction
The preparation and investigation of novel hybrid materials composed of inorganic nanostructures and biomolecules has evolved into a significant area of contemporary nanoscience.1–4 The increased interest in these systems is driven by a strong demand for the development and application of novel functional nanomaterials, especially in the fields of biotechnology, medicine and environment protection. Among a number of nano-biomaterials which have been reported recently, conjugates of biomolecules with nano-sized gold are the most extensively studied.5 It is well known that gold nanostructures exhibit size, shape and environment-dependent optical,6 electrical7 and photo-physical properties,8 while at the same time they are chemically stable and nontoxic.9 The hybridization of these nanostructures with biomolecules provides novel functionalities, but may also lead to the appearance of additional synergetic properties found in neither of the constituents. Based on this approach, several gold–biomolecule hybrids were developed and applied as contrasting agents for the labeling and visualizing of tissues,5 drug carriers for targeted delivery,10 the optical detection of biomolecules,11,12 or as heat sources in photothermal cancer therapy.13,14The application of the above-mentioned systems depends significantly on the type of biomolecule used as well as on the morphology and arrangement of the nanoparticles. All of these factors affect the surface plasmon resonance (SPR), the resonant absorption and the scattering of light that occurs due to the excitation of coherent electron oscillations at the surface of the particle.6 For this reason, the absorption spectrum can also provide, in combination with theoretical models, certain information about the size, shape and environment of the gold nanostructures. In the case of small spherical gold nanoparticles in water, the surface plasmon resonance occurs at approximately 525 nm, but nonspherical nanostructures or aggregates of nanoparticles15 exhibit absorption bands which are shifted to higher wavelengths. On the other hand, tuning the plasmon resonance via controlled aggregation and/or the self-assembling of nanoparticles holds great promise for many practical applications.16,17 The control of the resonant absorption of the gold nanostructures might also be of importance in photothermal cancer therapy.18 It was found that the nanoparticle-induced conversion of light into heat, and the consequent heat transfer to a malignant tissue, would be facilitated by an increase in the resonance wavelength due to the larger penetration depth of the incident radiation. In this study, we report on the preparation of bioconjugates, which allow the tunable positioning of the absorption band, that comprise gold nanoparticles and the carbohydrate polysaccharide glycogen.
Carbohydrates, along with proteins and amino acids, are essential components in a variety of metabolic processes. In nanotechnology, they have applications as reducing and templating agents for noble metal nanoparticles,19–21 while the preparation of functional glyconanoparticles has enabled the improved analysis of carbohydrate-based interactions with proteins.3,22,23 Polysaccharides (starch, chitosan, alginate, gum arabic, glycogen, etc.) are of particular interest and are the most-studied biomolecules for this purpose. Because of the large number of functional groups, these biopolymers have proved to be good environments for the growth of metal,19,24–36 semiconductor27,37–39 and oxide40,41 nanoparticles. Recently, they have been used as effective plasmonic platforms for SERS detection and tagging,32 and catalysis.33 In addition, the inherent biocompatibility of polysaccharides and the possibility of using green processing methods19 in the preparation of the nanoparticles are advantages in the medical applications of the obtained materials.34
Glycogen is a highly branched polymer of glucose which serves as the main glucose storage component in animal cells. Macromolecules of this polysaccharide are spherical structures, 20–40 nm in size, which are called β-particles. The well-defined morphology of glycogen is a consequence of the repetitive and uniform branching and linking of 13 glucose unit-long A-type and B-type polymer chains.42 According to the model proposed by Melendez-Hevia and co-workers,43,44 the structure of glycogen is optimal for the maximum glucose storage in the smallest possible volume, the maximum portion of the monosaccharide which can be released from the β-particles prior to debranching, and the efficient release of glucose. The dendrimer-like structure of glycogen also provides a large number of functional hydroxyl groups on its surface per volume unit of biopolymer, leading to as much as 34% of the glucose residues in the outermost layer of glycogen.44 Due to this highly developed surface, glycogen presents a convenient nanoreactor for the growth of metallic nanoparticles,28 and can act as a probe material for glucan–protein interactions.22,23 In our previous paper,28 we showed that glycogen–Ag nanoparticle conjugates can be successfully used as antimicrobial materials. Here we show that gold nanoparticles interact with glycogen molecules in a completely different way, which enables the control of the position of the SPR band.
Glycogen is native to human hepatic cells and using this biopolymer in biomedical applications can address the problems of biocompatibility and toxicity which sometimes arise in the applications of synthetic dendrimers.45 Furthermore, we used an in situ non-toxic chemical synthetic method for the preparation of the glycogen and gold nanoparticle hybrid nanosystems. The obtained hybrids were investigated by microscopic, structural and optical methods. The stability of the Au–glycogen colloids was tested in the presence of saline, phosphate buffered saline (PBS), alanine, histidine and D-glucose.
2 Experimental
2.1 Materials
Glycogen from bovine liver, soluble starch, gold(III) chloride hydrate (HAuCl4·4H2O), hydrogen peroxide (H2O2), alanine and histidine were purchased from Sigma-Aldrich and used as received. Saline and PBS were obtained from Hemofarm, Serbia. High purity water (specific resistance 108 Ω m) was used in all of the synthetic procedures.2.2 Synthesis of glycogen-stabilized Au nanoparticles
Gold nanoparticles were prepared in an aqueous glycogen solution, following the reaction of HAuCl4 with H2O2, and under the influence of ultrasonic irradiation. In a typical procedure, 0.2 mL of a 1.5 × 10−3 M (Au) aqueous solution of HAuCl4·4H2O was added to 10 mL of a 0.5% glycogen solution. The mixture was sonicated in an ultrasonic bath (Sonic, 45 kHz) for 5 min at 25 °C. After that, 0.2 mL of 1.5 × 10−3 M H2O2 was added and the solution was treated for an additional 30 min. Different samples were prepared by changing the initial concentration of gold ions [Au] and labeled AuGly1 ([Au] = 3 × 10−5 M), AuGly2 ([Au] = 4.5 × 10−5 M), AuGly3 ([Au] = 6 × 10−5 M) and AuGly4 ([Au] = 9 × 10−5 M). Prior to the investigations, all of the prepared samples were aged for 24 h under ambient conditions.2.3 Characterization methods
The morphology of the pure glycogen particles and the Au nanoparticles synthesized in the presence of glycogen was investigated by transmission electron microscopy (TEM) using a Philips CM100 instrument. The operating voltage was 80 kV. Aqueous solutions of the Au–glycogen samples were deposited on carbon coated copper grids and dried in air under ambient pressure and temperature. For the investigation of the morphology of the pure glycogen, the samples were stained with 2% uranyl nitrate solution.X-ray photoelectron spectroscopy (XPS) experiments were performed on a Physical Electronics Quantum 2000 instrument using Al Kα radiation (1486.6 eV). Prior to the analysis, the samples were sputtered by a 2 keV argon ion beam. The C 1s, O 1s, and Au 4f core levels were recorded at a 29.35 eV pass energy and a 55° take-off angle. The peak shift due to charge accumulation was corrected using the C 1s level at 284.44 eV as an internal standard.46 The XPS peaks were assumed to be linear combinations of Lorentzian and Gaussian line shapes and were resolved into individual components after the proper subtraction of the baseline, using the Shirley background subtraction method. The samples were prepared by the deposition of aqueous Au–glycogen solutions on the flat silicon wafers. They were subsequently dried in a vacuum at 40 °C.
A Nicolet 380 spectrophotometer was used for the FTIR spectroscopic analyses of the pure glycogen and glycogen–gold bioconjugates. The FTIR analyses were performed at room temperature in the spectral range of 4000–400 cm−1, with a resolution of 4 cm−1. The datasets were averaged over 200 scans.
The UV-vis absorption measurements of the Au–glycogen solutions were performed on a Thermo Evolution 600 spectrophotometer over a wavelength range of 200–900 nm.
The photoluminescence measurements of the pure glycogen and Au–glycogen aqueous solutions were performed on a Perkin-Elmer LS45 fluorescence spectrophotometer. For these analyses, we used a reference glycogen solution with the same concentration of biopolymer and pH values as the Au–glycogen colloids. The photoluminescence emission (PL) spectra were recorded in the wavelength range of 330–550 nm using an excitation wavelength λexc = 280 nm. The photoluminescence excitation (PLE) measurements were carried out at two emission wavelengths: λem = 360 nm and λem = 440 nm. The spectra were recorded over 250 nm–330 nm and 250 nm–400 nm intervals, respectively. The synchronous luminescence (SL) spectra were recorded over an excitation range of 240–500 nm and synchronous wavelength intervals (Δλ) from 30–140 nm. All of the measurements were performed in the right angle geometry with a scan speed of 100 nm min−1 and a slit size of 10 nm.
2.4 Chemical stability of the glycogen-stabilized Au nanoparticles
The chemical stability of the glycogen-capped Au nanoparticles in the presence of saline, PBS, alanine, histidine and D-glucose was investigated using UV-vis spectroscopy. UV-vis absorbance spectra were recorded over a 10-day period and the changes in the position and width of the surface plasmon resonance bands with time were analyzed. In these analyses, 2 mL of an as-prepared 4.5 × 10−5 M Au–glycogen solution was mixed with 2 mL of saline (0.9% NaCl), PBS, alanine (0.2 M in H2O), histidine (0.2 M in H2O) and D-glucose (0.1 M in H2O) and kept under ambient conditions. PBS was prepared by dissolving 8.0 g NaCl, 0.2 g KCl, 1.15 g Na2HPO4 and 0.2 g KH2PO4 into 800 mL of H2O (of pH = 7.4), followed by further addition of water to increase the volume to 1 L. The reference sample was prepared by adding 2 mL of water into 2 mL of a 4.5 × 10−5 M Au–glycogen solution. Each Au–glycogen system was prepared three times, and the mean values of the surface plasmon resonance band position and width were reported.3 Results and discussion
3.1 Morphology and composition
The TEM analysis and particle size distribution of the pure glycogen and the Au–glycogen samples are presented in Fig. 1. As predicted by the glycogen models, the image of the uranyl-stained biopolymer shows spheroidal particles of nanometer size (Fig. 1a). The diameter of the glycogen particles ranges from 20 to 80 nm with an average value of 45 nm and a polydispersity of 35%. The obtained size of the particles is in agreement with theoretical predictions for an optimal glycogen particle diameter of 42 nm,43,44 as well as with reported values of the average hydrodynamic radii for rabbit liver and oyster glycogen particles.47 On the other hand, the particles in Fig. 1a are larger than those observed for bovine liver glycogen by Ryu et al.48 The TEM image of the Au–glycogen sample obtained with an initial gold ion concentration of 3.0 × 10−5 M is shown in Fig. 1b. Due to the large difference in the mass densities of gold (19.5 g cm−3) and glycogen (1 g cm−3), the metallic phase can be easily distinguished from the biopolymer in the micrograph. The majority of the particles observed were between 10 and 25 nm in size with an average diameter Dc = 15.1 ± 5.4 nm. The particle size distribution in Fig. 1c follows a log-normal dependence with parameters DLN = 13.6 nm (mean) and σLN = 0.47 (standard deviation). Fig. 1d shows a typical TEM image of an Au–glycogen sample obtained for [Au] = 9.0 × 10−5 M. In contrast to the dispersion of the nanoparticles observed with a low gold ion concentration, a pronounced clustering of the nanoparticles can be seen. However, the size of the particles does not change significantly. The observed morphology of the sample suggests that the gold nanoparticles are attached to the biopolymer surface, presumably via the hydroxyl groups of A-type polymer chains which occupy the outer layers of the glycogen particle.43 Similar morphologies to that depicted in Fig. 1d were also observed for some of the other investigated gold ion concentrations, as can be seen in the TEM micrographs of the AuGly2 and AuGly3 samples (see ESI†).![(a) TEM image of uranyl-stained glycogen; (b) TEM image and (c) corresponding particle size distribution of the [Au] = 3.0 × 10−5 M Au–glycogen sample; (d) TEM image of the [Au] = 9.0 × 10−5 M Au–glycogen sample.](/image/article/2013/RA/c3ra40189h/c3ra40189h-f1.gif) | ||
| Fig. 1 (a) TEM image of uranyl-stained glycogen; (b) TEM image and (c) corresponding particle size distribution of the [Au] = 3.0 × 10−5 M Au–glycogen sample; (d) TEM image of the [Au] = 9.0 × 10−5 M Au–glycogen sample. | ||
X-ray photoelectron spectroscopy was used to determine the composition and chemical environment of the elements present in the material. An XPS survey scan (not shown) revealed the presence of carbon, oxygen and gold core levels (hydrogen, although present in the biomolecule, is not detectable by XPS). Fig. 2 shows the high-resolution XPS spectrum of the Au 4f core level of the sample prepared with an initial Au ion concentration of 4.5 × 10−5 M. Two distinct lines separated by 3.67 eV were observed, namely the Au 4f5/2 and Au 4f7/2 lines, which occur because of the spin–orbit splitting of the Au 4f level.49 The positions of these lines, estimated after the correction due to charge accumulation, were 87.91 eV and 84.24 eV, respectively. In comparison to the reference value of bulk metallic gold of 84.04 eV49 (the vertical line in Fig. 2), the Au 4f7/2 line was shifted towards higher binding energies by 0.2 eV. A shift of this type was also observed in the case of starch and polyvinyl pyrrolidone-capped silver nanoparticles,29,30,50 and was attributed to the cumulative effects of the adsorption of the polymer onto the nanoparticles and the higher number of surface atoms due to the nanoparticles’ small size.50
![XPS spectrum of the Au 4f level of glycogen-stabilized gold nanoparticles ([Au] = 4.5 × 10−5 M). The vertical line corresponds to the reference value for the Au 4f7/2 level of bulk gold (84.04 eV).](/image/article/2013/RA/c3ra40189h/c3ra40189h-f2.gif) | ||
| Fig. 2 XPS spectrum of the Au 4f level of glycogen-stabilized gold nanoparticles ([Au] = 4.5 × 10−5 M). The vertical line corresponds to the reference value for the Au 4f7/2 level of bulk gold (84.04 eV). | ||
The interaction between the glycogen biomacromolecules and gold nanoparticles was also investigated by FTIR spectroscopy, and no apparent differences were observed between the IR spectra of the pure glycogen and glycogen–Au conjugates. The former result implies that the biomolecule and the nanoparticles probably interact via OH groups.19,51
3.2 Optical properties
Fig. 3a shows the UV-vis absorption spectra of aqueous solutions of glycogen-stabilized Au nanoparticles prepared with different initial gold ion concentrations. In the case of the colloid with the lowest gold concentration (3.0 × 10−5 M Au), the maximum of the surface plasmon resonance band is positioned at 526 nm. Since Mie theory52 predicts a resonant absorption value of 525 nm for the 15 nm gold spheres in water (refractive index n = 1.33), it can be concluded that the position of the experimental band is unaffected by the adsorption of the biopolymer. At higher gold concentrations, a broadening of the bands and a shift towards higher wavelengths was observed (Fig. 3a). The intensity of the absorbance also increased with increasing gold content; however, the curves presented in Fig. 3a are normalized for clarity. The dependence of the position of the absorption maxima (λc([Au])) on the initial gold ion concentration is presented in Fig. 3b. The position of the band shifts linearly with the change in [Au] according to the following equation: λc([Au]) = 490(10) nm + 11(2) × 105 [Au] nm M−1 (R2 = 89.8%). This means that the presented preparation procedure offers the possibility to tune the position of the absorption maximum over a broad wavelength range of nearly 60 nm by a simple change to the initial gold ion concentration.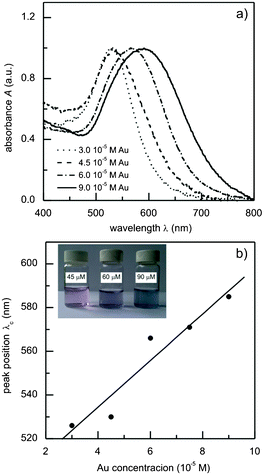 | ||
| Fig. 3 (a) UV-vis absorption spectra of the glycogen-stabilized gold nanoparticle colloids; (b) the dependence of the position of the absorption maxima on the initial gold ion concentration, and the linear fit. | ||
The observed differences in the band positions in the samples with different gold ion concentrations are a consequence of the interaction of individual glycogen molecules with multiple gold nanoparticles (see Fig. 1d). In particular, due to the nanometer size of the glycogen particles, the distances between the neighboring gold nanoparticles will be sufficiently small that the dipole–dipole interactions between them become significant. These interactions reduce the symmetry of the absorbers (with respect to that of the isolated spherical Au nanoparticles), which results in the appearance of additional modes at higher wavelengths53 and, consequently, results in the red shift of the absorption band of the colloid. At higher gold concentrations, the average number of nanoparticles per glycogen molecule increases and, due to smaller inter-particle distances, there is a further shift of the plasmon band towards higher wavelengths. It is worth mentioning that in a study on gold nanoparticles stabilized with poly(amidoamine) dendrimers, Srivastava et al.17 also managed to tune the position of the plasmon resonance by controlling the distance between the gold nanoparticles. However, instead of changing the concentration of the gold ions as in our case, the inter-particle distance was controlled by using different dendrimer generations (dendrimer sizes). The observed absorption behavior of the Au–glycogen colloids is closely related to the dendrimer-like structure of the glycogen β-particles and their nanometer size. To justify the former conclusion, we have prepared soluble starch-stabilized gold nanoparticles using the same synthetic procedure. The molecular structure of soluble starch is identical to that of glycogen, but its macromolecules are predominantly linear or have a lower extent of branching.40 In this case, it was found that the position of the absorption bands of the Au–starch colloids was not affected by the change in the gold ion concentration (see Fig. S2, ESI†). The concentration-dependent optical properties observed in Fig. 3 are also in agreement with the resonant light scattering study of Xiang et al.22 on glycogen–gold nanoparticle systems. They showed that the mixing of glycogen and a separately-prepared gold colloid resulted in the aggregation of the gold nanoparticles.
In order to confirm whether the proposed mechanism of interaction of the gold nanoparticles with glycogen molecules can indeed explain the changes in the plasmon absorption, we performed a series of calculations based on the effective medium theory. The employed theoretical method is based on the approach suggested by Schatz and co-workers54 in their investigations of the optical properties of nanoparticle aggregates. As a starting element in the present calculations, we considered a single glycogen molecule (represented by a sphere of diameter 2Rg = 40 nm) covered with N gold nanoparticles of diameter 2a = 15 nm. The dipole polarizability αag of such an aggregate is54
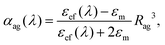 | (1) |
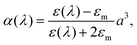 | (2) |
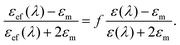 | (3) |
The value of the filling factor f represents the volume fraction of the Au nanoparticles in the aggregate and is represented by
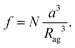 | (4) |
Obviously, for given values of a and Rg, the factor f depends solely on the number of particles N attached to the glycogen outer layer. The values of f are limited by percolation effects, i.e. by the maximum number of gold nanoparticles that can be positioned on the biomolecule surface without being in mutual contact. The experimental absorption spectra can be qualitatively compared to the theoretically estimated absorption coefficients Aabs(λ) by
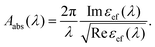 | (5) |
By changing the values of the filling factor f, it is possible to change the positioning of the band maxima of the Aabs(λ) curves until they match the positions of the experimental bands. These curves and their corresponding f-values are presented in Fig. 4. The calculations suggest that an increase in the initial gold concentration is followed by an increase in the filling factor. According to eqn (4), this means that, on average, a larger number of gold particles will be attached to each glycogen molecule. In the case of the colloid with the lowest concentration, we found N to be 1.9, and for the highest concentration, [Au] = 9.0 × 10−5 M, N to be 8.9. A schematic illustration of the changes in N with the change in gold concentration is shown in the insets of Fig. 4. It should be noted that the values of N are not absolute due to a number of other factors that were not included in the calculations. Nevertheless, the presented theoretical analysis clearly suggests a possible mechanism of gold nanoparticle–glycogen interaction which results in the red shift of the absorption maximum. The trend of the increase in the number of nanoparticles attached to the glycogen molecules with an increase in [Au] is in agreement with the TEM observations (Fig. 1).
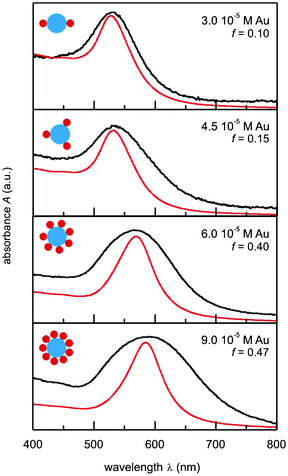 | ||
| Fig. 4 Comparison of the experimental (black) and theoretical (red) absorption spectra for the Au–glycogen colloids. Schematic representations of nanoparticle aggregates are provided in the insets. | ||
3.3 Photoluminescence spectroscopy
To confirm the interaction between the gold nanoparticles and glycogen, as well as to investigate the influence of the particles on the structure of the biomolecule, we used photoluminescence spectroscopy. The results of this analysis for glycogen and glycogen-stabilized gold nanoparticles ([Au] = 4.5 × 10−5 M) solutions are presented in Fig. 5. The PL spectrum of the glycogen solution (Fig. 5a), obtained at an excitation wavelength of λexc = 280 nm, shows two broad bands positioned at approximately 340 nm (peak A) and 426 nm (peak B). The PLE spectra of the same sample are shown in the inset of Fig. 5a. The spectra show that the excitation maximum for an emission wavelength of λem = 360 nm is at 280 nm, while two excitation peaks, at 267 nm and ∼360 nm, are obtained for λem = 440 nm. The emission of glycogen, in fact, originates from the protein glycogenin situated at the center of the macromolecule, which acts as a primer for glycogen synthesis.42 Specifically, the low-wavelength band is from the fluorescent tryptophan (λexc = 295 nm, λem = 353 nm) and tyrosine (λexc = 275 nm, λem = 304 nm) residues,58 while the peak at 426 nm can be attributed to the emission from their oxidation products.59 It is worth mentioning that although tryptophan is a less abundant amino acid in glycogenin than tyrosine,60 it is characterized by a larger extinction coefficient (εTrp = 5600 M−1 cm−1, εTyr = 1400 M−1 cm−1), and for this reason its emission dominates the protein fluorescence spectrum.58 The PL spectra of the Au–glycogen solutions (Fig. 5a) exhibit similar features as in the case of the neat biopolymer solution. However, the presence of gold in the system causes a decrease in the emission intensity due to the well-known quenching effect which metal ions and surfaces have on fluorophore emissions.61 A shift in the position of the bands (from 340 to 364 nm for peak A and from 426 to 441 nm for peak B) was also observed, which is a result of the change in the local polarity around the amino acid residues.58 Since the reduction in the intensity of the emitted light takes place when the distances between the quenchers and the emitter are sufficiently small62,63 the observed changes in the PL properties are also a consequence of the gold nanoparticle–glycogen interactions discussed above.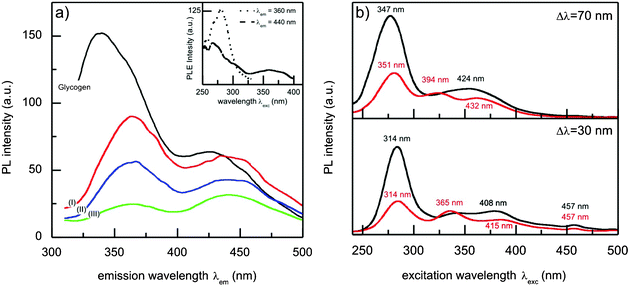 | ||
| Fig. 5 (a) Photoluminescence emission spectra (λexc = 280 nm) of glycogen (black line) and Au–glycogen (I: 0.03 mM Au, II: 0.06 mM Au and III: 0.09 mM Au) aqueous solutions. Photoluminescence excitation spectra (λem = 360 nm and 440 nm) of the glycogen solution are shown in the inset. (b) Synchronous photoluminescence emission spectra of glycogen (black line) and 0.045 mM Au–glycogen (red line) solutions for Δλ = 70 nm and 30 nm. The numbers in (b) correspond to the wavelengths of the emission maxima. | ||
The influence of gold on the PL emission of glycogen was further investigated by synchronous luminescence (SL) spectroscopy, since SL spectra provide information regarding the environment around the fluorophore functional groups.64 The surface plot of the SL data of glycogen-stabilized Au nanoparticles (concentration 4.5 × 10−5 M) shows different features to the spectrum of the glycogen solution (see Fig. S3, ESI†). The SL spectra of glycogen and Au–glycogen solutions obtained at scanning intervals of Δλ = 70 nm and 30 nm are shown in Fig. 5b. For these particular values of Δλ, the SL spectra reveal additional information about the effect of the nanoparticles on the emission of the tryptophan and tyrosine residues.64 In the Δλ = 70 nm case, a decrease in intensity and a red shift of the dominant band at λexc = 277 nm (λgm = 347 nm) in the presence of the gold nanoparticles was observed. This band corresponds to the fluorescence of tryptophan, and its shift towards higher wavelengths indicates that the nanoparticles induce an increase in the polarity of the environment around the tryptophan residues. The interaction of the gold nanoparticles with the glycogen leads to disaggregation of the hydrophobic cavities, thus forming a looser glycogenin structure. Fig. 5b also shows that the emission maximum (λem = 314 nm) from the tyrosine amino acid (Δλ = 30 nm) was not affected by the introduction of the nanoparticles, suggesting that the polarity of the environment around these residues did not change significantly. Similar effects were observed in the case of the SL spectra of human serum albumin treated with glycyrrhetinic acid.64 On the other hand, Kathiravan et al. showed that the conformational changes of bovine serum albumin induced by colloidal AgTiO2 particles should be associated with tyrosine and not with the tryptophan regions.65 The SL spectrum of the Au–glycogen sample for Δλ ≈ 100 nm, characteristic for fluorophores that produce peak B, did not show any additional features compared to the spectrum of the glycogen solution.
3.4 Chemical stability testing
The chemical stability of the gold nanoparticle–glycogen bioconjugates was studied by following the changes in the position and the width of the plasmon band. The stability was tested in environments commonly encountered in biomedical applications such as saline, PBS, amino acids and D-glucose. The results obtained for the AuGly2 sample are presented in Fig. 6. The position of the plasmon band of the reference sample (pure colloid) shifted negligibly, from 533 nm after one day of ageing to 531 nm after 10 days, indicating the good colloidal stability of the Au–glycogen system. It can also be seen that the treatment with all of the tested agents did not induce significant changes in the position of the absorption maxima after 10 days. The width of the resonance band in the case of the reference sample exhibited a mild increase from 59 nm after 24 h to 64 nm after 10 days. The presence of saline solution (Fig. 6a) induced a strong variation in the plasmon bandwidth in the early stages of testing. These changes can be attributed to charge transfer processes occurring between the surface of the nanoparticles and alkali ions present in the solution.66 A similar effect was observed when the colloid was mixed with the aromatic amino acid histidine (Fig. 6b). On the other hand, in the presence of alanine, broadening of the plasmon band, by approximately 5 nm, occurred. This effect is almost independent of the ageing time and can be attributed to the aggregation of the particles in the presence of alanine.67 Finally, there is a gradual decrease in the plasmon bandwidth with an increase in the ageing time in the presence of D-glucose (Fig. 6), which is probably a result of its reduction of the residual gold ions.20![Changes to the gold plasmon resonance position and width, of the glycogen-stabilized gold nanoparticles, with time, in the presence of saline (black squares), PBS (squares), 0.2 M alanine (black triangles), 0.2 M histidine (triangles) and 0.1 M d-glucose (black diamonds). The reference sample (circles) was prepared by diluting the initial colloid solution ([Au] = 4.5 × 10−5 M) with water.](/image/article/2013/RA/c3ra40189h/c3ra40189h-f6.gif) | ||
| Fig. 6 Changes to the gold plasmon resonance position and width, of the glycogen-stabilized gold nanoparticles, with time, in the presence of saline (black squares), PBS (squares), 0.2 M alanine (black triangles), 0.2 M histidine (triangles) and 0.1 M D-glucose (black diamonds). The reference sample (circles) was prepared by diluting the initial colloid solution ([Au] = 4.5 × 10−5 M) with water. | ||
It should be noted that Kattumuri et al.25 performed a similar procedure to test the chemical stability of the gold nanoparticles stabilized by the polysaccharide gum arabic. After seven days of monitoring the absorption properties of this system in the presence of 35% saline solution, they noticed a 15 nm red shift of the Au plasmon maximum and an increase in the band's width by approximately 10 nm. Also, the mixing of the Au–gum arabic bioconjugate with a 0.2 M histidine solution induced the broadening of the resonance band by nearly 25 nm.25 A comparison of these results with the results presented in Fig. 6 suggests that the stabilization of the gold nanoparticles by glycogen molecules results in the excellent chemical stability of the synthesized colloids.
4 Conclusion
Spherical, 15 nm diameter gold nanoparticles were synthesized in the presence of the polysaccharide glycogen by an in situ non-toxic chemical method. The experimental results on the morphology and optical properties of the Au–glycogen hydrocolloids, together with the theoretical analysis of the absorption spectra, suggest that the gold nanoparticles were attached to the surface of the glycogen particles. The interaction of the gold nanoparticles with the biopolymer was further confirmed by photoluminescence spectroscopy analysis of the glycogenin protein which was incorporated into the glycogen structure. The associations formed between the gold nanoparticles and glycogen resulted in the good chemical stability of the colloids in the presence of saline solutions, amino acids and D-glucose.The aggregation of the gold nanoparticles is related to the glycogen morphology (nanometer size of the β-particles), and has a profound influence on the absorption properties of these hybrid systems. Increasing the concentration of gold ions results in an increase in the number of nanoparticles per glycogen molecule and consequently there is a decrease in the distance between the nanoparticles. Since changes to the inter-particle distance induce a shift in the plasmon band, this effect can be used to control the position of the maximum absorption by changing the initial concentration of the gold ions. The gold–glycogen nanostructures studied here could be of interest in biomedical applications, particularly in photothermal cancer therapy. The good chemical stability of this colloidal system and the inherent biocompatibility of the glycogen biomolecules are certainly an advantage. However, for their successful application, further investigations are necessary, particularly on the influence of the nanoparticle size and loading capacity of the glycogen molecules on the position of the absorption band, as well as on the specific types of interaction which lead to their assembly.
Acknowledgements
This work was supported by the Ministry of Education and Science, Republic of Serbia (Project No. 172056 and 45020).References
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- C. Niemeyer and C. Mirkin, Nanobiotechnology: Concepts, Applications and Perspectives, Wiley-VCH, 2004 Search PubMed.
- M. Marradi, M. Martin-Lomas and S. Penades, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, 2010, vol. 64, pp. 211–290 Search PubMed.
- A. de la Escosura-Muniz, C. Parolo and A. Merkoci, Mater. Today, 2010, 13, 24–27 CrossRef CAS.
- R. A. Sperling, P. Rivera gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- V. Myroshnychenko, J. Rodriguez-Fernandez, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzan and F. J. Garcia de Abajo, Chem. Soc. Rev., 2008, 37, 1792–1805 RSC.
- G. Schmid and U. Simon, Chem. Commun., 2005, 697–710 RSC.
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef.
- E. J. Petersen and B. C. Nelson, Anal. Bioanal. Chem., 2010, 398, 613–650 CrossRef CAS.
- D. R. Bhumkar, H. M. Joshi, M. Sastry and V. B. Pokharkar, Pharm. Res., 2007, 24, 1415–1426 CrossRef CAS.
- C. Mirkin, R. Letsinger, R. Mucic and J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- A. Neely, C. Perry, B. Varisli, A. K. Singh, T. Arbneshi, D. Senapati, J. R. Kalluri and P. C. Ray, ACS Nano, 2009, 3, 2834–2840 CrossRef CAS.
- C. Loo, A. Lowery, N. Halas, J. West and R. Drezek, Nano Lett., 2005, 5, 709–711 CrossRef CAS.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639–13645 CrossRef CAS.
- S. Oldenburg, R. Averitt, S. Westcott and N. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- J. West and N. Halas, Annu. Rev. Biomed. Eng., 2003, 5, 285–292 CrossRef CAS.
- S. Srivastava, B. Frankamp and V. Rotello, Chem. Mater., 2005, 17, 487–490 CrossRef CAS.
- C. Pitsillides, E. Joe, X. Wei, R. Anderson and C. Lin, Biophys. J., 2003, 84, 4023–4032 CrossRef CAS.
- P. Raveendran, J. Fu and S. Wallen, J. Am. Chem. Soc., 2003, 125, 13940–13941 CrossRef CAS.
- P. Raveendran, J. Fu and S. Wallen, Green Chem., 2006, 8, 34–38 RSC.
- A. Panacek, L. Kvitek, R. Prucek, M. Kolar, R. Vecerova, N. Pizurova, V. K. Sharma, T. Nevecna and R. Zboril, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef CAS.
- M. Xiang, X. Xu, D. Li, F. Liu, N. Li and K. Li, Talanta, 2008, 76, 1207–1211 CrossRef CAS.
- M. Xiang, X. Xu, F. Liu, N. Li and K.-A. Li, J. Phys. Chem. B, 2009, 113, 2734–2738 CrossRef CAS.
- H. Huang and X. Yang, Biomacromolecules, 2004, 5, 2340–2346 CrossRef CAS.
- V. Kattumuri, K. Katti, S. Bhaskaran, E. J. Boote, S. W. Casteel, G. M. Fent, D. J. Robertson, M. Chandrasekhar, R. Kannan and K. V. Katti, Small, 2007, 3, 333–341 CrossRef CAS.
- R. Brayner, M.-J. Vaulay, F. Fievet and T. Coradin, Chem. Mater., 2007, 19, 1190–1198 CrossRef CAS.
- D. K. Bozanic, V. Djokovic, J. Blanusa, P. S. Nair, M. K. Georges and T. Radhakrishnan, Eur. Phys. J. E, 2007, 22, 51–59 CrossRef CAS.
- D. K. Bozanic, S. Dimitrijevic-Brankovic, N. Bibic, A. S. Luyt and V. Djokovic, Carbohydr. Polym., 2011, 83, 883–890 CrossRef CAS.
- V. Djokovic, R. Krsmanovic, D. K. Bozanic, M. McPherson, G. Van Tendeloo, P. S. Nair, M. K. Georges and T. Radhakrishnan, Colloids Surf., B, 2009, 73, 30–35 CrossRef CAS.
- D. K. Bozanic, V. Djokovic, S. Dimitrijevic-Brankovic, R. Krsmanovic, M. McPherson, P. S. Nair, M. K. Georges and T. Radhakrishnan, J. Biomater. Sci., Polym. Ed., 2011, 22, 2343–2355 CrossRef CAS.
- D. K. Bozanic, L. V. Trandafilovic, A. S. Luyt and V. Djokovic, React. Funct. Polym., 2010, 70, 869–873 CrossRef CAS.
- S. Suarasan, M. Focsan and S. Astilean, Colloid. Surf., B, 2013, 103, 475–481 CrossRef CAS.
- S. Hall, A. Collins, N. Wood, W. Ogasawara, M. Morad, P. Miedziak, M. Sankar, D. Knight and G. Hutchings, RSC Adv., 2012, 2, 2217–2220 RSC.
- I. Baginskiy, T. Lai, L. Cheng, Y. Chan, K. Yang, R. Liu, M. Hsiao, C. Chen, S. Hu, L. Her and D. Tsai, J. Phys. Chem. C, 2013, 117, 2396–2410 CAS.
- L. Turyanska, O. Makarovsky, A. Patane, N. Kozlova, Z. Liu, M. Li and S. Mann, Nanotechnology, 2012, 23, 045702 CrossRef CAS.
- P. Ferreira, M. Vivas, L. De Boni, D. dos Santos, D. Balogh, L. Misoguti and C. Mendonca, Opt. Express, 2012, 20, 518–523 CrossRef CAS.
- Q. Nie, W. Tan and Y. Zhang, Nanotechnology, 2006, 17, 140–144 CrossRef CAS.
- D. K. Bozanic, V. Djokovic, N. Bibic, P. S. Nair, M. K. Georges and T. Radhakrishnan, Carbohydr. Res., 2009, 344, 2383–2387 CrossRef CAS.
- T. Radhakrishnan, M. K. Georges, P. S. Nair, A. S. Luyt and V. Djokovic, J. Nanosci. Nanotechnol., 2007, 7, 986–993 CrossRef CAS.
- N. Vigneshwaran, S. Kumar, A. A. Kathe, P. V. Varadarajan and V. Prasad, Nanotechnology, 2006, 17, 5087–5095 CrossRef CAS.
- P. R. Chang, J. Yu, X. Ma and D. P. Anderson, Carbohydr. Polym., 2011, 83, 640–644 CrossRef CAS.
- E. Goldsmith, S. Sprang and R. Fletterick, J. Mol. Biol., 1982, 156, 411–427 CrossRef CAS.
- E. Melendez-Hevia, T. G. Waddell and E. D. Shelton, Biochem. J., 1993, 295, 477–483 CAS.
- R. Melendez, E. Melendez-Hevia, F. Mas, J. Mach and M. Cascante, Biophys. J., 1998, 75, 106–114 CrossRef CAS.
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS.
- C. Powell, Appl. Surf. Sci., 1995, 89, 141–149 CrossRef CAS.
- S. Zhang, F. Zhao, K. Li and S. Tong, Anal. Chim. Acta, 2001, 431, 133–139 CrossRef CAS.
- J.-H. Ryu, J. Drain, J. H. Kim, S. McGee, A. Gray-Weale, L. Waddington, G. J. Parker, M. Hargreaves, S.-H. Yoo and D. Stapleton, Int. J. Biol. Macromol., 2009, 45, 478–482 CrossRef CAS.
- T. Darrah Thomas and P. Weightman, Phys. Rev. B, 1986, 33, 5406–5413 CrossRef.
- H. Shin, H. Choi, Y. Jung, S. Kim, H. Song and H. Shin, Chem. Phys. Lett., 2004, 383, 418–422 CrossRef CAS.
- Z. H. Mbhele, M. G. Salemane, C. G. C. E. van Sittert, J. M. Nedeljkovi, V. Djokovi and A. S. Luyt, Chem. Mater., 2003, 15, 5019–5024 CrossRef CAS.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- T. C. Preston and R. Signorell, ACS Nano, 2009, 3, 3696–3706 CrossRef CAS.
- A. Lazarides, K. Kelly, T. Jensen and G. Schatz, THEOCHEM, 2000, 529, 59–63 CrossRef CAS.
- M. A. Sullivan, F. Vilaplana, R. A. Cave, D. Stapleton, A. A. Gray-Weale and R. G. Gilbert, Biomacromolecules, 2010, 11, 1094–1100 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- H. Hövel, S. Fritz, A. Hilger, U. Kreibig and M. Vollmer, Phys. Rev. B: Condens. Matter, 1993, 48, 18178–18188 CrossRef.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 2006 Search PubMed.
- S. Si and T. K. Mandal, Chem.–Eur. J., 2007, 13, 3160–3168 CrossRef CAS.
- D. G. Campbell and P. Cohen, Eur. J. Biochem., 1989, 185, 119–125 CrossRef CAS.
- J. Lakowicz, Anal. Biochem., 2001, 298, 1–24 CrossRef CAS.
- A. Campion, A. Gallo, C. Harris, H. Robota and P. Whitmore, Chem. Phys. Lett., 1980, 73, 447–450 CrossRef CAS.
- B. N. J. Persson and N. D. Lang, Phys. Rev. B, 1982, 26, 5409–5415 CrossRef CAS.
- J. Tang, F. Luan and X. Chen, Bioorg. Med. Chem., 2006, 14, 3210–3217 CrossRef CAS.
- A. Kathiravan, R. Renganathan and S. Anandan, Polyhedron, 2009, 28, 157–161 CrossRef CAS.
- U. Kreibig, G. Bour, A. Hilger and M. Gartz, Phys. Status Solidi A, 1999, 175, 351–366 CrossRef CAS.
- G. Mihailescu, L. Olenic, S. Pruneanu, I. Bratu and I. Kacso, J. Optoelectron. Adv. Mater., 2007, 9, 756–759 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of the Au–glycogen samples, as well as the UV-vis and SL spectra of the Au–glycogen and Au–starch solutions. See DOI: 10.1039/c3ra40189h |
| This journal is © The Royal Society of Chemistry 2013 |