The facile synthesis of 2-bromoindoles via Cs2CO3-promoted intramolecular cyclization of 2-(gem-dibromovinyl)anilines under transition-metal-free conditions†
Pinhua
Li
a,
Yong
Ji
b,
Wei
Chen
a,
Xiuli
Zhang
b and
Lei
Wang
*ac
aDepartment of Chemistry, Huaibei Normal University, Huaibei, Anhui 235000, P. R. China. E-mail: leiwang@chnu.edu.cn; Tel: +86-561-380-2069; Fax: +86-561-309-0518
bDepartment of Chemistry, Anhui Agricultural University, Hefei, Anhui 230062, P. R. China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 30th October 2012
Abstract
2-Bromoindoles were readily prepared through a facile Cs2CO3-promoted intramolecular cyclization of 2-(gem-bromovinyl)-N-methylsulfonylanilines in excellent yields under transition-metal-free conditions. This methodology could be extended to the synthesis of corresponding 2-chloroindoles. The reaction mechanism suggested that cyclization occurs through a key intermediate, phenylethynyl bromide, followed by cyclization in one-pot.
Introduction
The indole nucleus is an important structural motif frequently found in natural products, materials, and therapeutic agents, such as antibiotic, anticancer and anti HIV activity.1 Indole derivatives are also used as the starting materials for the synthesis of a large number of alkaloids. Many halogenated indoles are also found in nature, and several brominated indole natural products have been isolated.2 They are also present in biologically active compounds, for example, I (Convolutindole A, isolated from the marine bryozoan Amathia convolute, a very potent nematocide),2e and II, III and IV (isolated from the marine blue-green alga Riuularia firma) (Fig. 1).2f,g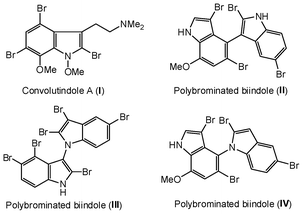 | ||
| Fig. 1 Representative biologically active 2-bromoindoles. | ||
Because of the electron-rich character of indoles, the synthesis of brominated indoles is a challenging project. The most straightforward method is electrophilic bromination, and 3-bromoindoles are obtained, along with oxidation side-products.3 While unprotected 2-bromoindoles are useful for numerous applications,4 their syntheses are rare and difficult. Although the synthesis of 2-bromoindole based on lithiation of the free indole, then bromination was reported, extension of its derivatives was less well investigated owing to the limitations of regioselectivity and functional group tolerance.5
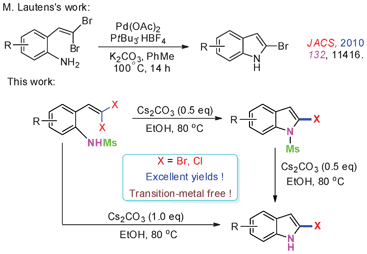
Gem-Dihaloolefins, as an important kind of synthetic intermediate due to their high reactivity and easy availability, have attracted considerable attention in recent years.6 Most importantly, the preparation of various substituted indoles from 2-(gem-dibromovinyl)anilines and 2-(gem-dibromovinyl)(N-substituted)anilines via transition-metal-catalyzed tandem cross-coupling strategies, such as C–N/C–C,7 C–N/C–N,8 C–N/C–P,7a C–N/C–H,9 C–N/carbonylation,10 and C–N/carbonylation/C–C reactions11 have been developed. Recently, an elegant method for the synthesis of 2-bromoindoles was developed by Lautens et al. via Pd/PtBu3-catalyzed intramolecular reactions of 2-(gem-dibromovinyl)anilines.12
In continuing our efforts on the organic reactions of gem-dihaloolefins under metal-free conditions,13 herein we describe an efficient Cs2CO3-promoted intramolecular cyclization of 2-(gem-dibromovinyl)-N-methylsulfonylanilines14 for the facile synthesis of 2-bromoindoles and 2-bromo-N-methylsulfonylindoles by controlling the amount of Cs2CO3 under transition-metal-free, fluoride-free, mild and environmentally friendly reaction conditions in excellent yields. In addition, this methodology can also be extended to the synthesis of 2-chloroindole derivatives (Scheme 1).
 | ||
| Scheme 1 Preparation of 2-bromo(chloro)indoles. | ||
Results and discussion
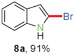
In our initial attempt to prepare 2-bromoindole (8a) from 2-(gem-dibromovinyl)aniline (1a) in the presence of Cs2CO3 (1 equiv.) in EtOH at 80 °C, however, no 8a was obtained (Table 1, entry 1). When N-substituted derivatives 2a–5a were used as substrates, there was also no desired 8a detected (Table 1, entries 2–5). To our delight, a 91% yield of 8a was obtained when 2-(gem-dibromovinyl)-N-methylsulfonylaniline (6a) was used (Table 1, entry 6). 2-(gem-Dibromovinyl)-N-(p-tolylsulfonyl)aniline (7a) was inferior and gave 8a in 70% yield (Table 1, entry 7). This result indicated that the cyclization depends on the nitrogen substituents of substrates. When the amine is activated by a strong electron-withdrawing substituent such as sulfonyl, the tandem reaction can proceed efficiently in one-pot. The sulfonyl group acts as a traceless dual-activating group to accomplish indole cyclization, and after indole formation, the sulfonyl group is deprotected via N–S bond cleavage.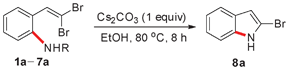
To further optimize the reaction conditions for the synthesis of 2-bromoindole (8a) through a base-mediated intramolecular reaction of 2-(gem-dibromovinyl)-N-methylsulfonylaniline (6a), a variety of bases were examined. As can be seen from Table 2, Cs2CO3 exhibited the highest reactivity in EtOH among the bases tested. t-BuOK, K2CO3, K3PO4, BuOLi, Na2CO3, KF and CsF were less effective (Table 2, entries 1–8). DBU, DIPEA, Et3N, DABCO and pyridine were ineffective (Table 2, entries 9–13). The solvent also played an important role in the reaction. EtOH was found to be the best medium for the reaction when Cs2CO3 was used as a promoter. Other solvents, DMSO, DMF, THF, MeCN, toluene and dioxane were less effective, and 18–85% yields of 8a were obtained (Table 2, entries 14–19). However, the reaction did not occur in H2O (Table 2, entry 20). When the reaction was performed in the presence of Cs2CO3 in EtOH at a temperature less than 50 °C, no reaction was observed. The temperature of 80 °C was found to be optimal (Table 2, entries 21–23). With respect to the amount of Cs2CO3 used in the reaction, 1.0 equiv. Cs2CO3 was found to be the best choice (Table 2, entry 24).
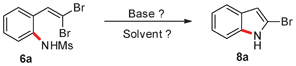
| Entry | Base | Solvent/T (°C) | Yield (%)b |
|---|---|---|---|
| a Reaction conditions: 2-(gem-dibromovinyl)-N-methylsulfonylaniline (6a, 0.50 mmol), base (0.50 mmol), solvent (2.0 mL) at the temperature indicated in Table 2 for 8 h. b Isolated yields. c Cs2CO3 (1.0 mmol) was used. | |||
| 1 | Cs2CO3 | EtOH/80 | 91 |
| 2 | t-BuOK | EtOH/80 | 80 |
| 3 | K2CO3 | EtOH/80 | 51 |
| 4 | K3PO4 | EtOH/80 | 35 |
| 5 | t-BuOLi | EtOH/80 | 24 |
| 6 | Na2CO3 | EtOH/80 | 18 |
| 7 | KF | EtOH/80 | 15 |
| 8 | CsF | EtOH/80 | 13 |
| 9 | DBU | EtOH/80 | NR |
| 10 | DIPEA | EtOH/80 | NR |
| 11 | Et3N | EtOH/80 | NR |
| 12 | DABCO | EtOH/80 | NR |
| 13 | Pyridine | EtOH/80 | NR |
| 14 | Cs2CO3 | DMSO/80 | 85 |
| 15 | Cs2CO3 | DMF/80 | 73 |
| 16 | Cs2CO3 | THF/70 | 67 |
| 17 | Cs2CO3 | MeCN/80 | 62 |
| 18 | Cs2CO3 | Toluene/80 | 22 |
| 19 | Cs2CO3 | Dioxane/80 | 18 |
| 20 | Cs2CO3 | H2O/80 | NR |
| 21 | Cs2CO3 | EtOH/50 | NR |
| 22 | Cs2CO3 | EtOH/60 | 12 |
| 23 | Cs2CO3 | EtOH/70 | 53 |
| 24 | Cs2CO3 | EtOH/80 | 92c |
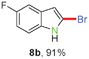
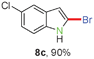
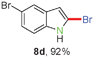
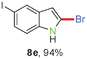
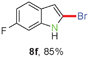
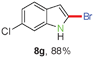
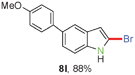
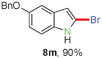
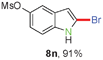
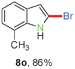
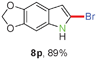
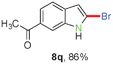
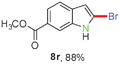
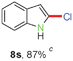
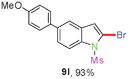
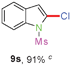
Having optimized the reaction conditions, the generality of this transformation was investigated and the results are listed in Table 3. A variety of 2-(gem-dibromovinyl)-N-methylsulfonylanilines containing substituents on the benzene rings were examined. The results indicated that a number of functional groups, including both electron-withdrawing and electron-donating ones were tolerated, and the corresponding 2-bromoindoles were obtained in good to excellent yields (Table 3, 8a–r). The reaction proceeded very well with halogen substituents F, Cl, Br and I on the para-, or meta-positions of anilines (6b–j) and afforded high yields of indoles 8b–j, which are potential synthetic intermediates in organic synthesis and could provide further transformation via transition-metal-catalyzed cross-coupling reactions. The product yields of substrates derived from para-haloanilines were superior to those from meta-haloanilines (Table 3, 8b–evs.8f–j). 2-(gem-Dibromovinyl)-N-methylsulfonylanilines with an electron-donating functionality, such as CH3, p-CH3OC6H4, C6H5CH2O, CH3SO2O and OCH2O, on the anilines also underwent the tandem cyclization smoothly to generate the corresponding products 8k–p in 86–91% yields. It should be noted that the reaction could tolerate an ortho-substituted group (8o). Substrates, 6q and 6r, with electron-withdrawing substituents CH3OCO and CH3CO also gave the corresponding products 8q and 8r in 86 and 88% yields, respectively. A more remarkable observation was that the reaction also proceeded with 2-(gem-dichlorovinyl)-N-methylsulfonylaniline (6s) to afford 2-chloroindole (8s) in good yield.
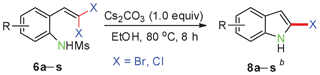
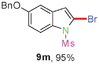
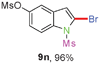
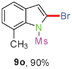
Sulfonamide is one of the most stable nitrogen protective groups. Generally, most sulfonamides are stable to alkaline hydrolysis, as well as to catalytic reduction. This property prompted us to prepare 2-bromo-N-methylsulfonylindoles from the corresponding 2-(gem-dibromovinyl)-N-methylsulfonylanilines. Although a facile synthesis of 2-bromo-N-methylsulfonylindoles via CuI-catalyzed intramolecular cross-coupling of gem-dibromoolefins was described,15 it is desirable to develop an efficient and practical method for their preparation under transition-metal-free conditions, which can overcome the drawbacks of its expensive, poisonous, and air-sensitive properties.16 During the investigation of the reaction of 6a under Cs2CO3/C2H5OH conditions, we were delighted to find that in the reaction of 6a in the presence of Cs2CO3 (0.50 equiv.) in EtOH at 80 °C, 2-bromo-N-methylsulfonylindole (9a) was isolated in 94% yield and no further deprotection product 6a was found. Then, a number of 2-(gem-dibromovinyl)-N-methylsulfonylanilines were examined to explore the generality of the reaction under Cs2CO3 (0.50 equiv.) in EtOH conditions. Satisfactorily, as shown in Table 4, substrates with both electron-withdrawing and electron-donating groups on the aromatic rings underwent the intramolecular tandem cyclization very cleanly to generate the corresponding 2-bromo-N-methylsulfonylindoles (9a–r) in excellent yields by controlling Cs2CO3 with 0.50 equiv. It is obvious that the yields of 9a–r are superior to the corresponding 8a–r, which are the further transformation products of 9a–rvia a deprecation process. Compared with the reaction of 6a–r to 8a–r, the similar steric and electronic effects of the substitutions on the anilines were also found in the reaction of 6a–r to 9a–r. It is noteworthy that 6s also could undergo the tandem reaction to generate the corresponding 2-chloro-N-methylsulfonyindole (9s) with good yield in DMF at 110 °C.
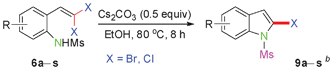
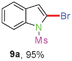
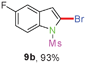
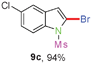
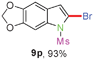
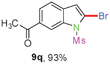
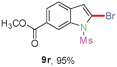
When the reaction scale was increased up to 10 mmol using Cs2CO3 and EtOH, 88% and 93% isolated yields of 8a and 9a were isolated from the starting material 6a, respectively, by increasing the reaction time to ensure completion (Scheme 2).
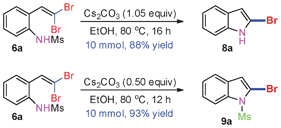 | ||
| Scheme 2 10 mmol scale reactions of 6a. | ||
2-Arylindoles exhibit good biological activities, such as antiestrogen, h5-HT2A antagonism, anti-inflammatory properties, and cytotoxicity.17 Palladium-catalyzed cross-coupling of 2-haloindoles with arylmetal species is a powerful method for preparing them,18 however, this application has been limited owing to the inaccessibility of 2-haloindoles.19 With the prepared 2-bromoindoles in our hands, converting them into the corresponding 2-arylindoles was investigated via palladium-catalyzed cross-coupling with boronic acids (Scheme 3). The results indicated that the corresponding products were obtained in excellent yields under Suzuki reaction conditions in the absence of ligand (Scheme 3). In the analogous studies, an essential ligand such as dppf, S-Phos, or PtBu3 is needed.7a,b,e In addition, 10d or 11d was obtained in a high yield while the reaction of 8d with 4-MeOC6H4B(OH)2 or PhB(OH)2 under controlled coupling conditions (Scheme 3, eqn (2) and (3)). This also implies that the reactivity of the 2-position C–Br bond of 2,5-dibromoindole (8d) is superior to its 5-position one towards palladium-catalyzed-coupling reactions.12
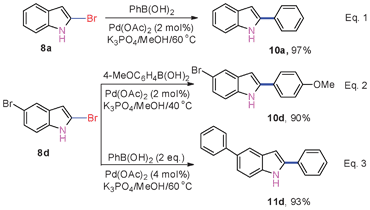 | ||
| Scheme 3 Suzuki coupling of 8a and 8d with boronic acids. | ||
To investigate the reaction mechanism, obtained 9a was further converted into 2-bromoindole (8a) in the presence of Cs2CO3 (0.5 equiv.), providing 95% yield (Scheme 4, eqn (1)). Meanwhile, 2-(gem-dibromovinyl)-N-(tert-butoxycarbonyl)aniline (3a) only afforded the corresponding phenylethynyl bromide intermediate A under Cs2CO3 (1.0 equiv.) conditions (Scheme 4, eqn (2)). When the reaction of 2-(1′-methyl-2′,2′-dichlorovinyl)-N-methylsulfonylanilines (6t) was performed in Cs2CO3/DMF, no product was detected and 6t was recovered in 98% yield (Scheme 4, eqn (3)). In addition, further isotope experiments indicated that when the reaction of deuterium-labeled 6a-D was performed under the present reaction conditions, 95% of the D-enriched element was lost in the product (Scheme 4, eqn (4)). These results suggested that the intramolecular tandem cyclization of 6a is through an intermediate phenylethynyl bromide,20 although it can not be obtained because of the fast reaction of B to 9a. The reaction pathway for the generation of 2-bromoindole (8a) was also shown in Scheme 4. Initially, an elimination of HBr from 6a to intermediate B proceeded smoothly in the presence of Cs2CO3, followed by an intramolecular nucleophilic addition of the nitrogen to the carbon–carbon triple bond of B, affording 2-bromo-N-methylsulfonylindole (9a) with the assistance of the carbonate anion. The as-obtained 9a then underwent a cleavage of the sulfonamide linkage to afford deprotection product 2-bromoindole (8a) under Cs2CO3 conditions.
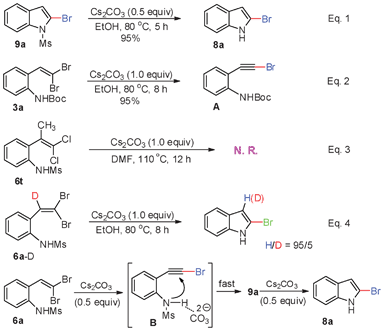 | ||
| Scheme 4 Possible reaction pathway and related experiments. | ||
Conclusion
In conclusion, we have developed a novel, efficient, economical, and facile Cs2CO3-promoted intramolecular cyclization of 2-(gem-dibromovinyl)-N-methylsulfonylanilines for the synthesis of 2-bromoindoles and 2-bromo-N-methylsulfonylindoles, respectively, under transition-metal-free conditions by controlling the amount of Cs2CO3 used in EtOH reaction media.21 Notably, this methodology can also be readily extended to the synthesis of 2-chloroindoles. The reaction mechanism investigation indicated that the tandem cyclization proceeds through a key intermediate phenylethynyl bromide, followed by the cyclization processes. The sulfonyl group acts as a traceless dual-activating group to undergo indole cyclization and easy deprotection in the presence of Cs2CO3. In addition, the further transformation of 2-bromoindoles via palladium-catalyzed Suzuki cross-coupling could be carried out in the absence of ligand. A detailed mechanistic study and further investigation on transition-metal-free reactions are currently underway in our laboratory.Acknowledgements
This work was financially supported by the National Science Foundation of China (Nos. 21172092, 20972057).References
- For selected reviews, see: (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS; (b) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS; (c) M. Somei and F. Yamada, Nat. Prod. Rep., 2005, 22, 73 RSC; (d) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278 RSC; (e) G. Collin and H. Höke, “Indole” In Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley-VCH, Weinheim Search PubMed; (f) Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, New York, 1996, vol. 2, p 259. For selected examples, see Search PubMed; (g) L. Berrade, B. Aisa, M. Ramirez, S. Galiano, S. Guccione, L. R. Moltzau, F. O. Levy, F. Nicoletti, G. Battaglia, G. Molinaro, I. Aldana, A. Monge and S. Perez-Silandes, J. Med. Chem., 2011, 54, 3086 CrossRef CAS; (h) K. Krajewski, Y. Zhang, D. Parrish, J. Deschamps, P. P. Rollera and V. K. Pathak, Bioorg. Med. Chem. Lett., 2006, 16, 3034 CrossRef CAS; (i) N. K. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2005, 127, 5970 CrossRef CAS; (j) J. F. Payack, E. Vazquez, L. Matty, M. H. Kress and J. McNamara, J. Org. Chem., 2005, 70, 175 CrossRef CAS.
- For selected reviews, see: (a) C. K. Narkowicz, A. J. Blackman, E. Lacey, J. H. Gill and K. Heiland, J. Nat. Prod., 2002, 65, 938 CrossRef CAS; (b) G. W. Gribble, J. Chem. Soc. Perkin Trans. 1, 2000, 1045 RSC; (c) G. W. Gribble, J. Nat. Prod., 1992, 55, 1353 CrossRef CAS; (d) Indoles, ed. R. J. Sundberg, Academic Press, San Diego, 1996. For selected examples, see Search PubMed; (e) C. C. Hughes, A. Prieto-Davo, P. R. Jensen and W. Fenical, Org. Lett., 2008, 10, 629 CrossRef CAS; (f) G. Bringmann, S. Tasler, H. Endress, J. Kraus, K. Messer, M. Wohlfarth and W. Lobin, J. Am. Chem. Soc., 2001, 123, 2703 CrossRef CAS; (g) R. S. Norton and R. J. Wells, J. Am. Chem. Soc., 1982, 104, 3628 CrossRef CAS.
- (a) J. C. Powers, in The Chemistry of Heterocyclic Compounds, ed. Houlihan, W. J., J. Wiley and Sons, New York, 1972, vol. 25, part 2, p. 128 Search PubMed; (b) W. C. Frank, Y. C. Kim and R. F. Heck, J. Org. Chem., 1978, 43, 2947 CrossRef CAS.
- (a) M. G. Bursavicha, N. Brooijmans, L. Feldberg, I. Hollander, S. Kim, S. Lombardi, K. Park, R. Mallon and A. M.Gilbert, Bioorg. Med. Chem. Lett., 2010, 20, 2586 CrossRef; (b) M. S. Techenor, J. D. Trzupek, D. B. Kastrinsky, F. Shiga, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 15683 CrossRef; (c) J. M. Herbert, Tetrahedron Lett., 2004, 45, 817 CrossRef CAS; (d) K. Dinnell, G. G. Chicchi, M. J. Dhar, J. M. Elliott, G. J. Hollingworth, M. M. Kurtz, M. P. Ridgill, W. Rycroft, K.-L. Tsao, A. R. Williams and C. J. Swaina, Bioorg. Med. Chem. Lett., 2001, 11, 1237 CrossRef CAS; (e) Y. Liu and G. W. Gribble, Tetrahedron Lett., 2000, 41, 8717 CrossRef CAS.
- (a) J. Bergman and L. Venemalm, J. Org. Chem., 1992, 57, 2495 CrossRef CAS.
- For recent reviews, see: (a) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS; (b) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC; (c) F. Legrand, K. Jouvin and G. Evano, Isr. J. Chem., 2010, 50, 588 CrossRef CAS; (d) J.-R. Wang and K. Manabe, Synthesis, 2009, 1405 Search PubMed.
- (a) S. Thielges, E. Meddah, P. Bisseret and J. Eustache, Tetrahedron Lett., 2004, 45, 907 CrossRef CAS; (b) Y.-Q. Fang and M. Lautens, J. Org. Chem., 2008, 73, 538 CrossRef CAS; (c) Y.-Q. Fang and M. Lautens, Org. Lett., 2005, 7, 3549 CrossRef CAS; (d) A. Fayol, Y.-Q. Fang and M. Lautens, Org. Lett., 2006, 8, 4203 CrossRef CAS; (e) M. Nagamochi, Y.-Q. Fang and M. Lautens, Org. Lett., 2007, 9, 2955 CrossRef CAS; (f) Y.-Q. Fang, J. Yuen and M. Lautens, J. Org. Chem., 2007, 72, 5152 CrossRef CAS; (g) Y.-Q. Fang, R. Karisch and M. Lautens, J. Org. Chem., 2007, 72, 1341 CrossRef CAS.
- (a) J. Yuen, Y.-Q. Fang and M. Lautens, Org. Lett., 2006, 8, 653 CrossRef CAS.
- (a) C. S. Bryan and M. Lautens, Org. Lett., 2008, 10, 4633 CrossRef CAS; (b) Z.-J. Wang, J.-G. Yang, F. Yang and W. Bao, Org. Lett., 2010, 12, 3034 CrossRef CAS; (c) Z.-J. Wang, F. Yang, X. Lv and W. Bao, J. Org. Chem., 2011, 76, 967 CrossRef CAS; (d) X.-R. Qin, X.-F. Cong, D.-B. Zhao, J.-S. You and J.-B. Lan, Chem. Commun., 2011, 47, 5611 RSC; (e) W. Chen, M. Wang, P. Li and L. Wang, Tetrahedron, 2011, 67, 5913 CrossRef CAS.
- (a) T. O. Vieira, L. A. Meaney, Y.-L. Shi and H. Alper, Org. Lett., 2008, 10, 4899 CrossRef CAS.
- (a) M. Arthuis, R. Pontikis and J.-C. Florent, Org. Lett., 2009, 11, 4608 CrossRef CAS.
- (a) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2010, 132, 11416 CrossRef CAS; (b) S. G. Newman, V. Aureggi, C. S. Bryan and M. Lautens, Chem. Commun., 2009, 5236 RSC.
- (a) W. Chen, Y. Zhang, L. Zhang, M. Wang and L. Wang, Chem. Commun., 2011, 47, 10476 RSC; (b) T. He, H. Li, P. Li and L. Wang, Chem. Commun., 2011, 47, 8946 RSC; (c) T. He, L. Yu, L. Zhang, L. Wang and M. Wang, Org. Lett., 2011, 13, 5016 CrossRef CAS; (d) X. Zhang and L. Wang, Green Chem., 2012, 14, 2141 RSC.
- (a) H.-C. Zhang, H. Ye, A. F. Moretto, K. K. Brumfield and B. E. Maryanoff, Org. Lett., 2000, 2, 89 CrossRef CAS; (b) H.-C. Zhang, H. Ye, K. B. White and B. E. Maryanoff, Tetrahedron Lett., 2001, 42, 4751 CrossRef CAS.
- (a) B. Jiang, K. Tao, W. Shen and J. Zhang, Tetrahedron Lett., 2010, 51, 6342 CrossRef CAS.
- (a) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS; (b) E. Shirakawa, K. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS; (c) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS; (d) J. Zhao, Y. Zhao and H. Fu, Angew. Chem., Int. Ed., 2011, 50, 3769 CrossRef CAS.
- (a) E. von Angerer, N. Knebel, M. Kager and B. Ganss, J. Med. Chem., 1990, 33, 2635 CrossRef CAS; (b) C. Biberger and E. von Angerer, J. Steroid Biochem. Mol. Biol., 1998, 64, 277 CrossRef CAS; (c) M. Medarde, A. C. Ramos, E. Caballero, R. P.-L. De Clairac, J. L. Lopez, D. G. Gravalos and A. S. Feliciano, Bioorg. Med. Chem. Lett., 1999, 9, 2303 CrossRef CAS; (d) G. I. Stevenson, A. L. Smith, S. Lewis, S. G. Michie, J. G. Neduvelil, S. Patel, R. Marwood, S. Patel and J. L. Castro, Bioorg. Med. Chem. Lett., 2000, 10, 2697 CrossRef CAS.
- (a) L. Chu, M. H. Fisher, M. T. Goulet and M. J. Wyvratt, Tetrahedron Lett., 1997, 38, 3871 CrossRef CAS; (b) C. A. Merlic and D. M. McInnes, Tetrahedron Lett., 1997, 38, 7661 CrossRef CAS; (c) A. L. Smith, G. I. Stevenson, S. Lewis, S. Patel and J. L. Castro, Bioorg. Med. Chem. Lett., 2000, 10, 2693 CrossRef CAS.
- (a) R. L. Hudkins, J. L. Diebold and F. D. Marsh, J. Org. Chem., 1995, 60, 6218 CrossRef CAS; (b) M. Amat, S. Hadida, G. Pshenichnyi and J. Bosch, J. Org. Chem., 1997, 62, 3158 CrossRef CAS; (c) C. N. Johnson, G. Stemp, N. Anand, S. C. Stephen and T. Gallagher, Synlett, 1998, 1025 CrossRef CAS.
- (a) H. Xu, S. Gu, W. Chen, D. Li and J. Dou, J. Org. Chem., 2011, 76, 2448 CrossRef CAS; (b) J. Liu, W. Chen, Y. Ji and L. Wang, Adv. Synth. Catal., 2012, 354, 1585 CrossRef CAS; (c) M. Okutani and Y. Mori, J. Org. Chem., 2009, 74, 442 CrossRef CAS.
- Cs2CO3 (99.995%) was purchased from Aldrich and contains Cu (0.30 ppm) and Pd (0.01 ppm); other elements including: Ni, Co, Fe, Ru, and Rh were not detected (ICP-MS determination data).
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures, analytical data, and 1H, 13C NMR and HRMS spectra of all intermediates and products or other electronic format. See DOI: 10.1039/c2ra22172a |
| This journal is © The Royal Society of Chemistry 2013 |