 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Immobilization of MacMillan catalyst via controlled radical polymerization: catalytic activity and reuse†
Beth L. Moorea, Annhelen Lua, Deborah A. Longbottomb and Rachel K. O'Reilly*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: Rachel.OReilly@Warwick.ac.uk
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 6th February 2013
Abstract
The MacMillan catalyst is an established organocatalyst capable of catalyzing a variety of organic reactions. Through the synthesis of a novel monomer containing the MacMillan catalytic functionality, a variety of copolymers have been synthesized with the comonomer, diethylene glycol methyl ether methacrylate (DEGMA). Reversible addition–fragmentation chain transfer (RAFT) polymerization was used for the synthesis of these functional polymers with good control over molecular weight, catalyst incorporation and polydispersity. These polymers showed lower critical solution temperature (LCST) behaviour where the cloud point was found to be dependent upon the degree of catalyst incorporation and catalyst loading was also found to have an effect on the Tg of the copolymers. The catalytic activity of the functional copolymers is demonstrated by the Diels–Alder reaction between cyclopentadiene and trans-hexen-1-al and shows enantioselectivity close to those previously reported by MacMillan. The polymers can be reused in multiple Diels–Alder reactions via a pseudo continuous process, maintaining high conversion and enantioselectivity throughout the cycles.
Introduction
Organocatalysis is a far reaching and important area of synthetic chemistry. One of the main problems, however, is the difficulty associated with reclaiming and reusing the catalyst. Often the catalyst is either the most expensive part of the reaction, most time consuming or difficult to prepare and as such, significant research has been dedicated to the immobilization of these catalysts.1–6 A review published in 2010 by Hansen and Kristensen demonstrated the various polymer supports available, including cross-linked polymer resins such as the Merrifield and JandaJel™ supports, as well as acrylic resins and linear polymers.7 They also contemplated the associated advantages and disadvantages of implementing either post-modification or copolymerization strategies. Another review in 2008 by Gruttadauria et al. described the techniques available to support the widely studied L-proline,8 including the use of polymeric resins,9–11 silica,12 ionic liquids,13 cyclodextrins14 and dendrimers.15Recent advances in polymerization techniques now allow the polymerization of functional monomers containing catalytically active groups with high levels of control, producing a myriad of polymer bound catalysts, which can often be recovered and recycled to some degree. For example, our group has reported the successful anchoring of L-proline and DMAP catalysts, incorporating them into polymeric frameworks which self-assemble into recyclable and reusable nanostructures.16–18 Huerta et al. have immobilized L-proline into a polymer able to fold into specific conformations which places the catalytic units in a hydrophobic domain.19 Ge et al. used a similar polymeric micellar system to support imidazole, which was demonstrated to efficiently catalyze ester hydrolysis reactions.20 Use of the MacMillan catalyst was first reported in 2000: it is a novel imine–enamine catalyst capable of accelerating the rate of the Diels–Alder reaction with good control over enantioselectivity and in the majority of cases, diastereoselectivity.21 The catalyst efficiently lowers the LUMO of the dienophile providing a substantial increase in rate of reaction, whilst its steric bulk controls the enantioselectivity. This work was then further extended in 2002 to a range of α,β-unsaturated ketone Diels–Alder substrates22 and beyond this, it has found use in a broad range of processes, including the Mukaiyama–Michael23 Friedel–Crafts alkylation,24 cascade reactions,25 transfer hydrogenation and even the first enantioselective organocatalytic hydride reduction.26 The range of reactions catalyzed by the MacMillan catalyst make it a very powerful, practical and truly versatile organocatalyst.
There have been several attempts to support the MacMillan catalyst, aiming to maintain its high selectivity but still allow for recovery and reuse (outlined in the review by Hansen and Kristensen).7 The first of these was reported by Cozzi et al., where the catalyst was immobilized on PEG supports. In most cases, comparable selectivities to those first reported by MacMillan were achieved, although a marginal loss of selectivity on recycling was reported.27 Other supports include a JandaJel™ system,28 mesoceullar foams29 and sulfonated polystyrene.30 One system, developed by Pericas and co-workers in 2012, utilized superparamagnetic Fe3O4 nanoparticles as the support where the catalytic functionality was introduced through a click reaction.31 The supported catalyst successfully catalyzed the Friedel–Crafts alkylation reaction and was recycled using its magnetic properties. Unchanged enantioselectivities were reported (∼90%) over six cycles. Hansen and co-workers immobilized the MacMillan catalyst in 2010 using a different approach: a polymerizable monomer was synthesized and copolymerized with poly(ethylene glycol) in a suspension polymerization.32 However, a decrease in enantioselectivity with recycling was reported (81% to 51% ee after four cycles). Their work bears the closest resemblance to the work discussed in this paper, as it moves away from the anchoring strategy using pre-made polymers (akin to solid-phase peptide synthesis) to instead use catalyst functionalized monomers to form a functional polymer scaffold. The advantages of this bottom-up strategy include greater control over catalyst loading compared to post-modification schemes. In addition, the catalyst position in the polymer chain can be controlled by employing sequential addition techniques, which may also be used to synthesize block copolymers. The polarity and hence solubility of the polymer is also easily tuned by simply changing the ratio of functionalized monomer to comonomer.
Recent advances in controlled radical polymerization (CRP) techniques have allowed for more complex and functional polymer architectures to be readily synthesized. This is achieved through the polymerization of monomers containing functional groups which were previously difficult to polymerize using conventional and sensitive living polymerization techniques. Reversible addition–fragmentation chain transfer (RAFT) polymerization has been used in this work, as this technique has been shown to be appropriate for a range of functional monomers and conditions.33–37 We herein report the synthesis of a novel MacMillan functionalized monomer and its subsequent copolymerization yielding well-defined copolymers. Efficient catalysis of a model Diels–Alder reaction between cyclopentadiene and trans-hexen-1-al is then demonstrated, followed by reuse of the copolymers in several catalytic cycles via a pseudo continuous process.
Experimental
Instrumentation
Both 1H and 13C NMR spectra were recorded on a 300 or 400 MHz Bruker DPX FT-NMR spectrometer using deuterated solvents. Chemical shifts are reported as δ in parts per million relative to the solvent used. Size exclusion chromatography/gel permeation chromatography (SEC/GPC) data were obtained using PLgel 5 μm mixed-D columns, plus one guard column and tetrahydrofuran (THF) with 2% triethylamine (TEA) as eluent, with a flow rate of 1.0 mL min−1. The data was analyzed using Cirrus GPC software and compared to poly(methylmethacrylate) (PMMA) standards. Lower critical solution temperature (LCST) measurements were performed on a Perkin Elmer UV/Vis spectrometer (Lambda 35) equipped with a Peltier temperature controller at 500 nm with a constant heating/cooling rate of 1 °C min−1. Enantiomeric excess (ee%) was measured by gas chromatography (GC) on a Varian 450-GC with a 25 m chirasil-Dex chiral column or by high performance liquid chromatography (HPLC) analysis on a Shimadzu Prominence HPLC with a Chiracel OD-H column 250 mm × 4.6 mm × 5 μm, with guard column (5 μm). The centrifuge used to reclaim the MacMillan monomer was a Sigma 2-16 P centrifuge operated at 7000 rpm. Glass transition temperatures (Tg) were determined using a Mettler Toledo DSC1-STAR Differential scanning calorimetry (DSC) was carried out on the sample (2–10 mg) in an aluminium sample holder where an empty holder was used as the reference. Changes in heat flow were recorded between 0 °C and 150 °C over two cycles with a scan rate of 5 °C min−1, under a nitrogen stream (50 mL min−1). The instrument was calibrated using indium metal standards supplied by Mettler Toledo and the data was analyzed using STARe software package (version 9.30).Methods and techniques
Azo-bis-isobutyronitrile (AIBN) was purchased from Sigma-Aldrich, recrystallized from methanol and stored in the dark at 4 °C. DEGMA was purchased from Sigma-Aldrich, filtered through an aluminium oxide column and stored at 4 °C before use. Cyclopentadiene was prepared by cracking dicyclopentadiene purchased from Sigma-Aldrich. All other chemicals were purchased from Sigma-Aldrich and used without further purification.Synthesis of MacMillan catalyst21
In order to determine if racemization of the catalyst stereocenter had occurred under the polymerization conditions, non-polymerizable MacMillan catalysts (both S and R versions) were synthesized from S- and R-phenylalanine. HPLC analysis (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :propan-2-ol, 90:10 using a Chiracel OD-H column 250 mm × 4.6 mm × 5 μm, with guard column (5 μm)) showed the following retention times: S-enantiomer, tR = 7.9 min and R-enantiomer, tR = 7.2 min.
:propan-2-ol, 90:10 using a Chiracel OD-H column 250 mm × 4.6 mm × 5 μm, with guard column (5 μm)) showed the following retention times: S-enantiomer, tR = 7.9 min and R-enantiomer, tR = 7.2 min.Synthesis of MacMillan functionalized monomer (M1)
S-Tyrosine methyl ester hydrochloride (25 g, 0.128 mol) was dissolved in a solution of methylamine in ethanol (33 wt% 81 mL, 0.64 mol) and stirred overnight. The resulting solution was diluted with Et2O (∼100 mL) and the solvent removed in vacuo. This process was repeated multiple times (∼5) until all excess amine had been removed. The resulting sticky yellow solid was dissolved in methanol (150 mL) and acetone (47 mL, 0.643 mol) and para-toluene sulfonic acid (0.22 g, 0.128 mol) was then added. The solution was stirred under reflux for 18 hours. Excess reagents and solvents were removed in vacuo. Care was taken to remove the methanol as remaining traces of the solvent affect the final crystallization step, due to the high solubility of the product in this solvent. Trifluoroacetic acid (30 mL) was added carefully to the resulting orange solid at 0 °C. The reaction was left to stir until the solid was dissolved. Trifluoromethanesulfonic acid (2.83 mL, 0.032 mol) was then added slowly and allowed to stir for 10 min before methacryloyl chloride (25 mL, 0.256 mol) was added slowly.38 The reaction mixture was then allowed to warm to room temperature and stirred overnight. Excess solvents and acids were removed by air flow and the resulting product thoroughly dried under vacuum with a sintered attachment to prevent loss of any solids. A sticky yellow solid resulted and to this, Et2O (300 mL) was added, resulting in a white precipitate which was collected, further washed with Et2O (∼100 mL) and dried in vacuo to give the desired MacMillan functionalized monomer M1 as an amorphous white solid (33.4 g, 87%). 1H NMR (300 MHz, CD3OD): δ 1.51 (3H, s, –NH–C(CH3)2–N(CH3)–), 1.65 (3H, s, –NH–C(CH3)2–N(CH3)–), 1.97 (3H, s, C(CH3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 2.85 (3H, s, N(CH3)), 2.94 (1H, dd, J = 10.5 Hz, 15 Hz, Ar–CHH–CH–), 3.48 (1H, dd, J = 3.6 Hz, 15 Hz, Ar–CHH–CH–), 4.55 (1H, dd, J = 3.6 Hz, 10.5 Hz, Ar–CHH–CH–), 5.76 (1H, s, C(CH3)CHH–), 6.25 (1H, s, C(CH3)CHH–), 7.09 (2H, d, J = 8.4 Hz, Ar), 7.39 (2H, d, J = 8.4 Hz, Ar). 13C NMR (75 MHz, CD3OD): δ 18.5 (CH(CH3)CH2), 22.2 (C(CH3)2), 24.5 (C(CH3)2), 25.7 (N(CH3)), 34.7 (Ar–CH2), 59.7 (Ar–CH2–CH), 78.9 (C(CH3)2), 123.5 (Ar), 128.1 (CH(CH3)CH2), 131.4 (Ar), 134.3 (Ar), 137.2 (CH(CH3)CH2), 151.9 (C(O)–CH(CH3)CH2), 168.1 (N(CH3)–C(O)), (ESI Fig. S1 and S2†). HR ESI-MS: found 303.1698 m/z [M + H]+ expected 303.1709.
CH2), 2.85 (3H, s, N(CH3)), 2.94 (1H, dd, J = 10.5 Hz, 15 Hz, Ar–CHH–CH–), 3.48 (1H, dd, J = 3.6 Hz, 15 Hz, Ar–CHH–CH–), 4.55 (1H, dd, J = 3.6 Hz, 10.5 Hz, Ar–CHH–CH–), 5.76 (1H, s, C(CH3)CHH–), 6.25 (1H, s, C(CH3)CHH–), 7.09 (2H, d, J = 8.4 Hz, Ar), 7.39 (2H, d, J = 8.4 Hz, Ar). 13C NMR (75 MHz, CD3OD): δ 18.5 (CH(CH3)CH2), 22.2 (C(CH3)2), 24.5 (C(CH3)2), 25.7 (N(CH3)), 34.7 (Ar–CH2), 59.7 (Ar–CH2–CH), 78.9 (C(CH3)2), 123.5 (Ar), 128.1 (CH(CH3)CH2), 131.4 (Ar), 134.3 (Ar), 137.2 (CH(CH3)CH2), 151.9 (C(O)–CH(CH3)CH2), 168.1 (N(CH3)–C(O)), (ESI Fig. S1 and S2†). HR ESI-MS: found 303.1698 m/z [M + H]+ expected 303.1709.Copolymerization of M1 and DEGMA
A typical polymerization was carried out as follows: to an oven-dried ampoule, M1 (0.087 g, 10 eq., 0.28 mmol), DEGMA (0.490 g, 90 eq., 2.6 mmol), AIBN (0.4 mg, 0.1 eq., 2.9 × 10−3 mmol), 2-cyano-2-propyl dodecyl trithiocarbonate (0.01 g, 1 eq., 2.9 × 10−2 mmol) and DMSO (1 mL) were added. The mixture was degassed via three freeze–pump–thaw cycles and backfilled with nitrogen, before being placed in a pre-heated oil bath at 80 °C. After 6 hours, the reaction was quenched by rapid cooling in liquid nitrogen and exposure to oxygen. The polymer was then extensively dialyzed against deionized water (MWCO = 3500 Da) before being freeze-dried. 1H NMR (300 MHz, DMSO): δ 0.8–2.1 (CH2 and CH3 polymer backbone), 3.2–3.7 (br m, DEGMA CH2 and CH3), 4.1 (br, C(O)O–CH2–CH2–O, DEGMA), 6.8–7.3 (br, Ar-MacMillan). Conversion by 1H NMR spectroscopy: M1 93% and DEGMA 84%. Mn (SEC, THF, PMMA calibration) = 11.8 kDa, Mw/Mn = 1.35, (ESI Fig. S3†).Synthesis of PDEGMA
DEGMA (0.5 g, 100 eq., 2.6 mmol), 2-cyano-2-propyl dodecyl trithiocarbonate (9.2 mg, 1 eq., 2.7 × 10−2 mmol), AIBN (0.4 mg, 0.1 eq., 2.7 × 10−3 mmol) and dioxane (0.5 mL) were weighed into an oven-dried ampoule and degassed via three freeze–pump–thaw cycles, backfilled with nitrogen and heated at 70 °C. After 6 hours, the reaction was quenched by rapid cooling in liquid nitrogen and exposure to oxygen. The polymer was then dialyzed extensively against deionized water (MWCO = 3500 Da) before being freeze-dried. 1H NMR (300 MHz, CDCl3): δ 0.8–1.1 (3H, br, CH3 polymer backbone), 1.7–2.0 (2H, br, CH2, polymer backbone), 3.41 (3H, br s, –O–CH3), 3.55 (2, br s, –O–CH2–CH2–O–CH3), 3.61 (2H, br s, –O–CH2–CH2–O–CH3), 3.66 (2H, br s, C(O)O–CH2–CH2–), 4.08 (2H, br s, C(O)O–CH2–CH2–). Conversion by 1H NMR spectroscopy: 95%, degree of polymerization (DP) = 89. Mn (1H NMR) = 16.7 kDa. Mn (SEC, THF, PMMA calibration) = 13.8 kDa, Mw/Mn = 1.36.Polymer end group removal39
P6 (50 mg, 1 eq., 4.4 × 10−3 mmol), AIBN (0.4 mg, 0.5 eq., 2.2 × 10−3 mmol), 1-ethylpiperidine hypophosphite (1-EPHP) (4 mg, 5 eq., 2.2 × 10−2 mmol) toluene (1 mL) and DMSO (0.5 mL) were weighed into an oven-dried ampoule and degassed via three freeze pump–thaw cycles, backfilled with nitrogen and heated at 100 °C. After 2 hours, toluene was removed under vacuum and the mixture dialyzed against deionized water (MWCO = 3500 Da). The polymer was then freeze-dried to yield a white solid. SEC analysis showed no absorbance in the UV-309 nm trace corresponding to the trithiocarbonate RAFT end group indicating that it was no longer present in significant quantities.Polymer chirality retention test
By exposing the non-polymerizable S-MacMillan catalyst to the same polymerization conditions as M1, possible racemization of the catalyst was investigated. The non-polymerizable S-MacMillan catalyst (0.250 g, 50 eq., 0.98 mmol), AIBN (0.3 mg, 0.1 eq., 2.0 × 10−3 mmol), CTA (2-cyano-2-propyl dodecyl trithiocarbonate, 6.8 mg, 1 eq., 2.0 × 10−2 mmol) and DMSO (0.5 mL) were weighed into an oven-dried ampoule and degassed via three freeze–pump–thaw cycles before being back-filled with nitrogen and heated to 80 °C. After 6 hours, the polymer was precipitated into cold stirring Et2O, giving a cloudy solution and the polymer was collected by centrifugation. The resultant solid was analyzed by HPLC (hexane:propan-2-ol, 90:10) and showed only the presence of the S-enantiomer, tR = 7.9 min (R-enantiomer, tR = 7.2 min, ESI Fig. S5†).Diels–Alder catalysis reaction40
A typical Diels–Alder reaction was carried out as follows: (N.B. Reactions were all carried out at identical reagent concentrations and the polymer concentration was varied to make up the same catalyst loading i.e. 5 mol%): the catalytically active polymer was weighed into a vial (5 mol% catalyst loading) and dissolved in the appropriate solvent (H2O or CH3OH/H2O mixture, 0.09 M of catalyst). TFA (0.013 mL, 1 eq.) was then added, followed by the dienophile (0.02 mL 1 eq.) and the solution allowed to stir for a few minutes before cyclopentadiene (0.015 mL, 1 eq.) was added. This resulted in a slightly turbid solution; particularly at high polymer concentrations and in the 100% water system. Analysis of the reaction was carried out directly when it was performed in H2O. Additional work up was required when CH3OH:H2O (95:5 v/v%) was used a solvent (to remove acetal side-products). The aliquot (∼0.1 mL) was stirred in H2O:TFA:CHCl3 (1:1:2) (∼4 mL) before being neutralized with NaHCO3 (∼2 mL) and then extracted into Et2O (2 × 5 mL). Conversion was determined by 1H NMR spectroscopy and ee% measured by GC, injection temperature 250 °C, column temperature 80 °C, ramp to 160 °C at 4.5 °C min−1, exo isomers tR = 12.8 and 13.2 min, endo isomers tR = 12.9 and 13.4 min. 1H NMR (300 MHz, CDCl3): δ 9.33 (1H, d, J = 3.0 Hz, C(O)H exo), 9.45 (1H, d, J = 8.1 Hz, C(O)H starting material), 9.75 (1H, d, J = 3.0 Hz, C(O)H endo). Data from M1 in H2O: conversion 94% (1H NMR spectroscopy), exo:endo ratio 1.00:1.05 (1H NMR spectroscopy), exo 73% ee, endo 88% ee (GC analysis), (ESI Fig. S7†).Polymer reuse by freeze-drying method
P6 (73% loading of M1) was weighed into a vial (30 mg, 0.05 eq. of catalyst, 5 mol%) and dissolved in CH3OH:H2O (95:5 v/v%, 1 mL, 0.09 M). TFA (0.130 mL, 1 eq.) and trans-hexen-1-al (0.200 mL, 1 eq.) were added to the polymer solution and left to stir for 5 min before cyclopentadiene (0.300 mL, 2 eq.) was added. After 4 hours an aliquot was taken for analysis and the reaction mixture was washed with Et2O (2 × 10 mL) and CHCl3 (2 × 10 mL) removing the organic starting materials and products. The remaining aqueous layer was then diluted with DMSO. The water–DMSO solution containing the polymer was then dialyzed extensively against deionized water (MWCO = 3500 Da) and freeze-dried to give a white solid. The recovered polymer was weighed and reused in a second cycle (ESI Table S5†).Polymer reuse through a pseudo-continuous method
P6 (73%) was weighed into a vial (30 mg, 0.05 eq. of catalyst, 5 mol%) and dissolved in CH3OH:H2O (95:5 v/v%, 1 mL, 0.09 M). TFA (0.13 mL, 1 eq.) and trans-hexen-1-al (0.2 mL, 1 eq.) were added to the polymer solution and left to stir for 5 min before cyclopentadiene (0.3 mL, 2 eq.) was added. The reaction being neutralized with NaHCO3 (2 mL) and extracted into Et2O (2 × 5 mL). The remaining polymer solution was washed with hexane, extracting the starting materials and products leaving the polymer in the acidic CH3OH:H2O solution. To this solution, more reagents (trans-hexen-1-al and cyclopentadiene) were added and the catalysis/reuse process repeated (ESI Table S6†).Results and discussion
Monomer synthesis21,38
In order to successfully control catalyst loading on the polymer, a novel monomer containing the catalytic functionality was synthesized for polymerization via RAFT. This was achieved using the three step synthetic route shown in Scheme 1, beginning with a derivative of the amino acid (S)-tyrosine, a naturally occurring, readily available source of chirality.41 The first two steps are based on the original synthesis by MacMillan using phenylalanine; reaction with methylamine followed by ring formation with acetone. The final step, coupling with methacryloyl chloride is based on well-known chemistries yielding M1 on a multi-gram scale.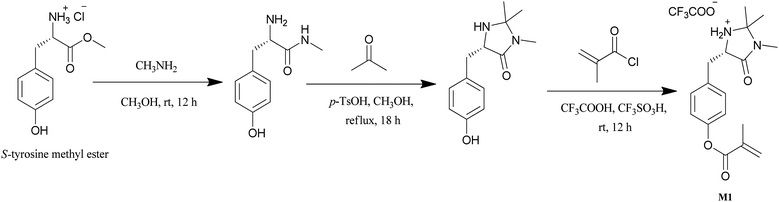 | ||
| Scheme 1 Synthesis of S-MacMillan functionalized monomer (M1) from S-tyrosine methyl ester. | ||
Polymer synthesis
A range of MacMillan catalyst functionalized copolymers were successfully synthesized using RAFT polymerization and M1 was found to polymerize well with the comonomer, DEGMA (Scheme 2). The degree of MacMillan catalyst incorporation was determined to be in the range 6–100%. At all degrees of incorporation, good control over molecular weight and polydispersity were achieved (Table 1). Molecular weights were difficult to ascertain by 1H NMR spectroscopy, as the end group signals from the polymers were concealed beneath the polymer signals. Therefore, molecular weights determined by SEC were used to determine the amount of polymer required for catalysis.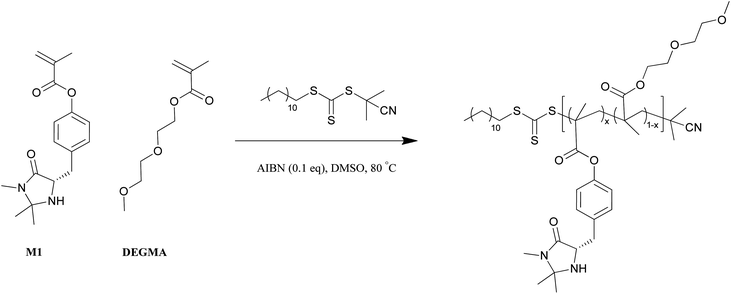 | ||
| Scheme 2 A representative RAFT polymerization scheme of M1 and DEGMA. | ||
| Polymer | Feed ratio (DEGMA:M1) | Mn,th (kDa) | Mna (kDa) | Mw/Mna | Catalyst incorporationb (%) |
|---|---|---|---|---|---|
| a Measured by THF SEC against PMMA standards.b Measured by 1H NMR spectroscopy. | |||||
| P1 | 92:8 | 17.0 | 11.8 | 1.35 | 6 |
| P2 | 76:24 | 21.7 | 11.6 | 1.39 | 26 |
| P3 | 69:31 | 20.7 | 11.8 | 1.33 | 33 |
| P4 | 58:42 | 23.3 | 12.0 | 1.39 | 38 |
| P5 | 37:63 | 22.7 | 11.5 | 1.47 | 57 |
| P6 | 13:87 | 26.8 | 11.4 | 1.46 | 73 |
| P7 | 0:100 | 27.8 | 5.8 | 1.31 | 100 |
To confirm that no racemization had taken place during the polymerization process, the standard non-polymerizable S-MacMillan catalyst was synthesized and subjected to the same polymerization conditions as M1. Following recovery, analysis by chiral-HPLC revealed only the S-enantiomer, suggesting no racemization occurred during polymerization and hence, it has been inferred that only the S-enantiomer of M1 is incorporated into the polymer (ESI Fig. S5†, R-enantiomer tR = 7.2 min and S-enantiomer tR = 7.9 min).
To determine the distribution of catalytic functionality along the polymer chain, monomer reactivity ratios were investigated. A number of polymerizations were carried out using a variety of monomer ratios, i.e. M1:DEGMA, 90:10, 70:30, 50:50, 30:70, 10:90. Conversions of both monomers were determined by 1H NMR spectroscopy with conversions kept low (between 5 and 15%). The mol fractions of the two monomers in the initial feed and in the final polymer were used in the Contour program, developed by van Herk;42 and the reactivity ratios were determined to be M1 = 0.892 and DEGMA = 0.575 (ESI Fig. S6†). These values suggest that whilst it is not a completely random system, the two monomers are well distributed throughout the resultant polymer.
Polymer properties
Notably, the physical appearance of the polymers is significantly different: at low M1 incorporation, rubber-like polymers are produced, whereas high incorporations give glassy polymers. A trend in Tg was determined where an increase in the degree of incorporation of M1 resulted in an increase in Tg (Table 2). The polymer with lowest incorporation, P1 (6% M1) has a Tg midpoint of 23.1 °C, which increases to 140.0 °C for P7 (100% M1). This increase in Tg may be due to the more rigid structure of M1 compared with DEGMA, resulting in the more glassy polymers observed at high incorporations. It is also possible that M1 undergoes intermolecular interactions such as H-bonding and π-stacking, which is more pronounced at the higher incorporations, increasing the order of the polymer and contributing to the higher Tg.The comonomer DEGMA is known to exhibit an LCST and thus the LCST of some of copolymers were examined (ESI Table S1†). The cloud points were found to be dependent on polymer concentration and were therefore examined at multiple concentrations (0.5, 1.0, 2.0 and 5.0 mg mL−1). The cloud points for the PDEGMA homopolymer (Mw = 16.7 kDa) were found to vary from 27.4 °C for 0.5 mg mL−1 to 24.4 °C for 5.0 mg mL−1. For P1 (6% loading), higher LCST cloud points were uniformly observed compared with DEGMA: from 37.7 °C for 0.5 mg mL−1 through to 26.5 °C for 5.0 mg mL−1, further increasing for P2 (26% loading): 58.8 °C for 0.5 mg mL−1 through to 42.4 °C for 5.0 mg mL−1. At lower concentrations of P2, the increase in absorption was found to be significantly smaller compared to the other polymers suggesting that less polymer is precipitated, potentially indicating that the polymer is losing its LCST behaviour. Analysis on polymers with higher incorporation proved inconclusive as the temperature range became too high. A possible explanation for the increase in LCST cloud point temperature is the ability of M1 to hydrogen-bond to itself as well as to water, and therefore increase the temperature at which entropic loss (due to formation of hydrogen-bonded structures) outweighs the enthalpic gain (from the formed bonds). Hence, as the incorporation of M1 increases, the LCST cloud point increases.
In 1998, Bergbreiter and co-workers reported the use of poly(N-isopropyl acrylamide) (PNIPAM), another polymer known to exhibit an LCST behaviour, as a recoverable polymeric system.43 The polymer was recovered and recycled by heating above its LCST, resulting in precipitation. The utility of LCST polymers for catalyst recovery has also been reported by our group in 2012, also using PNIPAM to efficiently recover a DMAP functionalized polymer.16 However, whilst some of the polymers reported here exhibit this behaviour, the temperature at which the polymer precipitates is strongly dependent on the degree of catalyst incorporation and polymer concentration. As the LCST gets higher (>40 °C) recovery via this method is less efficient and it is therefore not a general method for recycling in this case. Whilst P1 has an LCST cloud point that could potentially be utilized, due to small scale of the reactions this was difficult to investigate.
Diels–Alder catalysis with monomers and functionalized polymers
The reaction between cyclopentadiene and trans-hexen-1-al (R = C3H7) (Scheme 3) has been used previously to demonstrate the utility of the MacMillan catalyst and was therefore selected as our benchmark reaction. Catalysis with both the non-polymerizable MacMillan catalyst and its polymerizable derivative M1 was first carried out (Table 3) and the functionalized polymers were then examined under the same reaction conditions (Table 4). Ahrendt et al. reported a yield of 92%; an exo:endo ratio of 1.00:1.00; an exo ee% 84 and an endo ee% of 93.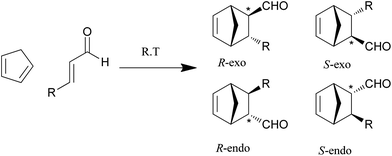 | ||
| Scheme 3 Model Diels–Alder reaction, where R = C3H7 and where there are four possible products: the endo and exo products of both enantiomers. The favoured enantiomer is likely to be the R-endo-enantiomer if the reaction follows the same pattern as suggested by MacMillan et al.,21 arising from attack from the opposite side to the phenyl group in the catalyst. | ||
:H2O 95:5 v/v% vs. H2O)
| Catalyst | Solvent | Conversiona (%) | exo:endoa | exo eeb% | endo eeb% |
|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy.b Determined by chiral GC analysis. | |||||
| M1 | CH3OH:H2O | 84 | 1.00:1.04 | 79 | 88 |
| H2O | 94 | 1.00:1.05 | 73 | 88 | |
| P1 | CH3OH:H2O | 87 | 1.00:1.00 | 79 | 83 |
| H2O | 75 | 1.00:1.08 | 69 | 84 | |
| P2 | CH3OH:H2O | 96 | 1.00:1.01 | 80 | 88 |
| H2O | 70 | 1.00:1.08 | 73 | 81 | |
| P3 | CH3OH:H2O | 100 | 1.00:1.12 | 81 | 86 |
| H2O | 86 | 1.00:1.11 | 74 | 85 | |
| P4 | CH3OH:H2O | 90 | 1.00:0.62 | 75 | 85 |
| H2O | 85 | 1.00:1.14 | 74 | 85 | |
| P5 | CH3OH:H2O | 94 | 1.00:0.77 | 78 | 74 |
| H2O | 95 | 1.00:1.07 | 74 | 88 | |
| P6 | CH3OH:H2O | 84 | 1.00:1.08 | 76 | 83 |
| H2O | 91 | 1.00:1.07 | 74 | 82 | |
| P7 | CH3OH:H2O | 92 | 1.00:1.08 | 77 | 84 |
| H2O | 86 | 1.00:1.13 | 76 | 85 | |
The results were similar for both monomers and polymers examined: conversions and the exo:endo ratios were similar across all the reactions (1.00:0.98–1.14) and the ees comparable but uniformly slightly lower than those reported in the literature for both endo and exo products. Moreover, changing the solvent from CH3OH:H2O (95:5 v/v%) to H2O appears to have little effect on the reaction. These results were interesting as the importance of catalyst site isolation has been previously demonstrated and it was anticipated that varying catalyst loading would have an effect on the catalytic activity.44 In the absence of any catalyst the reaction is significantly slower reaching 9.9% (H2O) and 0.99% (MeOH:H2O 95:5 v/v%) conversions after 4 hours.
Therefore, the reaction kinetics of two polymers were investigated to determine whether catalytic loading affected the rate of reaction. This was carried out using P2 (26% loading) and P6 (73% loading) (ESI Table S2†). Interestingly, this still did not lead to a tangible difference: the kinetics for both polymers were found to be very similar (25 min P6 = 66%, P2 = 53% and 240 min P6 = 96% and P2 = 93%). The exo:endo ratio (1.00:1.06–1.19) and enantioselectivities (74–80% ee for the major product) are also comparable for both polymers for the duration of the reaction. The comparable results for the two polymers suggest that in this case, catalyst site isolation does not affect polymer catalytic activity and that polymers with low catalyst loading (hence less expensive catalyst monomer is required) are still extremely efficient.
These initial catalysis results, carried out at room temperature, indicate that activity and selectivity are also unaffected by the Tg of the polymers. In order to confirm this, additional experiments were carried out at various temperatures (4–60 °C, ESI Table S3†). M1, P2 (Tg 36.1 °C) and P7 (Tg 140.0 °C) were used to catalyze the model Diels–Alder reaction (Scheme 3) and showed no significant differences: each displayed an increase in conversion and decrease in selectivity with increasing reaction temperature, confirming that the glass transition temperature does not have an effect.
Despite being a good handle for post-polymerization modifications,45 the RAFT end group present on the polymers after polymerization could potentially interfere with the catalysis reaction. In order to investigate the potential role of the end group in the Diels–Alder reaction it was removed from P6 using a radical induced end-group removal chemistry with 1-EPHP as the proton donor (ESI Fig. S8†). The activity of the polymer pre- and post-end group removal is comparable (91% vs. 91%), as is the enantioselectivity (endo 82 vs. 75% ee, ESI Table S4†). Therefore the presence of the end group, as well as the chemistries employed to remove it, have limited effect on the selectivity of the reaction.
The versatility of our polymer-supported MacMillan system was demonstrated by the catalytic efficiency of P2 in the Diels–Alder reaction of a range of substrates. Cyclopentadiene was reacted with a range of aldehydes (Table 5) structurally similar to the one presented in Scheme 3 but with a different R group. P2 catalyzed the reactions with great efficiency achieving conversions between 70 and 100% and good enantioselectivities (endo 72–89%), further demonstrating the catalytic ability of the polymers in a range of Diels–Alder reactions.
Polymer recycling
Facile recovery and reuse of supported catalysts is one of the main aims of catalyst immobilization and carries a great advantage over unsupported catalysts. In particular, the MacMillan catalyst is reported to be difficult to recycle and thus facilitation of successful recycling is of great significance.32As previously discussed, the use of PDEGMA as an LCST polymer has proven to be quite difficult as the LCST was found to be dependent on the degree of incorporation of M1, as well as the polymer concentration in solution (ESI Table S1†), thus, rendering this recovery route only applicable to certain polymers at certain concentrations.
Therefore, initially attempts were made to recover these MacMillan supporting polymers by other established polymer recovery techniques (i.e. via dialysis and freeze-drying). However, these resulted in low recovery yields (from mechanical loss), as well as a notable loss of enantioselectivity in subsequent reactions (ESI Table S5†). In light of this, a new pseudo continuous process was developed for polymer recovery and reuse: due to significant differences in solubility between the polymer catalyst and the reagents/products, starting materials and products may simply be extracted into hexane. The polymer, which is insoluble in hexane, remains in the reaction solvent, and can then be reused multiple times, achieving good results for each cycle (Table 6).
:H2O (95:5 v/v%) in multiple cycles via a pseudo continuous process
Conversion for all cycles has remained high, staying above 70% and pleasingly, enantioselectivities were also maintained (endo 79–88%). Crucially, it was found that if the acid (TFA) was not added in each new cycle, high conversions were maintained. However, if acid was added together with the new reagents, a significant drop in enantioselectivity was observed (ESI Table S6†). Therefore, for efficient recycling, it can be assumed that both acid and polymer remain in the CH3OH:H2O layer and extra acid should not be added during subsequent reuse reactions.
Conclusions
In conclusion, the synthesis of a novel polymerizable catalytic monomer based on the MacMillan catalyst is reported. The catalytic functionality was successfully incorporated into a range of copolymers using RAFT polymerization, achieving good control over the degree of incorporation. Catalytic activity of the copolymers was demonstrated using the Diels–Alder reaction with a range of substrates where high yields and selectivities were obtained. The polymerization procedure, end group removal and Tg were not found to have a detrimental effect on the polymers' catalytic ability and, in a significant step towards the sustained reuse of the MacMillan catalyst, polymer recycling was also demonstrated over several cycles. Work is ongoing to further develop this recycling procedure.Acknowledgements
Financial support from the EPSRC, Homerton College and the Universities of Warwick and Cambridge are gratefully acknowledged. The SEC and DSC equipment used in this research was obtained through Birmingham Science City: Innovative uses for Advanced Materials in the Modern World with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). Mr Daniel Wright is gratefully thanked for carrying out DSC analysis and Ms Dafni Moatsou for designing the graphical abstract.Notes and references
- D. E. Bergbreiter, Chem. Rev., 2002, 102, 3345–3384 CrossRef CAS.
- A. W. Bosman, R. Vestberg, A. Heumann, J. M. J. Fréchet and C. J. Hawker, J. Am. Chem. Soc., 2003, 125, 715–728 CrossRef CAS.
- Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322–6323 CrossRef CAS.
- S. M. Grayson and J. M. J. Fréchet, Chem. Rev., 2001, 101, 3819–3868 CrossRef CAS.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS.
- T. E. Kristensen and T. Hansen, Eur. J. Org. Chem., 2010, 3179–3204 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688 RSC.
- M. Benaglia, G. Celentano and F. Cozzi, Adv. Synth. Catal., 2001, 343, 171–173 CrossRef CAS.
- M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2002, 344, 533–542 CrossRef CAS.
- D. Font, C. Jimeno and M. A. Pericas, Org. Lett., 2006, 8, 4653–4655 CrossRef CAS.
- G. Sabitha, N. Fatima, E. V. Reddy and J. S. Yadav, Adv. Synth. Catal., 2005, 347, 1353–1355 CrossRef CAS.
- W. Miao and T. H. Chan, Adv. Synth. Catal., 2006, 348, 1711–1718 CrossRef CAS.
- Z. Shen, J. Ma, Y. Liu, C. Jiao, M. Li and Y. Zhang, Chirality, 2005, 17, 556–558 CrossRef CAS.
- E. Bellis and G. Kokotos, J. Mol. Catal. A: Chem., 2005, 241, 166–174 CrossRef CAS.
- P. Cotanda, A. Lu, J. P. Patterson, N. Petzetakis and R. K. O'Reilly, Macromolecules, 2012, 45, 2377–2384 CrossRef CAS.
- A. C. Evans, A. Lu, C. Ondeck, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2010, 43, 6374–6380 CrossRef CAS.
- A. Lu, T. P. Smart, T. H. Epps, D. A. Longbottom and R. K. O'Reilly, Macromolecules, 2011, 44, 7233–7241 CrossRef CAS.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2012 DOI:10.1002/anie.201207123.
- Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu and S. Liu, Macromolecules, 2007, 40, 3538–3546 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458–2460 CrossRef CAS.
- S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192–1194 CrossRef CAS.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370–4371 CrossRef CAS.
- Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051–15053 CrossRef CAS.
- S. P. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS.
- M. Benaglia, G. Celentano, M. Cinquini, A. Puglisi and F. Cozzi, Adv. Synth. Catal., 2002, 344, 149–152 CrossRef CAS.
- S. A. Selkälä, J. Tois, P. M. Pihko and A. M. P. Koskinen, Adv. Synth. Catal., 2002, 344, 941–945 CrossRef.
- Y. Zhang, L. Zhao, S. S. Lee and J. Y. Ying, Adv. Synth. Catal., 2006, 348, 2027–2032 CrossRef CAS.
- N. Haraguchi, Y. Takemura and S. Itsuno, Tetrahedron Lett., 2010, 51, 1205–1208 CrossRef CAS.
- P. Riente, J. Yadav and M. A. Pericas, Org. Lett., 2012, 14, 3668–3671 CrossRef CAS.
- T. E. Kristensen, K. Vestli, M. G. Jakobsen, F. K. Hansen and T. Hansen, J. Org. Chem., 2010, 75, 1620–1629 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- E. Rizzardo, M. Chen, B. Chong, G. Moad, M. Skidmore and S. H. Thang, Macromol. Symp., 2007, 248, 104–116 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- T. E. Kristensen, K. Vestli, K. A. Fredriksen, F. K. Hansen and T. Hansen, Org. Lett., 2009, 11, 2968–2971 CrossRef CAS.
- A. O. Moughton, K. Stubenrauch and R. K. O'Reilly, Soft Matter, 2009, 5, 2361–2370 RSC.
- M. Lemay and W. W. Ogilvie, Org. Lett., 2005, 7, 4141–4144 CrossRef CAS.
- R. K. O'Reilly, Polym. Int., 2010, 59, 568–573 CAS.
- A. M. van Herk, J. Chem. Educ., 1995, 72, 138 CrossRef CAS.
- D. E. Bergbreiter, B. L. Case, Y.-S. Liu and J. W. Caraway, Macromolecules, 1998, 31, 6053–6062 CrossRef CAS.
- J. Dzierzak, E. Bottinelli, G. Berlier, E. Gianotti, E. Stulz, R. M. Kowalczyk and R. Raja, Chem. Commun., 2010, 46, 2805–2807 RSC.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Further characterization of the synthesized monomer and polymers. HPLC chromatogram of the catalyst after polymerization. A typical GC spectrum for the chiral analysis of the Diels–Alder products. LCST cloud point data for polymers at different concentrations. Catalysis data for P2 and P7 at different temperatures; P2 and P6 kinetic data; P6 before and after end group removal and recycling by freeze-drying. Analysis of the recovered polymer after pseudo continuous experiments. See DOI: 10.1039/c3py21125h |
| This journal is © The Royal Society of Chemistry 2013 |