 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
QM/MM description of platinum–DNA interactions: comparison of binding and DNA distortion of five drugs†
Konstantinos
Gkionis
,
Shaun T.
Mutter
and
James A.
Platts
*
School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK. E-mail: platts@cardiff.ac.uk; Fax: +44-2920-874030; Tel: +44-2920-874950
First published on 23rd January 2013
Abstract
Hybrid QM/MM calculations on adducts of five platinum-based anti-cancer drugs, namely cisplatin, oxaliplatin, lobaplatin, and heptaplatin are reported. Starting from the NMR structure of a cisplatin–DNA octamer complex (PDB entry 1AU5), we compare DNA binding of drugs that differ in their carrier ligands, and hence in their potential interactions with DNA. It is shown that all drugs induce broadly similar changes to the regular helical structure of DNA, but that variations in ligand lead to subtle differences in complex geometry, with cisplatin exhibiting notably different properties to other drugs. Cisplatin is also the most weakly bound of drugs considered here, and heptaplatin the most strongly bound. Differences in binding appear to be due to changes in the pattern of non-covalent interactions between drug and DNA, especially hydrogen bonding to oxygen in guanine and phosphate groups. Despite adopting very similar geometries, two isomers of lobaplatin (RRS and SSS) are found to have quite different binding energies, the latter being bound by up to 30 kcal mol−1 more than the former.
Introduction
Cisplatin (1, cis-[PtCl2(NH3)2]) is one of the best-selling anti-cancer drugs of recent times.1,2 Despite its success, serious drawbacks related to toxicity and resistance3 mean that the search for deeper understanding of its mode of action, and for therapeutic alternatives, continues to be intense.4 To date, the only other drugs with global approval are carboplatin and oxaliplatin (2) (see Fig. 1) while others such as nedaplatin, heptaplatin (3) and lobaplatin (4) have found approval locally.5 All such complexes enter cells intact, and are then hydrolyzed to yield active aqua/hydroxo species via loss of anionic ligand(s). These active species then form adducts with nucleophilic sites in DNA within the cell nucleus, particularly the N7 position of guanine. Mono- adducts as well as inter- and intra-strand cross-links are possible, with the latter dominating, and the resulting distortion of the DNA helix give rise to recognition by repair proteins and ultimately cell apoptosis.3,5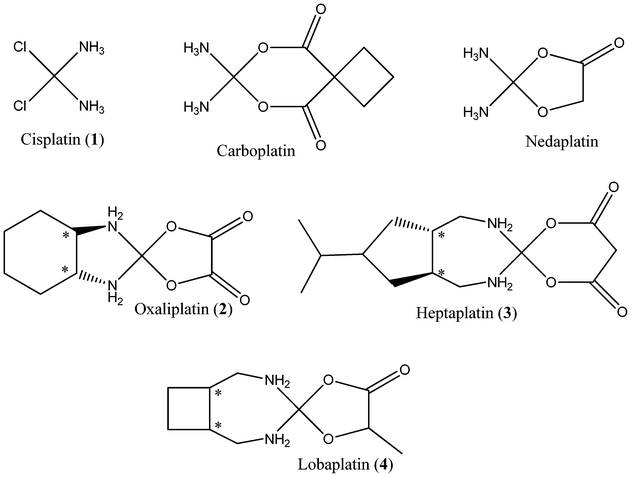 | ||
| Fig. 1 Molecular structures of current platinum based drugs, with chiral centres indicated by an asterisk. | ||
Carboplatin and nedaplatin yield the same active species as cisplatin on loss of anionic ligand, and hence are likely to share in fundamental mechanism of action, but have improved pharmacokinetics and reduce toxicity due to a chelate effect. Oxaliplatin is a third generation platinum anticancer drug with a 1,2-diaminocyclohexane (DACH) entity, predominantly used in the treatment of colorectal cancer.6 In this and related drugs like heptaplatin and lobaplatin, ammine carrier ligands are replaced by larger chelating amines that improve cell uptake by their greater hydrophobicity, as well as forming different interactions with DNA. Oxaliplatin forms fewer crosslinks than cisplatin at equimolar concentrations, as its adducts are bulkier and more hydrophobic than those formed from cisplatin or carboplatin, leading to different effects in the cell.7 Lobaplatin and heptaplatin similarly employ large, chelating amines. Oxaliplatin and heptaplatin are administered as single enantiomers, but lobaplatin is a nearly 50/50 mixture of two diastereoisomers (SSS and RRS configurations).
A recent NMR study of adducts of cisplatin and oxaliplatin with a DNA dodecamer showed subtle differences in the orientation of platinated bases that ultimately give rise to the different spectrum of activity of the drugs.8 Such experiments give detailed insight into the drug–DNA interaction, but can be expensive and time-consuming, and cannot be used for virtual screening of as-yet unsynthesized drugs. A reliable and general method to predict how a particular platinum complex may interact with DNA could be a valuable complement to the tools available in designing new and improved platinum-based anti-cancer drugs. A great many theoretical studies have addressed the structure and properties of platinum drugs,9 their mode(s) of activation and deactivation,10 and interaction with DNA.11 Density functional theory (DFT) is a very popular way to include electron correlation effects, and thus obtain accurate description of drugs, for relatively little computational expense. However, even with modern computing resources such studies are limited to perhaps a few hundred atoms, corresponding to small portions of DNA. Molecular mechanics (MM) methods allow study of much larger systems such as DNA oligomers, but require specific parameters for all atoms and bonds present. Such parameters have been reported and applied for cisplatin and oxaliplatin,12 but this approach is not generally applicable to all the drugs under study here.
Hybrid QM/MM approaches offer a middle way that preserves the accuracy of DFT in describing transition metals with the applicability to large systems of MM. QM/MM studies of nucleic acids are relatively rare, at least when compared to analogous studies of enzymes, and such studies of metal–DNA interactions are still rarer. Several pioneering studies employed Car–Parrinello molecular dynamics to probe binding of cisplatin and other Pt complexes to DNA,13 while others have recently addressed aspects of several transition metal drugs.14 In a recent study,15 we set out an efficient QM/MM scheme for simulation of platinum-DNA adducts, testing against crystal and NMR data for cisplatin complexed with a DNA dimer and octamer, respectively. This showed that DFT/MM gas-phase optimisation accurately reproduces dimer structures, but that explicit solvation is required to properly describe larger DNA segments. In this work, we apply the same methods to compare binding of drugs with varying carrier ligand.
Computational methods
Following our previous studies of cisplatin, QM/MM calculations were performed with the two-level ONIOM method16 within the Gaussian 09 suite of programs.17 Specifically, BHandH DFT method along with a 6-31+G(d,p) basis set on light atoms and SDD basis set and effective core potential on Pt 18,19 was used for the high layer, and AMBER (parm96) for the low layer.20 This approach has been shown to reproduce π-stacking potential energy curves and structures of DNA fragments and their metal complexes.15 Hydrogen link atoms replaced carbon at the base–sugar bond, such that bases directly attached to platinum are treated with DFT and all remaining atoms with AMBER. In ref. 15, we showed that generic parameters for metal–ligand bond lengths and angles satisfactorily describe atoms in the high layer: the same approach is used throughout this work. Replacement of the C–N bond in the full system with a C–H bond in the high layer of an ONIOM calculation is not expected to drastically alter the electronics of the system. An example of this partitioning scheme is shown in Fig. 2. Atomic charges were assigned manually to each atom in the low layer, and high layer charges determined by a restrained fit to the electrostatic potential (RESP) via the Merz–Kollman procedure.21 Electrostatic embedding, in which the QM region is polarised by the charges in the MM region was employed, following our findings in ref. 15 that lack of such embedding leads to poor reproduction of experimental data.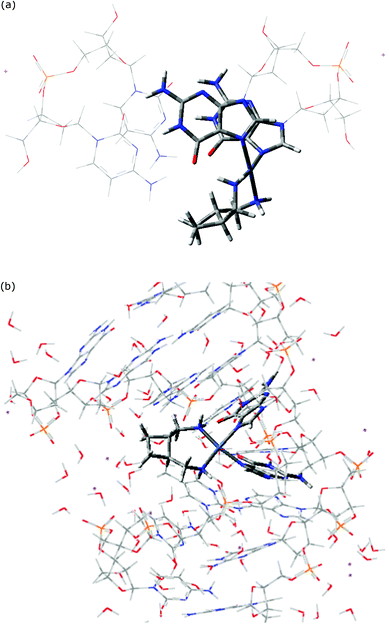 | ||
| Fig. 2 QM/MM optimized geometry of SSS 4 complexed to (a) GG/CC base-pair step, and (b) d(CCTGGTCC). | ||
Geometry optimisation was performed using the GDIIS (for smaller systems) and GEDIIS (for octamers) methods.22 Dimer and octamer structures were generated from the NMR structure of cisplatin with ds- CCTG*G*TCC (* indicating sites of platination: PDB entry 1AU5).23 Sodium ions were added ca. 2 Å from each phosphate, resulting in an overall complex charge of +2. Dimers were optimised in gas-phase, while octamer complexes were solvated by a large cubic box of water in Molecular Operating Environment (MOE) software,24 which was then truncated by use of a cut off of solute–solvent distance, above which solvent molecules are ignored, to retain approximately 100 solvent molecules, resulting in a total of approximately 800 atoms. Geometries were fully optimised in all cases, but vibrational frequency analysis is beyond our capability at present. Aqueous-phase binding energy calculations employed the polarizable continuum model (PCM) approach, and in particular the ONIOM-PCM method in which reaction field is computed only for the real-system at the low-level while the sub-calculations on the model-systems are performed assuming zero reaction field.25 Analysis of the resulting DNA structures was performed using the X3DNA software.26
Results and discussion
In order to have confidence in the ability of our QM/MM model to accurately describe drug–DNA interactions, we first summarize its ability to reproduce the experimental geometry of cisplatin–DNA dimer and octamers.15 A crystal structure of cisplatin bound to single stranded guanine-deoxyribose phosphate-guanine, d(GpG), that contains four independent molecules in the unit cell was used as the first test. QM/MM optimized geometry reproduces almost all experimental bond lengths and angles to within one standard deviation (sd) of experimental data (see ESI†). A key parameter for the disruption of native DNA structure by platination is the angle between mean planes of guanines: the experimental value for this is 81.2 ± 4.3°, while our predicted value is 86.5°.A more stringent test of our approach lies in its ability to reproduce the geometry of cisplatin bound to longer sections of double-stranded DNA. Table 1 reports such a comparison, concentrating on the base-pair step parameters obtained from X3DNA analysis. Agreement with NMR data is generally reasonable, especially when comparing average values across the entire sequence. Deviations from experiment are somewhat larger for the platinated GG/CC base pair step, but key aspects of platinated DNA and its deviation from the ideal B-form of DNA are reproduced. It is important to note that NMR experiments result in a family of structures that satisfy observed coupling and NOE data. For instance, ref. 23 reports the distance between H8 protons on G4 and G5 to lie between 2.52 and 3.92 Å, with an average value quoted in the PDB entry of 2.907 Å: our QM/MM optimized value of 3.757 Å is therefore significantly different from the reported value but actually lies comfortably within the range of acceptable distances. While the PDB entry used for the octamer complex (1AU5) reports just one structure, entry 2NQ0 (cisplatin–DNA dodecamer) reports 15 structures that satisfy NMR observations. Analysis of all 15 structures indicates that significant variation of base-pair step parameters exists within the family: shift exhibits variation of 0.45 Å, slide 1.12 Å, raise 0.71 Å, tilt 1.7°, roll 10.0° and twist 8.5°. In this context, the level of agreement between experimental and theoretical data in Table 1 is satisfactory.
| Shift | Slide | Rise | Tilt | Roll | Twist | |
|---|---|---|---|---|---|---|
| G4-G5 | ||||||
| Experiment | 1.20 | −1.55 | +5.50 | −13.18 | +58.72 | +23.24 |
| QM/MM | 0.92 | −1.94 | +4.97 | −5.64 | +48.00 | +35.17 |
| Average | ||||||
| Experiment | −0.14 | −0.02 | +3.63 | −0.46 | +4.13 | +33.24 |
| QM/MM | −0.12 | −0.20 | +3.55 | −3.16 | +5.56 | +35.04 |
Following these tests on cisplatin, similar methods were used to compare the DNA adducts of 1, 2, 3, and both isomers of 4. Initially, each drug was manually complexed with a GG/CC base pair step taken from PDB entry 1AU5, then QM/MM optimized. Representative examples of optimized structures are shown in Fig. 2, which also serves to illustrate the partition between QM and MMM layers employed. Coordination geometry varies little between drugs (see ESI†), with the exception of the N1–Pt–N2 angle for 2, which is around 10° less than in other complexes. The distortion of the helical DNA structure in these complexes, as determined by the angle between guanine planes, is much less in these double-stranded complexes than in the single-stranded example discussed above, with values between 25 and 28°. Analogous data for QM/MM optimised complexes with octameric DNA are also reported: Pt–N7 bond length to the 3′ guanine is slightly shorter (0.02–0.03 Å) in the octamer than the dimer, while in several cases the angle formed by the carrier ligand amines' coordination to Pt varies by as much as 7°. However, in general, the square-planar coordination geometry does not vary greatly between the two forms of DNA.
More detailed information on the DNA conformation is reported in Table 2. These data highlight the consistent deviations from the unbound DNA structure, optimised at the same level. In the dimers, platination gives rise to larger shift and smaller rise values than in free DNA, whereas slide values are barely affected. Similarly, roll values are significantly larger (more positive) and twist is slightly smaller in platinated complexes than in free DNA, but tilt values are close to their undistorted values. Within these overall trends, some changes due to the nature of the carrier ligand are evident: for instance, heptaplatin induces a noticeably smaller roll value than all other complexes considered here. Also, differences between RRS and SSS lobaplatin are rather large in some cases, especially shift and slide. However, on the whole Table 2 indicates that platination induces similar changes to DNA no matter what ligand is present. Base pair data (ESI†) show that shear values are uniformly more negative in the complexes than in free DNA, but stagger and especially stretch are close to the uncomplexed values. Similarly, propeller values differ from free DNA, but buckle and opening values are similar to free values. However, heptaplatin makes an exception from this overall trend, displaying markedly different stagger and propeller values in its 3′G–5′C base pair than other complexes or free DNA, indicating significant out-of-plane distortion of this base pair that is not present in others. Unlike in the base pair step data, differences in base pair parameters between chiral forms of lobaplatin are small.
| Shift | Slide | Rise | Tilt | Roll | Twist | |
|---|---|---|---|---|---|---|
| a Obtained from optimization of DNA using the same methods. | ||||||
| 1 | 1.43 | −1.75 | 3.36 | −0.4 | 13.6 | 31.3 |
| 1.14 | −1.11 | 3.18 | −8.0 | 28.5 | 22.9 | |
| 2 | 1.42 | −1.85 | 3.42 | −0.7 | 14.0 | 31.6 |
| 1.33 | −1.61 | 3.64 | +1.9 | 46.2 | 28.7 | |
| 3 | 1.47 | −1.71 | 3.39 | −1.8 | 6.9 | 28.8 |
| 0.99 | −1.35 | 3.51 | −3.1 | 42.6 | 22.1 | |
| 4 RRS | 1.39 | −1.77 | 3.32 | −1.8 | 12.7 | 28.2 |
| 1.12 | −1.13 | 3.71 | −0.8 | 43.6 | 24.8 | |
| 4 SSS | 1.54 | −1.94 | 3.40 | −1.7 | 11.2 | 30.8 |
| 1.00 | −1.13 | 3.61 | −4.7 | 45.0 | 20.3 | |
| DNAa | 0.87 | −1.73 | 3.83 | +2.3 | −1.0 | 36.2 |
| −0.40 | −0.57 | 3.09 | −8.3 | 7.9 | 34.3 | |
Analogous data for octameric complexes with drugs 1 to 4 are also reported in Table 2, and illustrated in Fig. 3, concentrating on the central GG/CC base pair. In all complexes, platination induces a bending of the helix towards the major groove, with a tendency to enclose the drug. This bending of the helix brings bulky Pt ligands spatially close to nucleobases other than those of the site of platination (G4, G5), therefore possibly giving rise to additional non-bonded interactions between atoms of the QM and MM regions. All complexes show large positive shift values, in contrast to the small negative value in free DNA, and slightly more negative slide values than free DNA. Twist parameters exhibit uniform displacement from the values in free DNA, and are slightly less positive than in free DNA. Tilt values change by only very small amounts across the series, and no clear trend is apparent. The clearest differences between drugs are found in the separation between base pairs: the cisplatin complex exhibits a smaller rise value than other drugs, similar to that in free DNA, whereas all other drugs give larger rise values. A similar trend is evident in the roll parameter: cisplatin has notably smaller roll values than the remaining complexes, although this is still much greater than in free DNA. Differences between the isomeric forms of 4 are generally very small in this data, and both exhibit broadly similar geometry to 3.
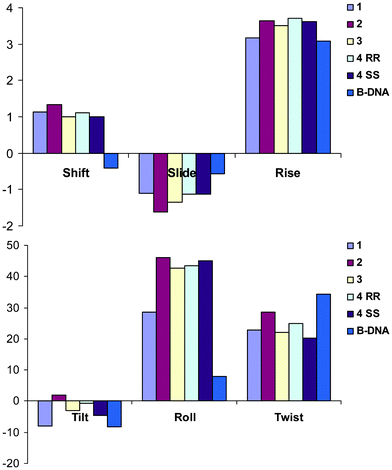 | ||
| Fig. 3 Central GG/CC base-pair step geometry of drug–octamer complexes (Å and °). | ||
The binding energy of each drug to its target is a key factor in the mode of action. Table 3 reports binding energies of each drug to DNA dimer and octamer, evaluated in gas- and simulated aqueous-phase. Binding to octamer is much more energetically favourable than to the dimer, perhaps due to additional interactions of ligand with flanking bases or to the greater number of anionic/cationic charge centres in the larger strand. Three of the four methods of estimating binding energy give the same trend, namely 1 < 2 ≅ 4RRS < 3 ≅ 4SSS. Only the gas-phase dimer values are out of step with the others; given the likely importance of solvation in binding of a cationic platinum complex to highly polar DNA as well as the larger-scale DNA structure, it seems likely that gas-phase dimer values do not capture some essential feature of binding. Differences between drugs are larger in gas-phase data than in simulated solvent, but the remaining three approaches yield the same trend of binding energy between drugs, with heptaplatin the most strongly bound and lobaplatin in its SSS configuration closely behind. 1 is the least strongly bound, while 2 and RRS 4 have binding energies intermediate between these extremes. This is illustrated for octamer data in Fig. 4. The weak binding of 1, and the significant difference between configurations of 4 (82 kcal mol−1 in gas-phase, 31 kcal mol−1 in PCM water), are somewhat surprising, and suggest that the larger drugs may undergo substantial non-covalent interactions with the receptor that are not present in the complex of 1 and/or not optimal in the RRS 4 complex.
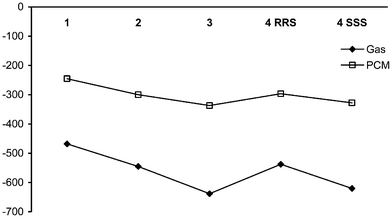 | ||
| Fig. 4 Binding energy of drugs to octameric DNA (kcal mol−1). | ||
| Dimer Gas | Aqueous | Octamer Gas | Aqueous | |
|---|---|---|---|---|
| 1 | −327.2 | −56.1 | −468.0 | −245.4 |
| 2 | −302.4 | −56.0 | −545.0 | −299.6 |
| 3 | −315.7 | −69.1 | −638.3 | −337.1 |
| 4 RRS | −304.2 | −64.5 | −537.7 | −296.6 |
| 4 SSS | −306.5 | −67.1 | −620.1 | −327.9 |
To examine the binding of each drug to DNA in more detail, we have carried out AIM analysis on model systems in which the optimized octamer structure was truncated to a single-stranded TGGT sequence,‡ since the full system cannot be evaluated at DFT level. The electron density for each such model was then evaluated with a full DFT calculation. Table 4 summarizes the covalent and non-covalent interactions between drug and DNA found for each complex. As expected, each complex has two covalent linkages between Pt and N7 of guanine, that are characterized by relatively large (>0.1 au) values of the electron density at the corresponding critical point ρc. Variation in ρc between drugs in these covalent bonds is relatively small, with all values lying in the range 0.121 to 0.137 au. However, all complexes except that of cisplatin show markedly greater ρc in the bond to 3′ guanine than to 5′, reflecting the shorter distances in the octamer noted above. Less expected is the presence of an interaction between Pt and O6 of the 5′ guanine, located over the plane defined by Pt–N bonds. The long-range nature of this contact (Pt⋯O = 3.01 Å in cisplatin complex), and the fact that ρc is an order of magnitude lower than in Pt–N bonds, suggest that this is an outer-shell effect and that the complexes are indeed best described as 4-coordinate square planar.
| Complex | Drug | DNA | No. | ρ c (au) | NCIa |
|---|---|---|---|---|---|
| a ss = strongly stabilizing; s = stabilizing; ws = weakly stabilizing. | |||||
| 1 | Pt | N7 (G) | 2 | 0.128, 0.128 | — |
| Pt | O6 (G5′) | 1 | 0.018 | s | |
| N–H | O6 (G3′) | 1 | 0.045 | ss | |
| N–H | O4 (T3′) | 1 | 0.020 | s | |
| N–H | π (T5′) | 1 | 0.011 | ws | |
| N–H | sugar 5′ | 1 | 0.003 | ws | |
| 2 | Pt | N7 (G) | 2 | 0.126, 0.137 | — |
| Pt | O6 (G5′) | 1 | 0.012 | s | |
| N–H | O6 (G3′) | 1 | 0.026 | ss | |
| N–H | O4 (T3′) | 1 | 0.018 | s | |
| N–H | π (T5′) | 1 | 0.015 | s | |
| N–H | O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P (5′) P (5′) |
1 | 0.045 | ss | |
| C–H | sugar 5′ | 4 | 0.013, 0.006, 0.005, 0.004 | ws | |
| C–H | O4 (T3′) | 1 | 0.006 | ws | |
| 3 | Pt | N7 (G) | 2 | 0.123, 0.132 | — |
| Pt | O6 (G5′) | 1 | 0.013 | s | |
| N–H | O6 (G3′) | 1 | 0.045 | ss | |
| N–H | O4 (T3′) | 1 | 0.018 | s | |
| N–H | π (T5′) | 1 | 0.009 | ws | |
| N–H | OP (5′) |
1 | 0.042 | ss | |
| C–H | T5′ | 5 | 0.009, 0.008, 0.007, 0.006, 0.004 | ws | |
| 4 RRS | Pt | N7 (G) | 2 | 0.121, 0.135 | — |
| Pt | O6 (G5′) | 1 | 0.012 | s | |
| N–H | O6 (G3′) | 1 | 0.047 | ss | |
| N–H | π (T3′) | 1 | 0.006 | s | |
| N–H | π (T5′) | 1 | 0.017 | s | |
| N–H | sugar 5′ | 1 | 0.014 | ws | |
| C–H | OP 5′ |
1 | 0.021 | s | |
| C–H | sugar 5′ | 1 | 0.008 | ws | |
| C–H | T 3′ | 2 | 0.013, 0.03 | ||
| 4 SSS | Pt | N7 (G) | 2 | 0.123, 0.132 | — |
| Pt | O6 (G5′) | 1 | 0.012 | s | |
| N–H | O6 (G3′) | 1 | 0.043 | ss | |
| N–H | O4 (T3′) | 1 | 0.016 | s | |
| N–H | OP (5′) |
1 | 0.036 | ss | |
| N–H | π (T5′) | 1 | 0.012 | ws | |
| N–H | sugar 5′ | 2 | 0.010, 0.009 | ws | |
| C–H | sugar 5′ | 1 | 0.008 | ws | |
| C–H | T5′ | 2 | 0.010. 0.007 | ws | |
| C–H | O4 (T3′) | 1 | 0.009 | ws | |
All complexes contain a relatively strong hydrogen bond linking N–H of coordinated am(m)ine ligand and O6 of the 3′ guanine. In most complexes this H-bond has consistent ρc values of around 0.045 au, but in the case of 2 the H-bond is rather weaker, with ρc = 0.026 au. This is reflected in H-bond distance, 1.96 Å in the complex of 2 compared with 1.72 to 1.75 Å in the remaining complexes. Hydrogen bonds from coordinated am(m)ine ligand to the 3′ thymine are also common, typically to O4 but in one case to the π-system of this base. An important difference between complexes appears to lie in the presence of N–H⋯OP H-bonds, which are present and rather strong in 2, 3 and 4 SSS complexes, i.e. the more strongly bound drugs, but absent in 1 and 4 RRS ones. Several of the complexes of larger drugs also contain numerous C–H⋯X interactions: this is most obvious in 2 and 3 complexes. In the former, many of these are of C–H⋯H–C type to the pentose sugar and thymine on the 5′ side of the drug, which might appear at first sight to be repulsive, steric contacts. In the latter, some such contacts are of C–H⋯H–C type, but others are C–H⋯π forming contacts to 5′ thymine, which seem likely to contribute to the overall stabilization of this complex. The (de)stabilizing nature of such contacts is discussed in more detail below.
Interestingly, there exists an approximate linear correlation between binding energy and the sum of ρc values found between drug and DNA, whether including the Pt–N and Pt⋯O contacts (R2 = 0.88) or excluding those and only using non-covalent interactions (R2 = 0.92). We have previously used such an approach to correlate density properties with binding energies for purely non-covalent interactions.27 The presence of such correlations in the current data supports the hypothesis that non-covalent interactions play an important role in determining the overall binding of a drug to DNA. However, it is not always clear from such AIM data whether an individual interaction acts to stabilize or destabilize. Indeed, there has been substantial discussion in literature over whether the presence of a bond critical point unambiguously indicates a stabilizing interaction.28
To complement AIM analysis, we have also employed the reduced density gradient s(r) and related properties, as defined by Johnson et al.'s program NCIPLOT.29 It has been shown that s(r) identifies non-covalent interactions, and that the sign(s) of the curvature of the density at relevant points can distinguish strongly stabilizing (e.g. hydrogen bonds), weakly stabilizing (e.g. van der Waals) or destabilizing (e.g. steric clash) contacts. This analysis finds non-covalent regions of reduced density gradient corresponding to all critical points located in AIM analysis (see Fig. 5 for an example plot of oxaliplatin), and indicates that N–H⋯O hydrogen bonds are strongly stabilizing, as expected. Weaker stabilization is indicated for C–H⋯O and N–H⋯π contacts, while even those that may seem like steric C–H⋯H–C clashes between drug and sugar are reported to be weakly stabilizing (Johnson et al. report similar results to ours for the methane dimer).30 NCI analysis also locates a region of s(r) corresponding to the Pt⋯O contact observed in AIM analysis, and assigns this as a stabilizing interaction, albeit not as strongly so as N–H⋯O hydrogen bonds. In this approach, we find no evidence of any destabilizing contacts between drug and DNA; this confirms the suitability of including all critical points in the correlations noted above.
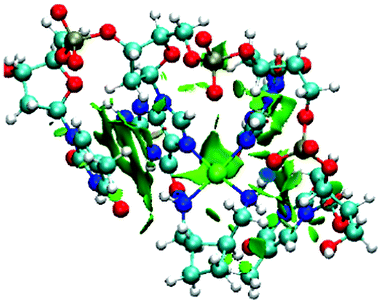 | ||
| Fig. 5 Plot of s(r) for 2-DNA complex, shown at 0.5 au isosurface. Strongly stabilizing interactions are shown in blue, and weakly stabilizing ones in green. | ||
As well as distortion of the regular DNA helix, another geometrical aspect of the drug complexes that may play a role in activity is the extent to which the carrier ligand is exposed and hence able to interact with solvent or be recognized by repair proteins. Table 5 reports the exposed surface area of each drug fragment when in complex with dimer and octamer DNA strands. Values are uniformly smaller in the larger complexes, albeit by significantly different amounts. The smallest drug, 1, unsurprisingly has the lowest exposed area in both complexes, but this barely differs between DNA strands. In contrast, 2 has a high exposed area when bound to a DNA dimer, but this is much reduced in the octamer which may suggest a close fit between the ligand and receptor. 4 in its RRS form shows a similar reduction in exposed area, whereas 3 and SSS 4 lie between these extremes. These trends are in accord with AIM data that show few drug–DNA contacts for 1 but a large number of C–H⋯X contacts for 2.
| Dimer | Octamer | |
|---|---|---|
| 1 | 67.3 | 63.2 |
| 2 | 157.0 | 117.1 |
| 3 | 208.1 | 189.7 |
| 4 RRS | 158.5 | 124.8 |
| 4 SSS | 155.8 | 131.2 |
Conclusions
We report hybrid QM/MM optimizations of the complexes of several platinum drugs with a DNA octamer, with the goal of examining and the effects of am(m)ine carrier on geometrical and energetic aspects of binding. In particular, known or promising drugs with larger chelating amine ligands are compared with the archetypal drug cisplatin. This allows, for instance, comparison of oxaliplatin (containing 1,2-DACH ligand), or of the diastereomeric forms of lobaplatin. All complexes considered distort DNA in broadly similar fashion, but some subtle differences are evident, for which the drugs considered fall into two broad categories, namely cisplatin and the rest. This is most evident in the rise and roll parameters, whereas other geometrical properties are either more uniformly shifted from free DNA values, or else no clear trend is apparent. The extent to which the drug is exposed to its environment when in complex with DNA has also been examined: unsurprisingly, this is smallest for cisplatin and largest for heptaplatin. Comparison of isomers shows that the RRS form of lobaplatin is less exposed than the SSS form.Binding energies of each drug to DNA have been calculated in gas-phase and simulated aqueous solvation, and show clear trends across amine ligands. Slightly surprisingly, cisplatin is the most weakly bound drug to octameric DNA in both gas- and aqueous-phase. Oxaliplatin shows slightly stronger binding, while heptaplatin is the most strongly bound of all drugs considered. The SSS configuration of lobaplatin is almost as strongly bound as heptaplatin, while its RRS diastereoisomer is more weakly bound and comparable to oxaliplatin.
To examine these trends in more detail, the electron density and related properties of a truncated model complex with single-strand TGGT was examined. This showed little or no difference in the covalent Pt–N7 binding to guanine, but major differences in the non-covalent interactions between drug and DNA. All complexes exhibit N–H⋯O6G3′ hydrogen bonding that are estimated on the basis of the electron density to have similar strength, except for oxaliplatin that has a much weaker and longer H-bond. Numerous other hydrogen bonds are present in all complexes, often involving coordinated N–H and O4 of T3′, the π-system of T5′ and phosphates in the DNA backbone. C–H⋯O, C–H⋯π and C–H⋯H–C contacts are also observed. This analysis also highlights contacts between the O6 of 5′ guanine and platinum in all complexes, approximately in the apical position over the square-plane of coordinated nitrogen. The long-range nature of this contact, and the low electron density at the corresponding critical point, suggest that this is not a fifth coordination site, but rather an outer-sphere effect that makes only a small contribution to overall binding.
By means of examination of the reduced density gradient and the signs of the curvature of the electron density, such contacts can be classified as strongly stabilizing, stabilizing, or weakly stabilizing; no evidence of unfavorable steric clashes is found in any complex, indicating that all non-covalent interactions identified contribute to the overall stability of binding of the drug to its target. Further support for this view comes from approximate linear correlations between the sum of the electron density at all bond critical points located and the calculated binding energy. Thus, the importance of non-covalent interactions in determining the covalent binding of platinum drugs to DNA cannot be discounted.
Acknowledgements
JAP is grateful to the Leverhulme Trust for a Research Fellowship. We also thank the UK National Service for Computational Chemistry Software for a generous allocation of resources.References
- B. Rosenberg, L.V. Van Camp and T. Krigas, Nature, 1965, 205, 698 CrossRef CAS.
- B. Rosenberg, L.V. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451 CrossRef CAS.
- J. Reedijk, Chem. Commun., 1996, 801 RSC.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075 CrossRef CAS.
- M. A. Jakupec, M. Galanski and B. K. Keppler, Rev. Physiol., Biochem., Pharmacol., 2003, 146, 1 CrossRef CAS.
- J. M. Woynarowski, S. Faivre and M. C. Herzig, Mol. Pharmacol., 2000, 58, 920–7 CAS.
- Y. Wu, D. Bhattacharyya, C. L. King, I. Baskerville-Abraham, S.-H. Huh, G. Boysen, J. A. Swenberg, B. Temple, S. L. Campbell and S. G. Chaney, Biochemistry, 2007, 46, 6477 CrossRef CAS.
- (a) P. Carloni, W. Andreoni, J. Hutter, A. Curioni, P. Giannozzi and M. Parinello, Chem. Phys. Lett., 1995, 234, 50–56 CrossRef CAS; (b) P. N. V. Pavankumar, P. Seetharamulu, S. Yao, J. D. Saxe, D. G. Reddy and F. H. Hausheer, J. Comput. Chem., 1999, 20, 365–382 CrossRef CAS.
- (a) Y. Zhang, Z. Guo and X.-Z. You, J. Am. Chem. Soc., 2001, 123, 9378–9387 CrossRef CAS; (b) J. K.-C. Lau and D. V. Deubel, J. Chem. Theory Comput., 2006, 2, 103–106 CrossRef CAS; (c) J. Raber, C. Zhu and L. A. Eriksson, Mol. Phys., 2004, 102, 2537–2544 CrossRef CAS; (d) J. V. Burda and M. Zeizinger, J. Comput. Chem., 2005, 26, 907–914 CrossRef CAS.
- (a) H. Basch, M. Krauss, W. J. Stevens and D. Cohen, Inorg. Chem., 1986, 25, 684–688 CrossRef CAS; (b) M.-H. Baik, R. A. Friesner and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14082–14092 CrossRef CAS; (c) J. V. Burda and J. Leszczynski, Inorg. Chem., 2003, 42, 7162–7172 CrossRef CAS; (d) A. Robertazzi and J. A. Platts, Inorg. Chem., 2005, 44, 267–274 CrossRef CAS; (e) Y. Mantri, S. J. Lippard and M.-H. Baik, J. Am. Chem. Soc., 2007, 129, 5023–5030 CrossRef CAS.
- E. D. Scheeff, J. M. Briggs and S. B. Howell, Mol. Pharm., 1999, 56, 633–643 CAS.
- (a) K. Spiegel, U. Rothlisberger and P. Carloni, J. Phys. Chem. B, 2004, 108, 2699–2707 CrossRef CAS; (b) K. Spiegel and A. Magistrato, Org. Biomol. Chem., 2006, 4, 2507–2517 RSC; (c) A. Magistrato, P. Ruggerone, K. Spiegel, P. Carloni and J. Reedijk, J. Phys. Chem. B, 2006, 110, 3604–3613 CrossRef CAS; (d) K. Spiegel, A. Magistrato, P. Maurer, P. Ruggerone, U. Rothlisberger, P. Carloni, J. Reedijk and M. L. Klein, J. Comput. Chem., 2008, 29, 38–49 CrossRef CAS.
- (a) C. Gossens, I. Tavernelli and U. Rothlisberger, Chimia, 2005, 59, 81–84 CrossRef CAS; (b) A. Robertazzi and J. A. Platts, Chem.–Eur. J., 2006, 12, 5747–5756 CrossRef CAS; (c) T. Matsui, Y. Shigeta and K. Hirao, J. Phys. Chem. B, 2007, 111, 1176–1181 CrossRef CAS; (d) M.-E. Moret, I. Tavernelli and U. Rothlisberger, J. Phys. Chem. B, 2009, 113, 7737–7744 CrossRef CAS; (e) D. Ambrosek, P.-F. Loos, X. Assfeld and C. Daniel, J. Inorg. Biochem., 2010, 104, 893–901 CrossRef CAS; (f) J. P. Ceron-Carrasco, D. Jacquemin and E. Cauet, Phys. Chem. Chem. Phys., 2012, 14, 12457–12464 RSC; (g) A. V. Vargiu and A. Magistrato, Inorg. Chem., 2012, 51, 2046–2057 CrossRef CAS; (h) Z. Futera, J. A. Platts and J. V. Burda, J. Comput. Chem., 2012, 33, 2092–2101 CrossRef CAS.
- K. Gkionis and J. A. Platts, Comput. Theor. Chem., 2012, 993, 60–65 CrossRef CAS.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery Jr, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815 CrossRef CAS.
- M. J. Frisch et al., Gaussian09 Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS; (b) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS; (c) A. A. Haußerman, U. Dolg, M. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123 CrossRef.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- B. H. Besler, K. M. Merz Jr and P. A. Kollman, J. Comput. Chem., 1990, 11, 431 CrossRef CAS.
- P. Csaszar and P. Pulay, J. Mol. Struct., 1984, 114, 31 CrossRef CAS.
- D. Yang, S. S. van Boom, J. Reedijk, J. H. van Boom and A. H. Wang, Biochemistry, 1995, 34, 12912 CrossRef CAS.
- MOE 2009.10, Chemical Computing Group Inc., Montreal, 2009 Search PubMed.
- (a) T. Vreven, B. Mennucci, C. O. da Silva, K. Morokuma and J. J. Tomasi, Chem. Phys., 2001, 115, 62–72 CAS; (b) S. J. Mo, T. Vreven, B. Mennucci, K. Morokuma and J. Tomasi, Theor. Chem. Acc., 2004, 111, 154–61 CrossRef CAS.
- X.-J. Lu and W. K. Olson, Nucleic Acids Res., 2003, 31, 5108–21 CrossRef CAS.
- K. Gkionis, J. G. Hill, S. P. Oldfield and J. A. Platts, J. Mol. Model., 2009, 15, 1051 CrossRef CAS.
- (a) R. W. F. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS; (b) R. W. F. Bader, Found. Chem., 2011, 13, 11–37 CrossRef; (c) P. Dem'yanov and P. Polestshuk, Chem.–Eur. J., 2012, 18, 4982–4993 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. J. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef.
- E. R. Johnson, R. A. Wolkow and G. A. Dilabio, Chem. Phys. Lett., 2004, 394, 334–338 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra23041d |
| ‡ Tests on larger fragments with cisplatin, such as double-stranded TGGT, showed no difference in drug-DNA critical points found. |
| This journal is © The Royal Society of Chemistry 2013 |