 Open Access Article
Open Access ArticleReversible addition–fragmentation chain transfer polymerization of 2-chloro-1,3-butadiene†
Nikki
Pullan
a,
Max
Liu
b and
Paul D.
Topham
*a
aDepartment of Chemical Engineering and Applied Chemistry, Aston University, Aston Triangle, Birmingham, B4 7ET, UK. E-mail: p.d.topham@aston.ac.uk; Tel: +44 (0)1212043413
bRobinson Brothers Ltd., Phoenix Street, West Bromwich B70 0AH, UK. E-mail: mliu@robinsonbrothers.co.uk; Tel: +44 (0)1215002402
First published on 29th January 2013
Abstract
Controlled polymerization of 2-chloro-1,3-butadiene using reversible addition–fragmentation chain transfer (RAFT) polymerization has been demonstrated for the first time. 2-Chloro-1,3-butadiene, more commonly known as chloroprene, has significant industrial relevance as a crosslinked rubber, with uses ranging from adhesives to integral automotive components. However, problems surrounding the inherent toxicity of the lifecycle of the thiourea-vulcanized rubber have led to the need for control over the synthesis of poly(2-chloro-1,3-butadiene). To this end, four chain transfer agents in two different solvents have been trialed and the kinetics are discussed. 2-Cyano-2-propylbenzodithioate (CPD) is shown to polymerize 2-chloro-1,3-butadiene in THF, using AIBN as an initiator, with complete control over the target molecular weight, producing polymers with low polydispersities (Mw/Mn < 1.25 in all cases).
Introduction
2-Chloro-1,3-butadiene (CB), commonly referred to as chloroprene, has been widely used in the rubber industry for decades.1 Poly(2-chloro-1,3-butadiene) (PCB) in its crosslinked form was the first synthetic rubber to find widespread commercial acceptance. PCB rubber (also known as neoprene) possesses a unique combination of properties leading to its use in many diverse high value markets from adhesives and sealants through to automotive applications.2 Currently, PCB, the uncrosslinked precursor to chloroprene rubber (CR), is synthesized via uncontrolled free-radical emulsion polymerization in the presence of a thiol-based chain transfer agent (CTA) to regulate the molecular weight.3,4 Typically, this feedstock resin has a variable molecular weight and an extremely broad molecular weight distribution (Mw/Mn > 2). For industrial purposes, this does not present a significant problem. After crosslinking, the material properties are similar regardless of precursor batch quality. However, advances in CR technology are sought regarding the inherent toxicity surrounding the curing process and the subsequent degradation routes of the rubber.5–7 This high toxicity is due to the presence of a thiourea-based accelerator that is employed in the crosslinking process. To replace this accelerator with a non-toxic equivalent, mechanistic studies of the crosslinking of PCB form an essential component towards understanding, and significantly enhancing, the entire lifecycle of the material. This is not possible using the conventional high molecular weight PCB gumstock resin, due to its inherent insolubility. To probe the mechanism, PCB with controllably lower molecular weight is essential.Controlled polymerization processes have been revolutionary for the production of well-defined macromolecules with predetermined molecular weights. However, techniques which offer the greatest control, such as anionic polymerization,8 often carry the disadvantage of extremely stringent experimental procedures. Controlled radical polymerization (CRP),9 on the other hand, offers a range of more industrially relevant techniques, which are more forgiving in terms of cost and labor intensity, yet still deliver excellent control over molecular weight. The most common CRP techniques are Atom Transfer Radical Polymerization (ATRP),10–13 Nitroxide-Mediated Polymerization (NMP)14 and Reversible Addition–Fragmentation chain Transfer (RAFT) polymerization.15,16 Collectively, an extremely wide range of monomers has been polymerized by these techniques. Recently, this range has been extended to include monomers that are similar, in part, to CB, such as vinyl chloride,17 divinylimidazole,18 butadiene19 and 2-methyl-1,3-butadiene19–21 using RAFT polymerization. However, to date, the only report on the controlled polymerization of CB is through its copolymerization with a phosphonated diene monomer using NMP.22 In the present work, we describe the controlled polymerization of CB alone, for the first time. RAFT polymerization has been employed and an optimum system for producing well-defined PCB with predetermined molecular weights has been identified. The structures of the RAFT chain transfer agents (CTAs) employed in this study are shown in Fig. 1 alongside the polymerization scheme showing the major commercial polymer isomer.23
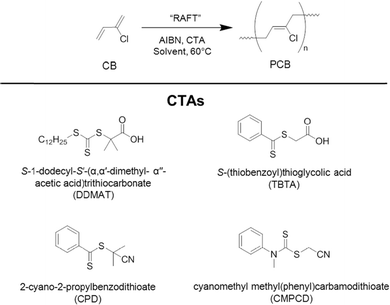 | ||
| Fig. 1 Polymerization reaction scheme for 2-chloro-1,3-butadiene (CB) monomer (top), showing the major 1,4-trans isomer of the product and the chain transfer agents (CTAs) that have been trialed in this study (bottom). | ||
Results and discussion
Four different RAFT CTAs have been investigated for the controlled polymerization of CB; DDMAT, TBTA, CPD and CMPCD (see Fig. 1 for CTA structures). These CTAs were selected because they have been successfully employed to polymerize 2-methyl-1,3-butadiene,20 styrene,24 methyl methacrylate25 and vinyl chloride,17 respectively. The latter (CMPCD) was selected as a negative control as this CTA is known not to be effective for polymerizing more activated monomers.16 TBTA showed limited control achieved for styrene (PDI 1.38), particularly when compared to the control over MMA and BA.24 Our CTA range comprises a trithiocarbonate (DDMAT), two dithiobenzoates (TBTA and CPD) and a dithiocarbamate (CMPCD). Fig. 2 shows the progression of molecular weight (Mn) versus monomer conversion for each CTA during the polymerization of CB at 60 °C using a commercial solution of monomer (50% in xylene) and AIBN as the initiator.Firstly, Fig. 2 clearly shows that the molecular weight increased with monomer conversion for all CTAs trialed with the exception of CMPCD. Here, a high molecular weight polymer was produced in the very early stages of the polymerization, followed by the formation of lower molecular weight species to yield a bimodal distribution in the GPC trace, as shown in the ESI (Fig. S1†). The bimodal distribution became more enhanced as the polymerization progressed. It should be noted that CMPCD has been previously shown to be an effective CTA (albeit conducted in THF rather than xylene) for the polymerization of a different chlorinated monomer, vinyl chloride,17 however this monomer has significantly different reactivity to CB. Conversely, DDMAT, TBTA and CPD all exhibited an increase in polymer molecular weight with monomer conversion. For the dithiobenzoate CTAs, growth of molecular weight continued beyond the target, suggesting that it is not possible to synthesize pre-defined PCB using TBTA or CPD in xylene under these conditions. The target molecular weight was achieved at high conversion (96%) with DDMAT, but the PDI values exceeded 1.2 throughout the polymerization, reaching ∼1.5 at higher conversions. Overall, most promise resided with CPD, as PDI values did not exceed 1.2 up to 71% conversion (26 h) and at this point Mthn had been reached. It is important to note that all measured MGPCn values are relative to polystyrene standards and are therefore used as a guide to the final absolute molecular weight only.
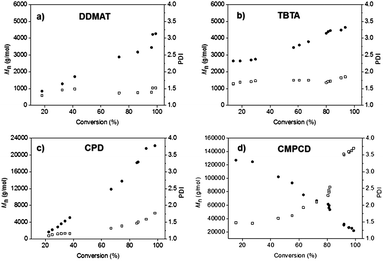 | ||
| Fig. 2 Plots of molecular weight, Mn (●), and polydispersity, PDI (□), versus monomer conversion utilizing different CTAs in xylene; (a) S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate, DDMAT, (b) S-(thiobenzoyl)thioglycolic acid, TBTA, (c) 2-cyano-2-propylbenzodithioate, CPD, and (d) cyanomethyl methyl(phenyl)carbamodithioate, CMPCD. | ||
Our four CTAs were subsequently investigated in a more polar solvent. THF was chosen for comparison against xylene, as this has been employed previously in the controlled RAFT polymerization of vinyl chloride.17 Again, polymerization reactions were performed under comparable conditions and were monitored over time (Fig. 3). Generally, enhanced control was observed in THF for each system. There are very few reports on the precise role of the solvent in the RAFT process.26,27 In fact, most accounts suggest that overall solvent effects are relatively minor and that the aromaticity and polarity of the solvent have no effect on molar mass, molecular weight distribution or growth rate.28,29 Clearly, our data (and those of Abreu for vinyl chloride17) reveal contrasting trends, with the solvent having a profound effect over the control and rate of PCB synthesis. This forms part of an ongoing investigation in our group to identify the effect of solvent on the RAFT polymerization.
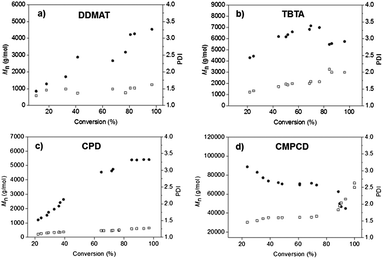 | ||
| Fig. 3 Plots of molecular weight, Mn (●), and polydispersity, PDI (□), versus monomer conversion utilizing different CTAs in tetrahydrofuran (THF); (a) S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate, DDMAT, (b) S-(thiobenzoyl)thioglycolic acid, TBTA, (c) 2-cyano-2-propylbenzodithioate, CPD, and (d) cyanomethyl methyl(phenyl)carbamodithioate, CMPCD. | ||
Although CMPCD has been shown to be an ideal CTA for vinyl chloride,17 our studies show that this is not the case for CB when using AIBN as the initiator. Similarly to the polymerization performed in xylene (Fig. 2 and ESI, Fig. S1†), a large molecular weight species was produced in the early stages of the reaction. In THF, however, a bimodal distribution was not formed, but the molecular weight distribution did broaden significantly over the course of the polymerization (see ESI, Fig. S5†). The next CTA which showed poor control for the polymerization of CB in THF was the dithiobenzoate with carboxylic acid functionality, TBTA. PDIs started relatively high (ca. 1.5) and increased throughout the polymerization to a final value of around 2. The molecular weight (Mn) evolution was far from linear, showing the formation of polymeric species around the target molecular weight after merely 20% monomer conversion and reaching a plateau at approximately 60% conversion. Furthermore, this average molecular weight appears to decrease above 80% conversion, due to a significant increase in the polydispersities in the latter stages of the polymerization (see Fig. S8†). The two CTAs which showed most promise in terms of linear progression of molecular weight with monomer conversion and relatively low PDIs were DDMAT (aliphatic trithiocarbonate CTA bearing carboxylic acid functionality) and CPD (aromatic dithiocarbonate bearing cyano functionality). Both systems reached the approximate target molecular weight. In the case of CPD, there was a marked improvement in PDI; PDI values did not exceed 1.3 up to 97% monomer conversion, whereby the system using DDMAT produced PCB with PDI ∼1.5, seemingly independent of monomer conversion.
Fig. 4 summarizes the experimental results from the most effective system, comprising CPD in THF. Control over molecular weight is evident due to the low PDIs, linear increase of molecular weight with monomer conversion and the final MGPCn value being close to Mthn. Furthermore, the first-order semi-logarithmic plot (Fig. 4) is linear up to ∼95% monomer conversion, with an apparent rate constant, kapp, of 0.050 h−1, implying that the number of propagating radicals is constant over the course of the polymerization (N.B. our systems were monitored beyond 60 hours polymerization time, significantly longer than the expected constant radical flux, therefore any increase in molecular weight is attributed to radical combination reactions, as reflected in the GPC data, shown in the ESI†). In comparison, DDMAT, TBTA and CMPCD (in THF) gave rise to apparent rate constants of 0.034, 0.022 and 0.338 h−1, respectively. In short, the rate constants for propagation using CPD, DDMAT and TBTA are comparable, whereas the CMPCD system proceeded at a much faster rate, which is reflected in the complete lack of control over the PCB growth. The CPD system was probed further in an attempt to successfully synthesize higher molecular weight PCB with narrow molecular weight distribution. The monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CPD ratio (target Dp) was altered to afford polymers of 20000 and 50000 g mol−1 (Dp 230 and 560, respectively). Fig. 5 shows the success of this system for producing PCB over a range of predetermined target molecular weights, all with good control of the final molecular weight and low polydispersities (Mw/Mn < 1.25 in all cases).
:CPD ratio (target Dp) was altered to afford polymers of 20000 and 50000 g mol−1 (Dp 230 and 560, respectively). Fig. 5 shows the success of this system for producing PCB over a range of predetermined target molecular weights, all with good control of the final molecular weight and low polydispersities (Mw/Mn < 1.25 in all cases).
![Kinetic plots for the RAFT polymerization of CB under the following conditions: [AIBN]0/[CPD]0/[CB]0 = 0.2/1/45 at 60 °C in THF (50 wt%).](/image/article/2013/PY/c3py21151g/c3py21151g-f4.gif) | ||
| Fig. 4 Kinetic plots for the RAFT polymerization of CB under the following conditions: [AIBN]0/[CPD]0/[CB]0 = 0.2/1/45 at 60 °C in THF (50 wt%). | ||
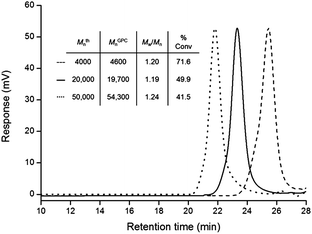 | ||
| Fig. 5 GPC traces of PCB with varying target molecular weights, synthesized by RAFT using CPD in THF. MGPCn values are calculated relative to polystyrene standards. | ||
To demonstrate the “living” nature of the CB RAFT polymerization using CPD, a self-blocking experiment was performed, where PCB was used as a macroCTA (Mn = 1500 g mol−1, PDI 1.16) for the polymerization of CB monomer. As shown in Fig. 6, a near-monomodal increase in molecular weight was observed in the GPC trace of PCB following chain extension (Mn = 3900 g mol−1, PDI 1.27), with only approximately 0.2% macroCTA remaining in the extended PCB (as calculated by GPC). It is noteworthy that the relatively insignificant peaks at high retention time arise due to a small amount of low molecular weight impurities outside of the range of the calibration standards (i.e. <162 g mol−1).
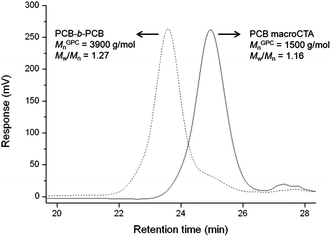 | ||
| Fig. 6 GPC traces of the PCB macroCTA and the corresponding PCB-b-PCB following chain extension. | ||
Conclusions
The controlled polymerization of chlorinated divinyl monomer, 2-chloro-1,3-butadiene (CB), has been demonstrated for the first time using RAFT chemistry. Four different RAFT chain transfer agents (CTAs) were investigated for their suitability towards complete control over the molecular weight characteristics of the final polymer. The two CTAs which showed most promise were S-1-dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DDMAT, an aliphatic CTA bearing carboxylic acid functionality) and 2-cyano-2-propylbenzodithioate (CPD, an aromatic CTA with cyano-functionality), both producing well defined PCB homopolymers when syntheses were conducted in THF. CPD was identified as the most effective CTA for CB polymerization, showing pseudo first-order kinetics and complete control over target molecular weight (up to 50000 g mol−1). The “living” character was further demonstrated through the success of a self-blocking experiment.
Experimental part
Materials
CB monomer (50% in xylene, ABCR) was distilled before use to remove the inhibitor. 2,2′-Azobis(isobutyronitrile) (AIBN, Molekula), 3,4-dichloro-1-butene (>99%, TCI) and sodium hydroxide (NaOH, 97%, Fisher Scientific) were used as received. Tetrabutylammonium bromide (PTC, 99%) and phenothiazine (99%) were purchased from Acros and used as received. S-(Thiobenzoyl)thioglycolic acid (TBTA, 99%), 2-cyano-2-propylbenzodithioate (CPD, >97%) and cyanomethyl methyl(phenyl)carbamodithioate (CMPCD, 98%) were purchased from Sigma-Aldrich and used as received. S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (DDMAT) was initially purchased from Sigma-Aldrich (98%) and subsequently synthesized according to the literature.30 THF and methanol (Fisher Scientific, Laboratory Grade) were used as received.Methods
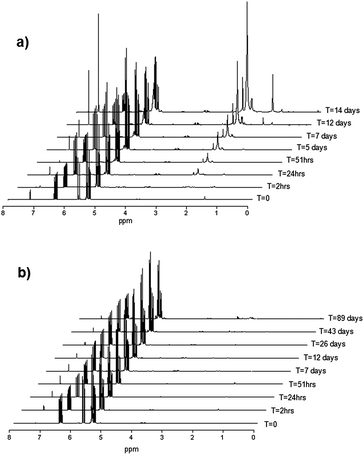 | ||
| Fig. 7 1H NMR spectra showing the self-polymerization of (a) neat 2-chloro-1,3-butadiene (CB) and (b) CB with 0.1% (w/w) phenothiazine under ambient conditions. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1444, 1430 (d, CH2), 1303 (w, CH2), 1215, 1115 (s, C–C), 827, 759 (r, CH2), 667 (s, C–Cl). 1H NMR (300 MHz, CDCl3, δ ppm from TMS): reference CDCl3 = 7.28 ppm, δ = 5.90–5.03 (br, –CH2C(Cl)C(H)CH2–), 2.56 (br, –CH2C(Cl)C(H)CH2–), 2.55–2.35 (br, –CH2C(Cl)C(H)CH2–). 13C NMR (75 MHz, CDCl3, δ ppm): reference CDCl3 = 77.17 ppm, δ = 134.86 (–CH2C(Cl)C(H)CH2–), 124.08 (–CH2C(Cl)C(H)CH2–), 38.29 (–CH2C(Cl)C(H)CH2–), 26.75 (–CH2C(Cl)C(H)CH2–).
C), 1444, 1430 (d, CH2), 1303 (w, CH2), 1215, 1115 (s, C–C), 827, 759 (r, CH2), 667 (s, C–Cl). 1H NMR (300 MHz, CDCl3, δ ppm from TMS): reference CDCl3 = 7.28 ppm, δ = 5.90–5.03 (br, –CH2C(Cl)C(H)CH2–), 2.56 (br, –CH2C(Cl)C(H)CH2–), 2.55–2.35 (br, –CH2C(Cl)C(H)CH2–). 13C NMR (75 MHz, CDCl3, δ ppm): reference CDCl3 = 77.17 ppm, δ = 134.86 (–CH2C(Cl)C(H)CH2–), 124.08 (–CH2C(Cl)C(H)CH2–), 38.29 (–CH2C(Cl)C(H)CH2–), 26.75 (–CH2C(Cl)C(H)CH2–).
Characterization
NMR spectroscopy (Bruker Avance, 1H 300 MHz, 13C 75 MHz, CDCl3) confirmed monomer and polymer structures and was used to determine monomer conversion. Polymer molecular weight (Mn) and polydispersity (Mw/Mn, PDI) were measured using gel permeation chromatography (GPC) (flow rate 1 mL min−1) through three PL gel 5 μm 300 × 7.5 mm mixed-C columns using a degassed THF eluent system containing 2% (v/v) TEA and 0.05% (w/v) BHT. The system, operating at 40 °C, was calibrated with narrow polystyrene standards (Mp range = 162 to 6035000 g mol−1). All data were analyzed using PL Cirrus software (version 2.0) supplied by Agilent Technologies (previously Polymer Laboratories). Fourier transform infrared (FTIR) spectra were obtained on a Perkin Elmer Spectrum One spectrophotometer over the range 4000–500 cm−1 for 16 scans with a resolution of 4 cm−1. The samples were recorded as thin films held between two NaCl windows.
Acknowledgements
The authors thank Pera Technology, the European Commission and the EPSRC for supporting this work through a CASE studentship with Robinson Brothers Ltd. and through funding as part of the Framework Programme 7 SafeRubber project (243756).Notes and references
- W. H. Carothers, I. Williams, A. M. Collins and J. E. Kirby, J. Am. Chem. Soc., 1931, 53, 4203–4225 Search PubMed.
- M. Lynch, Chem.-Biol. Interact., 2001, 135–136, 155–167 Search PubMed.
- K. Itoyama, N. Hirashima, J. Hirano and T. Kadowaki, Polym. J., 1991, 23, 859–864 Search PubMed.
- K. Itoyama, N. Shimizu and S. Matsuzawa, Polym. J., 1991, 23, 1139–1142 Search PubMed.
- P. Y. Le Gac, V. Le Saux, M. Paris and Y. Marco, Polym. Degrad. Stab., 2012, 97, 288–296 Search PubMed.
- K. T. Gillen, R. Bernstein and D. K. Derzon, Polym. Degrad. Stab., 2005, 87, 57–67 Search PubMed.
- H. Plugge and R. J. Jaeger, Toxicol. Appl. Pharmacol., 1979, 50, 565–572 Search PubMed.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- T. Ando, M. Kato, M. Kamigaito and M. Sawamoto, Macromolecules, 1996, 29, 1070–1072 CrossRef CAS.
- E. J. Ashford, V. Naldi, R. O'Dell, N. C. Billingham and S. P. Armes, Chem. Commun., 1999, 1285–1286 RSC.
- J. Lad, S. Harrisson, G. Mantovani and D. M. Haddleton, Dalton Trans., 2003, 4175–4180 RSC.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Mejis, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- C. M. R. Abreu, P. V. Mendonça, A. C. Serra, J. F. J. Coelho, A. V. Popov, G. Gryn'ova, M. L. Coote and T. Guliashvili, Macromolecules, 2012, 45, 2200–2208 CrossRef CAS.
- M. H. Allen, S. T. Hemp, A. E. Smith and T. E. Long, Macromolecules, 2012, 45, 3669–3676 Search PubMed.
- G. Bar-Nes, R. Hall, V. Sharma, M. Gaborieau, D. Lucas, P. Castignolles and R. G. Gilbert, Eur. Polym. J., 2009, 45, 3149–3163 CrossRef CAS.
- D. S. Germack and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4100–4108 CrossRef CAS.
- V. Jitchum and S. Perrier, Macromolecules, 2007, 40, 1408–1412 CrossRef CAS.
- N. Ajellal, C. M. Thomas and J.-F. Carpentier, Polymer, 2008, 49, 4344–4349 CrossRef CAS.
- R. C. Ferguson, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 4735–4741 Search PubMed.
- S. C. Farmer and T. E. Patten, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 555–563 Search PubMed.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- M. G. Frohlich, M. M. Nardai, N. Forster, P. Vana and G. Zifferer, Polymer, 2010, 51, 5122–5134 CrossRef.
- J. Ferreira, J. Syrett, M. Whittaker, D. Haddleton, T. P. Davis and C. Boyer, Polym. Chem., 2011, 2, 1671–1677 RSC.
- M. R. Wood, D. J. Duncalf, P. Findlay, S. P. Rannard and S. Perrier, Aust. J. Chem., 2007, 60, 772–778 Search PubMed.
- M. Benaglia, E. Rizzardo, A. Alberti and M. Guerra, Macromolecules, 2005, 38, 3129–3140 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme showing CB synthesis, GPC traces taken periodically throughout each RAFT polymerization and spectral data for PCB synthesized using CPD CTA. See DOI: 10.1039/c3py21151g |
| This journal is © The Royal Society of Chemistry 2013 |