 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution structure of hyperactive type I antifreeze protein†
Luuk L. C.
Olijve
a,
Tianjun
Sun
b,
Theyencheri
Narayanan
c,
Corinne
Jud
d,
Peter L.
Davies
b and
Ilja K.
Voets
*a
aLaboratory for Macromolecular and Organic Chemistry and Institute for Complex Molecular Systems, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: I.Voets@tue.nl
bDepartment of Biomedical and Molecular Sciences, Queen's University, Kingston, Ontario, K7L 3N6, Canada
cESRF, 6 rue Jules Horowitz, F-38043, Grenoble, France
dAdolphe Merkle Institute, University of Fribourg, Route de l'Ancienne Papeterie, P.O. Box 209, CH-1723 Marly 1, Switzerland
First published on 14th February 2013
Abstract
Antifreeze proteins (AFPs) protect freeze-intolerant fish species living in icy polar waters against freeze damage. A 34 kDa dimeric type I antifreeze protein (wfAFP 1h) with unusually high activity in comparison to all other antifreeze proteins in fish was recently discovered in the winter flounder. We have measured the size and shape of this hyperactive AFP by using small angle X-ray scattering. Our results show that wfAFP 1h adopts a long cylindrical shape with a length of 19 ± 2 nm and a diameter of 2.3 ± 0.2 nm, which means that wfAFP 1h does not form a fully extended helical dimer in solution. These findings call for a revision of the structural model of wfAFP 1h and the concept of a flat, threonine-rich ice-binding site extending down the length of the protein. Instead, the hyperactive nature of wfAFP 1h may be derived from a unique 3D arrangement of the helices —yet to be resolved— by which it is able to bind to ice surfaces.
Introduction
The freezing point of seawater (−1.9 °C) is more than 1 °C below the equilibrium freezing point of the body fluids of fish surviving in icy polar seawaters. To protect themselves in this undercooled state from rapidly freezing upon contact with ice, these fish produce antifreeze plasma proteins that lower the freezing point by stopping the growth of ice crystals.1 Antifreeze proteins (AFPs) have specific structural properties that enable them to adsorb to the surface of ice crystals thereby restricting the addition of water molecules to the growing ice surface. The ice surface is forced to grow with a thermodynamically unfavorable curvature, leading to a local depression of the non-equilibrium freezing point, which results in an arrest of further ice growth.2 Over the years, a wide diversity of AFP structures have been found in many different organisms ranging from fish3 to insects,4 bacteria5 and fungi.6In 2004, Marshall and coworkers reported an unusually potent type I AFP isoform in winter flounder (wfAFP 1h), that was 10 to 100 times more active than the previously discovered type I AFP in the same fish species.7 Circular dichroism measurements at 4 °C showed that this alanine-rich, 195 residue-long (16.7 kDa) protein is almost entirely α-helical.8 Analytical ultracentrifugation analysis and gel permeation chromatography furthermore indicated that this protein forms a dimer in solution and that it has an extreme asymmetry with an estimated axial ratio of L/d = 18.9 Based on these data, Marshall and coworkers concluded that wfAFP 1h is a helical dimeric rod of 29 nm in length and 1.6 nm in diameter, consisting of two long helices associated side-by-side. A coiled-coil structure was ruled out based on the paucity of hydrophobic amino acid residues (e.g., Leu, Ile, Val), thereby explaining the instability of the protein (Tdenat ∼9 °C).8,10 Also, a coiled-coil structure would interfere with regularly positioned threonine residues appearing on a flat ice-binding site along the length of the protein. The complex was therefore modelled as an antiparallel dimer composed of two straight helices with relatively weak interactions between the alanine (Ala) side chains, i.e., as an Ala-zipper, resulting in a closer packing with L = 27.5 nm and d = 1.6 nm giving an axial ratio of L/d = 17.10 In this model both subunits could simultaneously engage in ice-binding.
To obtain reliable evidence on the exact dimensions and multimeric state of wfAFP 1h, we have performed small angle X-ray scattering (SAXS) experiments on the protein in solution. SAXS is a powerful tool for characterizing the structure of macromolecules in their native environment.11 Coherent scattering of X-rays by randomly oriented proteins gives information on their size, shape and molar mass. In contrast to X-ray diffraction, SAXS does not require crystalline samples which greatly broadens its application perspective, but reduces the attainable resolution.
The results presented here confirm the dimeric state of wfAFP 1h and provide accurate values for its dimensions. The length of wfAFP 1h as determined from the SAXS experiments is found to be smaller than hypothesized based on the previous model,10 which implies that the helical dimer is arranged differently and not fully stretched out.
Materials and methods
Sample preparation
Recombinant wfAFP 1h was produced as previously described12 with the following modification. Instead of ice affinity purification, three rounds of ammonium sulfate precipitation were used. This is an effective method for purification because wfAFP 1h precipitates from solution at a relatively low percent saturation of ammonium sulfate (28%) while most Escherichia coliproteins remain soluble. The wfAFP 1h pellet from the last precipitation was re-suspended and dialyzed overnight in 50 mM HEPES buffer (pH 6.5) to remove ammonium sulfate. Purity was estimated by SDS-PAGE, and the protein concentration was measured by amino acid analysis. Recombinant wfAFP 1h at 15 mg mL−1 was stored at 4 °C and used within 2–3 weeks of its preparation.Small angle X-ray scattering
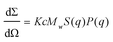 | (1) |
First, the scattering data were analyzed using a Guinier approximation to extract the radius of gyration, Rg, and the forward scattering intensity, I0, which is dΣ/dΩ(q → 0). For monodisperse globular proteins, the Guinier approximation is valid for qRg ≤ 1.3 and Rg and I0 were determined from the slope and y-intercept of the Guinier plot ln(dΣ/dΩ(q)) vs. q2 using PRIMUS from the ATSAS software package.15 For a rod-like particle, a modified Guinier approximation can be used in the q-range where dΣ/dΩ(q) ∝ q−α with α ∼ 1 to extract the cross-sectional radius of the cylinder from the determined cross-sectional radius of gyration, with 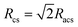 .
.
Using the obtained radius of gyration and the cross-sectional radius from the Guinier approximation, the length of a cylinder can be calculated by
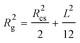 | (2) |
Subsequently, I0 = dΣ/dΩ(q → 0) was used to calculate the molar mass of the protein (complex), which is listed in Table 1, according to
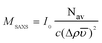 | (3) |
![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) = 0.7302 in cm3 g−1 calculated using SEDNTERP.17
= 0.7302 in cm3 g−1 calculated using SEDNTERP.17
| a Reported for 1.9 mg mL−1 measurement. b D max is a model parameter in the P(r) calculation and no variance associated with Dmax can be calculated. | |
|---|---|
| Data-collection parameters | |
| Beamline | ID02 (ESRF, Grenoble, France) |
| Operating energy (keV) | 12.46 |
| Flux (photons s−1) | 1012 |
| Wavelength (nm) | 0.1 |
| q range (nm−1) | 0.064–3.85 |
| Sample-to-detector distance (m) | 1.5 and 3 |
| Total exposure time (s) | 3 |
| Concentration (mg mL−1) | 2–15 |
| T (°C) | 5 |
| Sample holder | Poly carbonate flow-through capillary, ø = 1.9 mm |
| Structural parameters a | |
| I 0 (cm−1) [Guinier] | 0.055 ± 0.002 |
| R g (nm) [Guinier] | 5.0 ± 0.09 |
| I(0) (cm−1) [P(r)] | 0.0557 ± 0.001 |
| R g (nm) [P(r)] | 5.7 ± 0.1 |
| D max (nm)b | 22 |
| Molecular mass determination | |
| Partial specific volume (cm3 g−1) | 0.7302 |
| Contrast (Δρ × 1010 cm−2) | 3.060 |
| Molecular mass Mw from I(0) (Da) | 34700 ± 1100 |
| Calculated monomeric Mw from sequence (Da) | 16685 |
| Software employed | |
| Primary data reduction | SAXSutilities |
| Data processing | PRIMUS |
| Ab initio analysis | DAMMIF |
| Validation and averaging | DAMAVER |
| Computation of model intensities | CRYSOL |
| Rigid-body modelling | SASREF |
| Graphical representation | PyMol and SITUS |
Finally, we have analyzed the scattering profiles in the entire recorded q-range with various form factor models using the software package SASfit18 assuming that wfAFP 1h was measured in an ideal dilute solution where interparticle interactions can be neglected (i.e., S(q) = 1). Two form factors were used; one describing a cylindrical object19 and the other describing a worm-like chain model according to the equations reported by Pedersen and co-workers.20
Molecular shape reconstruction
To derive information on the solution structure of the protein, the molecular shape was reconstructed using simulated annealing methods. First, the radial distribution function (RDF), P(r), which describes the probable frequency of interatomic vector lengths (r) within the scattering particle, was obtained upon indirect inverse Fourier transformation of the scattering data using GNOM.21 The maximum linear dimension (Dmax) was set to approximately 3 × Rg and adjusted to give the best fit to the experimental data. The RDF was considered to be zero at r = 0 Å and approaches zero at Dmax.The GNOM output files were used as input for simulated annealing calculations over the range 0.08 < q < 3.8 nm−1 using the online version of DAMMIF.22 For each scattering curve, 10 independent bead models were generated without predefined shape or symmetry. The 10 different models were superimposed using DAMSEL and DAMSUP. Next, DAMAVER was used to average the aligned models and compute the probability map.23 Finally, DAMFILT was used to filter the averaged model to give a structure that has high densities on the probability map.
Results and discussion
Guinier and form factor analysis
Fig. 1A displays the small angle X-ray scattering profiles obtained at four different concentrations (2–15 mg mL−1) at the high-brilliance beamline ID02 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The accessible q-range covers the full SAXS profile of wfAFP 1h, which extends from the Guinier regime at low q-values up to the form factor oscillation at high q-values with a slope of ∼q−1 typical of cylindrical structures at intermediate q-values. A Guinier approximation (Fig. 1B) was used to obtain the radius of gyration Rg and molecular weight Mw. The estimated molecular weight of Mw = 34.7 ± 2.5 kDa corresponds to the formation of a dimer in solution. By using a modified Guinier approximation for a rod (Fig. 1C) the cross-sectional radius Rcs is obtained. Using these structural parameters, the length of the protein complex was calculated using eqn (2), confirming that wfAFP 1h adopts a highly elongated structure with a length L = 17.6 ± 0.9 nm and a cross-sectional radius of Rcs = 1.12 ± 0.01 nm, giving an axial ratio of L/d = 7.9 ± 0.4 (Table 2).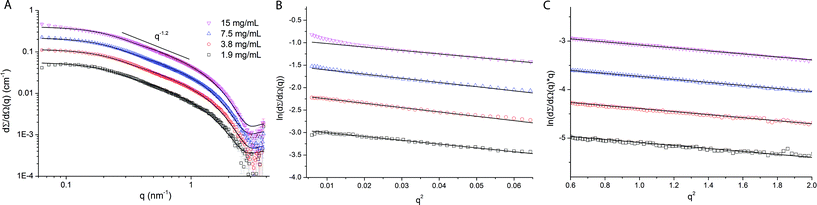 | ||
| Fig. 1 (A) Small angle X-ray scattering profiles of 2–15 mg mL−1 wfAFP 1h in 50 mM HEPES at pH 6.5 and T = 5 °C. The experimental data (symbols) are well described by a cylindrical form factor (solid lines). (B) Fit of the Guinier approximation of the experimental data of wfAFP 1h. The used q-ranges are 0.08 ≤ q ≤ 0.23 nm−1 (1.9 mg mL−1), 0.09 ≤ q ≤ 0.21 nm−1 (3.8 mg mL−1), 0.13 ≤= q ≤ 0.21 nm−1 (7.5 mg mL−1) and 0.17 ≤ q ≤ 0.23 nm−1 (15 mg mL−1). (C) Fit of a modified Guinier approximation for a rod to the experimental data. The used q-ranges are 0.65 ≤ q ≤ 1.49 nm−1 (1.9 mg mL−1), 0.65 ≤ q ≤ 1.62 nm−1 (3.8 mg mL−1), 0.65 ≤= q ≤ 1.57 nm−1 (7.5 mg mL−1) and 0.73 ≤ q ≤ 1.50 nm−1 (15 mg mL−1). | ||
| Conc. (mg mL−1) | R g (nm) | M w (kDa) | R cs (nm) | L (nm) | L/d |
|---|---|---|---|---|---|
| 1.9 | 5.0 | 34.7 | 1.11 | 17.3 | 7.8 |
| 3.8 | 5.4 | 37.3 | 1.12 | 18.5 | 8.2 |
| 7.5 | 5.3 | 35.4 | 1.12 | 18.1 | 8.1 |
| 15 | 4.8 | 31.4 | 1.11 | 16.4 | 7.4 |
| Average | 5.1 | 34.7 | 1.12 | 17.6 | 7.9 |
| Std | 0.3 | 2.5 | 0.01 | 0.9 | 0.4 |
The experimental data are also well described by a rigid cylindrical form factor (Fig. 1A and Table 3) and a worm-like chain model (Fig. S1 and Table S1 of the ESI†). The cylindrical model assumes a rigid cylinder with uniform scattering length density. The structural parameters extracted from fitting the data with a cylindrical form factor are the cross-sectional radius Rcs = 1.26 ± 0.02 nm that is close to the Rcs obtained from the Guinier analysis, and the length L = 19.7 ± 0.2 nm, which is slightly larger.
| Conc. (mg mL−1) | R g (nm) | R cs (nm) | L (nm) | L/d | χ 2 |
|---|---|---|---|---|---|
| 1.9 | 5.8 | 1.28 | 19.9 | 7.7 | 0.98 |
| 3.8 | 5.7 | 1.23 | 19.5 | 7.9 | 0.85 |
| 7.5 | 5.8 | 1.26 | 19.8 | 7.9 | 0.98 |
| 15 | 5.7 | 1.25 | 19.6 | 7.8 | 0.92 |
| Average | 5.8 | 1.26 | 19.7 | 7.8 | 0.93 |
| Std | 0.1 | 0.02 | 0.2 | 0.1 | 0.06 |
Structural model of the protein complex
From the results of the Guinier and form factor analyses it is evident that wfAFP 1h has a highly extended shape with a length of L = 19 ± 2 nm and diameter of d = 2.3 ± 0.2 nm. These dimensions imply that wfAFP 1h cannot form a dimer of two fully extended helices, since the latter would correspond to a length of ∼27.5 nm. To derive information on the shape of the dimer, we have reconstructed the ab initio shape of the protein from simulated annealing calculations using DAMMIF, which features an unlimited and adapting search volume that avoids boundary effects, especially in highly elongated objects.21 The averaged and filtered volumetric map of wfAFP 1h shows a rod-like shape with a length of 18.5 nm (Fig. 2D), resembling the dimensions found in the Guinier and form factor analysis. Given the dimensions and high helical content of wfAFP 1h, it is evident that the molecular structure of this hyperactive antifreeze protein is unlike any antifreeze protein found so far. The β-helical folds of hyperactive AFPs from insects display a flat β-sheet ice-binding surface with regularly positioned threonine residues.4 Hence, a likely arrangement of the dimer would be two helices associated side-by-side, enabling a uniform presentation of threonine residues along the length of the protein.10 Yet, this structure is not consistent with the SAXS results, which means that a different conformation of the helices might confer the hyperactive antifreeze activity.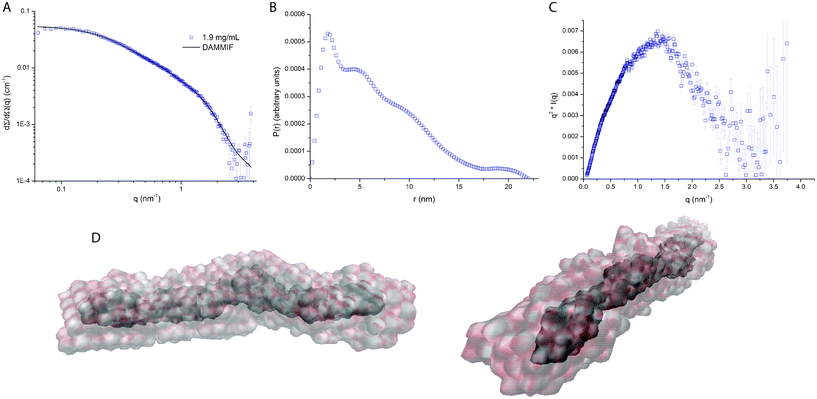 | ||
| Fig. 2 (A) Fit of simulated annealing with DAMMIF, (B) radial distribution function (RDF) obtained after inverse Fourier transformation of the experimental data, (C) Kratky plot showing a parabolic shape indicative of a folded state of the protein,24 and (D) 10 calculations with DAMMIF were performed to produce the average and filtered volumetric map of wfAFP 1h, showing a rod-like shape with a length of 18.5 nm (final χ2 = 2.57). SITUS was used to create the volumetric maps of the bead models. | ||
To get more insight into the 3D arrangement of the helices in the protein complex of wfAFP 1h, we have performed rigid body modelling using SASREF.25 SASREF uses multiple rigid subunits with known atomic coordinates to perform quaternary structure modelling of a protein complex against the experimental data. Since no crystallographic data of the subdomains in the protein complex of wfAFP 1h is available, we have constructed two long α-helices with a length of 18.5 nm and two short helices to account for the remaining number of residues of the protein complex. These subunits and contact conditions (Fig. 3A) were provided as input for SASREF. The result of the rigid body modelling (Fig. 3C) seems consistent with the results of the more coarse-grained modelling approach and comes very close to the hypothesized structure in which the protein complex is able to display regularly positioned threonine residues on its ice-binding face, enabling a large protein surface area to engage in ice-binding which could explain the hyperactive nature.
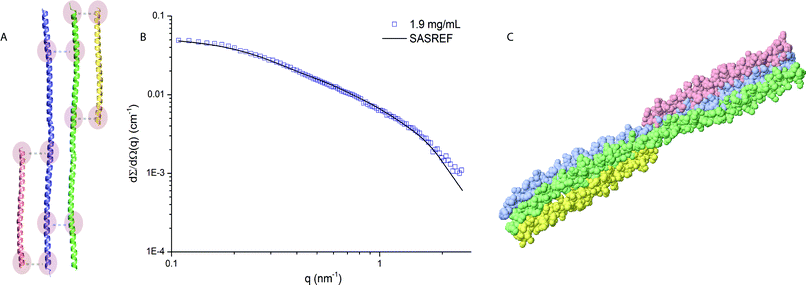 | ||
| Fig. 3 (A) Subdomains with provided contact conditions used for the rigid body modelling. The subdomains were constructed from the PDB entry 3VEM. (B) Fit of the model to the experimental data (final χ2 = 3.79), using P1 symmetry. Data range used for the rigid body modelling 0.08 ≤ q ≥ 2.5 nm−1. (C) Resulting structural model from the rigid body modelling. | ||
However, we need to draw attention to the limitations of the resulting structural model. Firstly, the initial contact conditions influence the SASREF results. Therefore, about six contact conditions per complex were set and various contact conditions were tested (Fig. S3–5 of the ESI†). Reducing the number of contact conditions resulted in one of the subunits losing contact with the complex. Secondly, we propose the structural model in Fig. 3C on the basis of additional information rather than the goodness of fit of the different SASREF models. Thirdly, the subunits are adapted from PDB entries of other proteins. Hence, the validity of the model needs to be tested with additional biochemical experiments.
Protein complex has unique molecular structure
Given the extraordinary shape of the protein that is very distinct from any other AFP found so far, we have searched in the Protein Database (PDB) for proteins with similar shape based on four criteria: (1) high α-helicity, (2) dimeric state, (3) number of residues close to 195 as found for wfAFP 1h and (4) length and axial ratio. We have compared the experimental data with theoretical scattering profiles of 10 different crystal structures, and computed the theoretical scattering curves from the electron density profiles of the selected proteins using the known atomic coordinates deposited in the PDB by using CRYSOL.26 The theoretical scattering curves were then compared to the experimental scattering data (see Fig. S6–7 of the ESI†). Only a handful of the selected α-helix rich dimers of the protein database have a structure that is comparable yet not identical to that of wfAFP 1h. Most of the matching protein structures originate from the cytoskeleton or muscle. Clearly, the protein complex adopts a structure with high asymmetry, which is unique for a plasma protein.Conclusions
Structural data form the basis for understanding the hyperactive nature of wfAFP 1h. Our SAXS experiments provide the first reliable and independent estimation of the length, diameter and molecular weight of wfAFP 1h in solution. We find that wfAFP 1h self-assembles into a dimer with a highly extended (i.e., non-globular) conformation that is unusual for a plasma protein and mostly found in muscle and cytoskeleton proteins. The protein complex has a rigid cylindrical shape with a length and diameter of 19 ± 2 nm and 2.3 ± 0.2 nm, respectively. This demonstrates unequivocally that the two α-helices in the dimer are not fully extended (i.e., L = 27.5 nm) contrary to what was hypothesized previously, irrespective of whether they are arranged in a coiled-coil or parallel conformation. Furthermore, if the helical dimers would be held together via an interaction of the alanine side chains, the packing would be too close to match the experimentally observed diameter suggesting that wfAFP 1h adopts a more complex conformation. Further evidence from biochemical methods and/or diffraction studies is necessary to gain more insight into the 3D arrangement of the two monomers in the protein complex and the positioning of regularly placed threonine residues at the ice-binding site.Acknowledgements
The authors acknowledge valuable suggestions by Dr Maxim Petoukhov. The small angle X-ray experiments were performed at the high-brilliance beamline ID02 of the European Synchrotron Radiation Facility, Grenoble, France. I.K.V. is grateful for financial support from the Netherlands Organisation for Scientific Research (NWO – VENI Grant: 700.10.406) and the European Union through the Marie Curie Fellowship program FP7-PEOPLE-2011-CIG (Contract No. 293788). P.L.D. holds a Canada Research Chair in Protein Engineering and acknowledges financial support from the Canadian Institutes for Health Research.References
- A. L. DeVries and D. E. Wohlschlag, Science, 1969, 163, 1073–1075 CAS.
- J. A. Raymond and A. L. DeVries, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2589–2593 CrossRef CAS.
- G. L. Fletcher, C. L. Hew and P. L. Davies, Annu. Rev. Physiol., 2001, 63, 359–390 CrossRef CAS.
- (a) B. L. Pentelute, Z. P. Gates, V. Tereshko, J. L. Dashnau, J. M. Vanderkooi, A. A. Kossiakoff and S. B. Kent, J. Am. Chem. Soc., 2008, 130, 9695–9701 CrossRef CAS; (b) S. P. Greather, M. J. Kuipers, S. M. Gagné, V. K. Walker, Z. Jia, B. D. Sykes and P. L. Davies, Nature, 2000, 406, 325–328 CrossRef.
- C. P. Garnham, R. L. Campbell and P. L. Davies, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7363–7367 CrossRef CAS.
- J. H. Lee, A. K. Park, H. Do, K. S. Park, S. H. Moh, Y. M. Chi and H. J. Kim, J. Biol. Chem., 2012, 287, 11460–11468 CrossRef CAS.
- C. B. Marshall, G. L. Fletcher and P. L. Davies, Nature, 2004, 429, 153 CrossRef CAS.
- S. Y. Gauthier, C. B. Marshall, G. L. Fletcher and P. L. Davies, FEBS J., 2005, 272, 4439–4449 CrossRef CAS.
- C. B. Marshall, A. Chakrabartty and P. L. Davies, J. Biol. Chem., 2005, 280, 17920–17929 CrossRef CAS.
- L. A. Graham, C. B. Marshall, F.-H. Lin, R. L. Campbell and P. L. Davies, Biochemistry, 2008, 47, 2051–2063 CrossRef CAS.
- (a) D. I. Svergun and M. H. J. Koch, Rep. Prog. Phys., 2003, 66, 1735–1782 CrossRef CAS; (b) C. D. Putnam, M. Hammel, G. L. Hura and J. A. Tainer, Q. Rev. Biophys., 2007, 40, 191–285 CrossRef CAS.
- F. H. Lin, T. Sun, G. L. Fletcher and P. L. Davies, Protein Expression Purif., 2012, 82, 75–82 CrossRef CAS.
- M. Sztucki, E. Di Cola and T. Narayanan, J. Appl. Crystallogr., 2010, 43, 1479–1487 CrossRef CAS.
- (a) Small-angle X-ray Scattering, ed. O. Glatter and O. Kratky, Academic Press, London, 1982 Search PubMed; (b) E. Mylonas and D. I. Svergun, J. Appl. Crystallogr., 2007, 40, s245–s249 CrossRef CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- Online scattering length density calculator of NIST http://www.ncnr.nist.gov/resources/sldcalc.html.
- D. A. Jacques and J. Trewhella, Protein Sci., 2010, 19, 642–657 CrossRef CAS.
- SASfit software package for fitting small angle scattering curves http://kur.web.psi.ch/sans1/SANSSoft/sasfit.html.
- Neutron, X-rays and light: scattering methods applied to soft condensed matter, ed. P. Lindner and T. Zemb, Elsevier, Amsterdam, 2002 Search PubMed.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
- D. I. Svergun, J. Appl. Crystallogr., 1992, 25, 495–503 CrossRef.
- D. Franke and D. I. Svergun, J. Appl. Crystallogr., 2009, 42, 342–346 CrossRef CAS.
- V. V. Volkov and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 860–864 CrossRef CAS.
- D. A. Jacques, J. M. Guss, D. I. Svergun and J. Trewhella, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 620–626 CrossRef.
- M. V. Petoukhov and D. I. Svergun, Biophys. J., 2005, 89, 1237–1250 CrossRef CAS.
- D. I. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3ra22729d |
| This journal is © The Royal Society of Chemistry 2013 |