 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of variable conformation pH responsive block co-polymers for nucleic acid delivery and targeted cell entry†
Teresa
Matini
a,
Nora
Francini
ac,
Anna
Battocchio
ac,
Sebastian G.
Spain
a,
Giuseppe
Mantovani
a,
Maria J.
Vicent
*b,
Joaquin
Sanchis
b,
Elena
Gallon
c,
Francesca
Mastrotto
ac,
Stefano
Salmaso
*c,
Paolo
Caliceti
c and
Cameron
Alexander
*a
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bCentro de Investigation Principe Felipe (CIPF), Polymer Therapeutics Laboratory, Av. Eduardo Primo Yúfera 3', E-46012, Valencia, Spain
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, 35131, Padova, Italy
First published on 12th July 2013
Abstract
Responsive materials that change conformation with varying pH have been prepared from a range of amphiphilic block co-polymers. The individual blocks are composed of (a) permanently hydrophilic chains with neutral functionality and (b) acrylate polymers with weakly basic side-chains. Variation in co-monomer content, molar mass and block ratios/compositions leads to a range of pH-responses, manifest through reversible self-assembly into micelles and/or polymersomes. These transitions can be tuned to achieve environmental responses in a pH range from 5–7, as shown by turbidimetric analysis, NMR and dynamic light scattering measurements (DLS). Further characterization by transmission electron microscopy (TEM) indicates that polymersomes with diameters of 100–200 nm can be formed under certain pH-ranges where the weakly basic side-chains are deprotonated. The ability of the systems assembled with these polymers to act as pH-responsive containers is shown by DNA encapsulation and release studies, and their potential for application as vehicle for drug delivery is proved by cell metabolic activity and cell uptake measurements.
Introduction
Polymeric carriers offer many advantages for controlled release applications, drug delivery and nanomedicine.1–4 A number of macromolecular carrier systems are currently under investigation in order to improve the delivery of potent but delicate drugs such as proteins and nucleic acids.5,6 These systems need to be water-soluble, non-toxic and non-immunogenic, as well as compatible with serum components. For most applications, polymer based delivery systems must also be capable of being either degraded to harmless breakdown products or eliminated entirely from the body. Furthermore, these carriers need functional groups that allow them to interact with or encapsulate a drug of interest, and preferably should contain recognition motifs, which target disease-related antigens or receptors. Not surprisingly, the combination of these factors is hard to achieve with existing materials, leading to an urgent need for new highly functional and active biomedical polymers.1The aim of these studies was to prepare a modular and effective controlled release system, based on polymers designed to respond to the pH-changes that occur in tissues affected by peculiar disease.7,8 Microenvironments in solid tumours are generally heterogeneous and hypoxic, and cancer cells also lack a nutrient supply. This in turn leads to the changes in the energy production machinery towards the use of glycolytic pathways for energy production, resulting in an increase of lactic acid production and a decrease in pH in the tissue. Intracellular pH is similar in both solid tumour and normal tissues, but extracellular pH is lower in solid tumours than in normal tissue. These differences in pH provide a biological rationale for selective cancer chemotherapy.9 A specific further requirement is that agents for cancer drug delivery are taken-up selectively by cancer cells and efficiently release their drug payloads to yield action at the desired molecular targets.
A number of prior reports have indicated that block co-polymers with acidic or basic side chains can alter their conformation in response to biologically-relevant pH changes.10–15 However, the systematic variation of specific components contributing to the side-chain pH-response over the key 5–8 pH range have been rather less studied. In this work, new pH-sensitive polymers (Fig. 1) have been designed, incorporating pH-response imidazolic units at varying distance from a polymer backbone, as a first stage in a programme to generate materials that can be tuned to exploit specific local conditions for therapy.
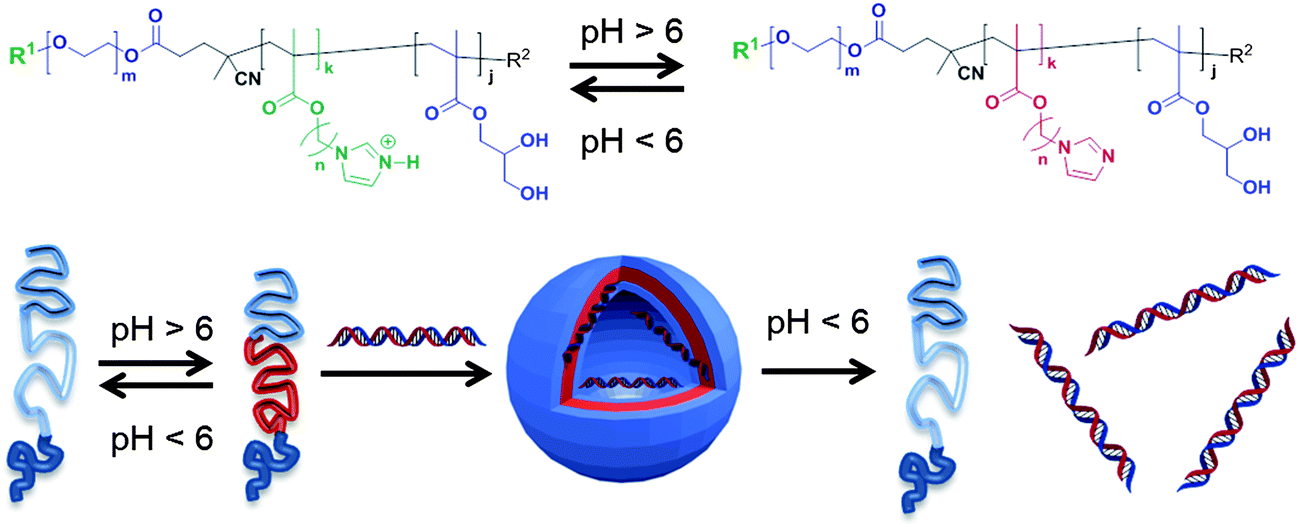 | ||
| Fig. 1 Self-assembly of pH-responsive triblock co-polymers. Deprotonation of the central imidazole-containing blocks above a critical pH range causes self-association of hydrophobic components and formation of polymer nanoparticles. Assembly of nanoparticles in the presence of short DNA strands results in encapsulation of nucleic acids, which can be released subsequently by a pH switch. | ||
Ultimately we aim to generate polymeric carriers with prolonged circulation in the bloodstream, high retention in the heterogeneous, acid and hypoxic tumor environments, and a variety of response profiles to enable release of drugs across a range of disease settings. We report here the initial synthesis and characterisation of a family of polymers and evaluate their pH-response through biophysical measurements, dye and DNA encapsulation/release studies and cytotoxicity assays. While the primary goal of this work was not to achieve fully biocompatible polymers in the first instance, nevertheless, the experiments provide insights into the behaviour of polymers containing pH-responsive side-chains, and suggest design criteria for future materials that may be used in vitro as well as in vivo.
Materials and methods
Chemicals and analysis
Reagents and solvents for synthesis were obtained at the highest purity available and used without further purification unless stated.Tetrahydrofuran anhydrous (THF), azobisisobutyronitrile (AIBN), imidazole, sodium hydride (NaH), sodium hydroxide (NaOH), methacryloyl chloride, 6-chloro-1-hexanol, 4-chloro-1-butanol, acetic anhydride, magnesium sulfate, potassium carbonate (K2CO3), glycerol methacrylate (GMA), N,N′-dimethylamino pyridine (DMAP) dimethylsulfoxide anhydrous (DMSO), N,N′-dimethylacetamide (DMAC), dichloromethane (DCM), ethyl acetate (EtOAc), methanol (MeOH), diethylether (Et2O), petroleum ether (b.p. 40–60 °C), N,N′-dicyclohexylcarbodiimide (DCC), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB), triethylamine (Et3N), trifluoroacetic acid (TFA), N-hydroxysuccinimide (NHS) and t-Boc–NH–PEG3500Da were obtained from Sigma-Aldrich, Alfa Aesar, JenKem and Fisher Scientific companies and used without further purification. Methoxy-PEG1900Da was purchased from Polysciences Inc. and dried azeotropically from toluene.
Polymerizations were carried out using standard Schlenk techniques under a nitrogen atmosphere. Thin layer chromatography (TLC) was performed using pre-coated plates (silica gel 60 ALUGRAM SIL G/UV254) and eluted in the solvent system indicated. Compounds were visualized by use of UV light (254 nm) or a basic solution (10% w/w K2CO3 in water) of KMnO4. Acros Organic 60 Å (0.035–0.070 mm) silica gel was used for column chromatography.
1H and 13C NMR spectra were obtained on a Bruker DPX400 Ultrashield spectrometer and pH studies were performed on a Bruker AV(III)500 spectrometer. All NMR data were processed using MestreNova 6.2.1 Software. All chemical shifts are reported in ppm (δ) relative to tetramethylsilane, referenced to the chemical shifts of residual solvent resonances. The following abbreviations were used for NMR peak multiplicities: s = singlet, bs = broad singlet, d = doublet, t = triplet, m = multiplet.
Mass spectrometry was carried out using a Waters LCT-TOF mass spectrometer fitted with a Waters 2795 separations unit and electrospray ionisation.
Turbidimetry assays were performed using a VARIAN Cary 50Bio UV-visible spectrophotometer.
Dynamic light scattering (DLS) and ζ-potential measurements were performed at 25 °C and 37 °C using a Malvern Zetasizer (Nano-ZS) and data acquired using Zetasizer Software (version 6.12).
Transmission electron microscopy (TEM) was performed using a Tecnai G2 (FEI, Oregon, USA). Samples were placed on copper grid, the excess was removed with filter paper and then stained with uranyl acetate (1% in deionized water).
Fluorimetry analyses were performed using a LS 50 B Perkin-Elmer fluorimeter.
Synthesis of monomers and intermediates
4-Chlorobutyl acetate (1a). A solution of 4-chloro-1-butanol (39.0 g, 360 mmol), acetic anhydride (55.1 g, 536 mmol), Et3N (109.0 g, 1077 mmol) and DMAP (4.40 g, 36.0 mmol) in DCM (200 mL) was prepared and stirred at 0 °C for 30 minutes and then left to react for an hour at room temperature. The volume of DCM was reduced to ∼70 mL, then the mixture was poured in a separating funnel containing 200 mL of H2O. The aqueous layer was extracted twice with Et2O (2 × 150 mL) and the organic layers were combined and washed with water (2 × 150 mL) then dried over MgSO4. The mixture was filtered and the solvent was removed under reduced pressure to yield 49.0 g (325 mmol, 90%) of 1a as a yellow oily liquid which was used for the next step without further purification.
1H NMR (400 MHz, CDCl3): δ 4.00 (t, J = 6.6 Hz, 2H, CH2), 3.48 (t, J = 7.1 Hz, 2H, CH2), 1.95 (s, 3H, CH3), 1.80–1.66 (m, 4H, CH2).
13C NMR (101 MHz, CDCl3): δ 180.81 (1C), 63.41 (1C), 44.56 (1C), 29.25 (1C), 26.02 (1C), 20.92 (1C).
4-(1H-imidazol-1-yl)butan-1-ol (2a). NaH (60% (w/w, stabilized in mineral oil)) (7.76 g, 195 mmol) was suspended in anhydrous DMSO (200 mL). Imidazole (22.0 g, 323 mmol) was added under stirring, at room temperature. The mixture was heated to 100 °C and then 4-chlorobutyl acetate (1a) (49.0 g, 325 mmol) was added. The reaction was stirred for 3 hours at 100 °C and monitored by 1H NMR in DMSO-d6. The reaction mixture was added to a solution (500 mL) of K2CO3 (111 g, 803 mmol) at room temperature, under vigorous stirring. The product was extracted (5 × 100 mL) with EtOAc, washed with aqueous base and dried over MgSO4. After removal of MgSO4 by filtration, the product was recovered from EtOAc solution by rotary evaporation of solvent.
The product was added to 250 mL of NaOHaq 10% (w/v) and the mixture heated to 70 °C and reaction was continued for 2 hours at 70 °C, until complete deacetylation of the alcohol was confirmed by 1H NMR.
The mixture was then extracted with DCM (3 × 200 mL) and dried over MgSO4. After removal of MgSO4 by filtration, the solvent was removed under reduced pressure and the crude product was purified by flash column chromatography (100% EtOAc).
The obtained yield of 2a was 10 g (71.4 mmol, 37%).
1H NMR (400 MHz, CDCl3): δ 7.57 (s, 1H, H-aromatic), 7.11 (s, 1H, H-aromatic), 6.85 (s, 1H, H-aromatic), 4.00 (t, J = 7.1 Hz, 2H, CH2N), 3.50 (t, J = 6.4 Hz, 2H, CH2OH), 1.72 (m, 2H, CH2), 1.44 (m, 2H, CH2).
13C NMR (101 MHz, DMSO-d6): δ 137.61 (1C), 128.71 (1C), 119.64 (1C), 60.61 (1C), 46.35 (1C), 39.98 (1C), 29.86 (1C), 27.89 (1C).
ESI-ToF mass spectrometry: expected for m/z [M − H]+1 141.10 Da, found 141.75 Da.
4-(1H-imidazol-1-yl)butyl methacrylate hydrochloride (ImBuMA, 3a). The imidazolic alcohol (2a) (10.0 g, 71.4 mmol) was dissolved in DCM (20 mL) and the resulting solution cooled to −20 °C. A solution of methacryloyl chloride (16.0 g, 153 mmol, 15 mL) in DCM (40 mL) was added dropwise under stirring over ca. 1 hour. The mixture was stirred at this temperature for ca. 30 minutes, then left at room temperature overnight. DCM was evaporated under reduced pressure to reduce the total solvent volume, the monomer was precipitated in petroleum ether and the resulting residue purified by two flash column chromatography on silica gel, eluting with 100% EtOAc, then EtOAc–MeOH 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The volatile components were removed under reduced pressure and product 3a (7.46 g, 30.5 mmol, 43%) was stored at −20 °C.
:1. The volatile components were removed under reduced pressure and product 3a (7.46 g, 30.5 mmol, 43%) was stored at −20 °C.
1H NMR (400 MHz, CDCl3): δ 9.27 (s, 1H, H-aromatic), 7.37 (s, 1H, H-aromatic), 7.15 (s, 1H, H-aromatic), 6.10 (s, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHH), 5.59 (s, 1H, CCHH), 4.35 (t, J = 6.5 Hz, 2H, OCH2), 4.21 (t, J = 7.1 Hz, 2H, NCH2), 2.02–1.94 (m, 2H, CH2), 1.95 (s, 3H, CH3), 1.77–1.65 (m, 2H, CH2).
CHH), 5.59 (s, 1H, CCHH), 4.35 (t, J = 6.5 Hz, 2H, OCH2), 4.21 (t, J = 7.1 Hz, 2H, NCH2), 2.02–1.94 (m, 2H, CH2), 1.95 (s, 3H, CH3), 1.77–1.65 (m, 2H, CH2).
13C NMR (101 MHz, DMSO-d6): δ 166.29 (1C), 135.66 (1C), 125.20 (1C), 121.96 (1C), 119.38 (1C), 78.72 (1C), 62.56 (1C), 47.79 (1C), 25.62 (1C), 24.80 (1C), 17.81 (1C).
ESI-Tof mass spectrometry: expected for m/z [M − H]+1 209.13 Da, found 209.68 Da.
Synthesis of 6-chlorohexyl acetate (1b). A solution of 6-chloro-1-hexanol (40.0 g, 293 mmol), acetic anhydride (44.0 g, 428 mmol), Et3N (88.0 g, 870 mmol) and DMAP (3.5 g, 29 mmol) in DCM (100 mL) were reacted and purified as described for 4-chlorobutyl acetate 1a, to give 1b (49.4 g, 276 mmol, 94%) as a pale yellow oil that was used for the next step without further purification.
1H NMR (400 MHz, CDCl3): δ 3.98 (t, J = 6.6 Hz, 2H, CH2), 3.47 (t, J = 7.1 Hz, 2H, CH2), 1.97 (s, 3H, CH3), 1.71 (m, 2H, CH2), 1.57 (m, 2H, CH2), 1.40 (m, 2H, CH2), 1.31 (m, 2H, CH2).
13C NMR (101 MHz, CDCl3): δ 171.48 (1C) 64.68 (1C), 45.25 (1C), 32.72 (1C), 28.65 (1C), 26.64 (1C), 25.40 (1C), 21.43 (1C).
6-(1H-imidazol-1-yl)-hexan-1-ol (2b). NaH (6.59 g, 276 mmol) was suspended in 200 mL of DMSO anhydrous. Imidazole (18.0 g, 264 mmol) was added under stirring, at room temperature. The mixture was heated to 100 °C and then 6-chlorohexyl acetate (1b) (49.4 g, 276 mmol) was added. The reaction was carried out as described for 2a and the crude product (21.0 g, 125 mmol, 45%) was used for the next step without further purification.
1H NMR (400 MHz, CDCl3): δ 7.39 (s, 1H, H-aromatic), 6.96 (s, 1H, H-aromatic), 6.85 (s, 1H, H-aromatic), 3.89 (t, J = 7.1 Hz, 2H, CH2N), 3.55 (t, J = 6.4 Hz, 2H, CH2OH), 1.72 (m, 2H, CH2), 1.49 (m, 2H, CH2), 1.34 (m, 2H, CH2), 1.25 (m, 2H, CH2).
13C NMR (101 MHz, DMSO-d6); δ 137.63 (1C), 128.70 (1C), 119.66 (1C), 61.01 (1C), 46.35 (1C), 32.82 (1C), 31.10 (1C), 26.27 (1C), 25.42 (1C).
ESI-Tof mass spectrometry: expected for m/z [M − H]+1 169.25 Da, found 169.85 Da.
6-(1H-imidazol-1-yl)hexyl methacrylate hydrochloride (ImHeMA, 3b). The imidazolic alcohol (2b) (21 g, 125 mmol) was dissolved in DCM (40 mL) and kept at −20 °C. A solution of methacryloyl chloride (26.0 g, 250 mmol, 24.5 mL) in DCM (50 mL) was added dropwise under stirring over ca. 1 hour. The reaction was stirred at this temperature ca. 30 minutes, then left at room temperature overnight.
Purification was carried out firstly by precipitation of the monomer in petroleum ether and then by flash column chromatography on silica gel eluting with 100% EtOAc and subsequently with EtOAc–MeOH 3:1. The volatiles were removed under reduced pressure and the product (3b) (21.3 g, 78.0 mmol, 63%) was stored at −20 °C.
1H NMR (400 MHz, CDCl3): δ 9.22 (s, 1H, H-aromatic), 7.81 (s, 1H, H-aromatic), 7.67 (s, 1H, H-aromatic), 5.99 (m, 1H, CCHH), 5.66 (m, 1H, CCHH), 4.19 (t, J = 6.6 Hz, 2H, OCH2), 4.05 (t, J = 7.1 Hz, 2H, NCH2), 1.89 (s, 3H, CH3), 1.81 (m, 2H, CH2), 1.60 (m, 2H, CH2), 1.35–1.23 (m, 4H, CH2).
13C NMR (101 MHz, DMSO): δ 166.38 (1C), 136.17 (1C), 135.27 (1C), 125.78 (1C), 121.85 (1C), 119.71 (1C), 63.98 (1C), 48.18 (1C), 29.46 (1C), 27.83 (1C), 25.16 (1C), 24.70 (1C), 18.17 (1C).
ESI-Tof mass spectrometry: expected for m/z [M − H]+1 238.17 Da, found 238.92 Da.
PEG1900 macroCTA (4). mPEG1900 (2.20 g, 1.16 mmol) was dried from water by azeotropic distillation with toluene under reduced pressure. mPEG1900 was dissolved in DCM (30 mL) and CPADB (969 mg, 3.47 mmol), DMAP (0.05 g, 0.4 mmol) and DCC (716 mg, 3.47 mmol) were added. The mixture was left to react overnight. The precipitate was filtered to remove the DCU co-product and the solution added dropwise under stirring to 100 mL of petroleum ether to give (4) as a pink powder.
1H NMR (400 MHz, D2O): δ 7.90 (s, 2H, CH-Ar), 7.57 (m, 1H, CH-Ar), 7.40 (m, 2H, CH-Ar), 4.26 (t, 2H, CH2O), 3.66 (bs, 204H, PEG repeating unit), 3.38 (s, 3H, OCH3), 2.72–2.60 (m, 4H, CH2CH2C), 1.94 (s, 3H, CN(C)CH3).
Synthesis of t-Boc–NH–PEG3500 macroCTA (5). t-Boc–NH–PEG3500 (400 mg, 0.114 mmol) was dissolved in DCM (10 mL) and CPADB (0.096 g, 0.350 mmol), DMAP (0.009 g, 0.07 mmol) and DCC (0.071 g, 0.34 mmol) were added. The mixture was left to react overnight, the resultant precipitate was filtered to remove the DCU co-product and the solution added dropwise under stirring to 100 mL of petroleum ether to give (5) as a pink powder.
1H NMR (400 MHz, D2O): δ 7.90 (s, 2H, CH-Ar), 7.57 (m, 1H, CH-Ar), 7.40 (m, 2H, CH-Ar), 4.27 (t, 2H, CH2O), 3.65 (bs, 342H, PEG repeat unit), 2.63 (m, 4H, COCH2CH2C), 1.94 (s, 3H, CN(C)CH3), 1.72 (s, 9H, t-Boc).
Synthesis of diblock co-polymer PEG1900-b-p(3a) (6). ImBuMA (3a) (2.0 g, 8.1 mmol), PEG1900 macroCTA (4) (182 mg, 0.08 mmol) and AIBN (7.00 mg, 0.04 mmol) in DMAC (8 mL) were sealed in a Schlenk tube, deoxygenated by argon bubbling for 30 minutes and then heated at 65 °C.
The conversion of monomer to polymer was calculated by 1H NMR following the decrease of the integrals of the monomer in the vinyl region (6.10 and 5.59 ppm) relative to the broad singlet of the PEG1900 repeat unit protons (3.71 ppm). Polymerization was stopped at 43% conversion. Polymers were obtained by precipitation in Et2O–petroleum ether (1:1 v/v).
The dithioester group (RAFT functionality) of the block copolymer was removed following a standard protocol16 in presence of AIBN (molar ratio polymer 6/AIBN = 1/20) at 80 °C in DMSO for 3 hours. The final polymer was purified by precipitation in THF and isolated as a white solid.
1H NMR (400 MHz, D2O): δ 8.82 (bs, 1H, H-Ar), 7.51 (bs, 2H, H-Ar), 4.32 (bs, 2H, CH2OC), 4.04 (bs, 2H, NCH2), 3.71 (bs, 204H, PEG repeat unit), 3.40 (s, 3H, CH3), 2.00–1.70 (bs, 5H, CH2 + CH3).
Synthesis of diblock co-polymer PEG1900-b-p(3b) (7). ImHeMA (3b) (3.570 g, 12.69 mmol), PEG1900 macro-CTA (4) (330 mg, 0.151 mmol) and AIBN (12 mg, 0.08 mmol) in DMAC (10 mL) were sealed in a Schlenk tube, deoxygenated by argon bubbling for 30 minutes and then heated at 65 °C.
The conversion of the polymer was calculated via1H NMR following the decrease of the integrals of the monomer vinyl signals (5.99 and 5.66 ppm) relative to the broad singlet of the PEG1900 repeat unit protons (3.71 ppm). Polymerization was stopped at 50% of conversion. The desired polymer was recovered by precipitation in Et2O–petroleum ether (1:1 v/v) and used for the next reaction as a macro-chain-transfer agent (CTA) without further purification.
1H NMR (400 MHz, D2O): δ 8.82 (bs, 1H, H-Ar), 7.51 (bs, 2H, H-Ar), 4.32 (bs, 2H, CH2OC), 4.04 (bs, 2H, NCH2), 3.71 (bs, 204H, PEG repeat unit), 3.66 (t, 2H, CH2O), 3.35 (s, 3H, CH3), 2.00 (bs, 2H, CH2), 1.70 (bs, 3H, CH3).
Synthesis of diblock co-polymer t-Boc–NH–PEG3500-b-p(3b) (8). ImHeMA (3a) (910 mg, 3.30 mmol), PEG3500 macroCTA (5) (150 mg, 0.04 mmol) and AIBN (3.00 mg, 0.02 mmol) in DMAC (5 mL) were sealed in a Schlenk tube, deoxygenated by argon bubbling for 30 minutes and then heated at 65 °C.
The conversion of the polymer was calculated via1H NMR following the decrease of the integrals of the monomer vinyl signals (5.99 and 5.66 ppm) relative to the broad singlet of the BocNHPEG3500 repeat unit protons (3.71 ppm). Polymerization was stopped at 54% of conversion. Polymers were obtained by precipitation in Et2O–petroleum ether (1:1 v/v) and used in the next reaction as a macro-CTA.
1H NMR (400 MHz, D2O); δ 8.57 (bs, 1H, H-Ar), 7.43 (bs, 2H, H-Ar), 4.20 (bs, 2H, CH2OC), 4.00 (bs, 2H, NCH2), 3.71 (bs, 342H, PEG repeat unit), 2.23–1.84 (bs, 2H, CH2), 1.65 (s, 9H, t-Boc), 1.44–1.28, 1.02–0.86 (bs, 2H, CH2).
Synthesis of triblock co-polymer PEG1900-b-p(3b)-p(GMA) (9). Block copolymer macro-CTA (7) (2.18 g, 0.11 mmol), GMA (1.13 g, 7.08 mmol) and AIBN (9.6 mg, 0.06 mmol) in DMAC (5 mL) were sealed in a Schlenk tube, deoxygenated by argon bubbling for 30 minutes and then heated at 65 °C. The conversion of the polymer was calculated via1H NMR following the decrease of the integrals of the monomer vinyl signals (6.35 and 5.68 ppm) using the broad singlet (8.82 ppm) of the aromatic repeat unit protons of the imidazole ring as an internal standard. Polymerization was stopped at 50% conversion and the desired material obtained by precipitation in Et2O–petroleum ether (1
:1 v/v). The dithioester end-group was removed by reaction in with AIBN (molar ratio polymer 9/AIBN = 1/20) at 80 °C in DMSO for 3 hours and the resulting polymer recovered by repeated precipitations in THF.
1H NMR (400 MHz, D2O): δ 7.64 (bs, 1H, H-Ar), 7.13 (bs, 1H, H-Ar), 6.88 (bs, 1H, H-Ar), 3.91 (bs, 2H, CH2OC), 3.83 (bs, 2H, NCH2), 3.66 (t, 2H, CH2O), 3.50 (bs, 204H, PEG repeat unit), 3.35 (bs, 3H + 2H, CH3 + CH2), 1.63 (bs, 2H, CH2), 1.48 (bs, 2H, CH2), 1.29 (bs, 3H, CH3), 1.19 (bs, 3H, CH3).
Synthesis of triblock co-polymers t-Boc–NH–PEG3500-b-p(3b)-b-p(GMA) (10). Block copolymer macroCTA 8 (586 mg, 0.04 mmol), GMA (370 mg, 2.43 mmol) and AIBN (164 mg, 0.02 mmol) in DMAC (5 mL) were sealed in a Schlenk tube, deoxygenated by argon bubbling for 30 minutes and then heated at 65 °C.
The conversion of the polymer was calculated via1H NMR following the decreasing of the integrals of the monomer vinyl signals (6.35 and 5.68 ppm) using the broad singlet (8.57 ppm) of the aromatic repeat unit protons of the imidazole ring as internal standard. Polymerization was stopped at 60% conversion. Polymers were obtained by precipitation in Et2O–petroleum ether (1:1 v/v). The dithioester end-group of the block copolymer was removed by reaction with AIBN (molar ratio polymer 10/AIBN = 1/20) at 80 °C in DMSO for 3 hours and the polymer was recovered by precipitation in THF.
1H NMR (400 MHz, D2O): δ 8.32 (bs, 1H, H-Ar), 7.37 (bs, 2H, H-Ar), 4.15 (bs, 4H, CH2 + CH2), 3.99 (bs, 4H, CH2 + CH2), 3.71 (bs, 342H, PEG repeat unit), 2.30–1.93 (bs, 2H + 2H, CH2 + CH2), 1.70 (s, 9H, t-Boc), 1.20–0.83 (bs, 6H, CH3 + CH3).
Synthesis of folate-terminated triblock co-polymers folate–NH–PEG3500-b-(3b)20-b-(GMA)58 (11). I – polymer 10 (20 mg, 0.0011 mmol) was dissolved in a 1
:1 (v/v) CF3COOH–DCM mixture (1 mL/1 mL) at room temperature. The solution was stirred at room temperature for 2 hours and then TFA and DCM were removed by evaporation under reduced pressure. The reaction was monitored by 1H NMR in MeOD by following the disappearance of the t-Boc protons at 1.44 ppm.
1H NMR (400 MHz, MeOD): δ 9.02 (bs, 1H, H-Ar), 7.70 (bs, 1H, H-Ar), 7.61 (bs, 1H, H-Ar), 4.48 (bs, 4H, CH2 + CH2), 4.29 (bs, 3H, CH2 + CH), 3.91 (bs, 2H, CH2), 3.64 (bs, 342H, PEG repeat unit), 1.94 (bs, 2H + 2H, CH2 + CH2), 1.42–0.93 (bs, 6H, CH3 + CH3).
II – folic acid (100.0 mg, 0.226 mmol) was dissolved in anhydrous DMSO (2 mL). NHS (26 mg, 0.23 mmol) and DCC (47 mg, 0.23 mmol) were added to the folic acid solution (folic acid–NHS–DCC = 1:1:1 mol/mol/mol). The reaction was carried out overnight at room temperature in the dark. N-Hydroxysuccinimidyl-ester-activated folic acid was precipitated by drop-wise addition to 40 mL of cold Et2O under stirring. The obtained yellow precipitate was washed with Et2O (3 × 30 mL) and then dried under vacuum.
III – NH2–PEG3500-b-(3b)20-b-(GMA)58 was dissolved in anhydrous DMSO and N-hydroxysuccinimidyl-ester-activated folic acid (1.8 mg, 0.0034 mmol) was added to the polymer solution (1:4 polymer–activated folate). The reaction was performed overnight under stirring at room temperature in the dark. The product was recovered by dropwise precipitation in Et2O and dried after solvent removal. The resulting dried product was dissolved in high purity (resistivity > 18 MΩ) water (2 mL); the solution was acidified by addition of HCl (5 M) in order to precipitate the free folic acid (which was not soluble under acidic conditions). The polymer-containing solution was recovered by centrifugation (5 minutes, 14000 rpm) then dialyzed (MWCO 3500 Da) and the solvent removed by lyophilization.
Folate–PEG3500-b-(3b)20-b-(GMA)58 was characterized by spectrophotometric methods. The conjugate was dissolved in high purity water, then diluted to 0.5 mg mL−1 in 20 mM phosphate buffer, NaCl 150 mM, at pH 7 and the absorbance at 363 nm was measured to quantify the folate concentration in the modified polymer. The folate concentration was derived using the εM of folic acid reported in the literature (6197 mol−1 cm−1).3 The solution was also diluted in water and tested by an iodine assay in order to determine the PEG concentration based on a previously prepared calibration curve. The quantification tests showed a conjugation yield of folic acid of 96% and thus a 1:1 folate–polymer molar ratio.
The presence of residual free folic acid in the synthesized conjugate was tested by reverse phase high-performance liquid chromatography (RP-HPLC). The system was equipped with a RP-C18 column eluted with 10 mM ammonium acetate buffer pH 6.5 (eluent A) and acetonitrile (eluent B), in a gradient mode from 10 to 40% of eluent B in 40 minutes.4 The UV detector was set to 363 nm. Free folic acid was not detected in the chromatogram confirming the high degree of purity of the conjugate folate–PEG3500-b-(3b)20-b-GMA58.
Potentiometric titration
Block co-polymers (20 mg) were dissolved in 150 mM NaCl (1 mg mL−1) in deionized water. Potentiometric titration was carried out by addition of 10 μL aliquots of NaOH 0.1 M under stirring over a pH range of 3–10. The back titration was started from the pH value reached at the end of the titration by addition of 10 μL aliquots of HCl 0.1 M until pH 3 was achieved. Variations of pH were recorded after each addition.Polymersome preparation
Polymersome formulations were prepared using: (a) polymer 9; (b) different polymer 9–10 mixtures [99:1, 95:5, 90:10 w/w polymer 9–10]; (c) 90:5:5 w/w polymer 9–10–11 mixture following a protocol adapted from prior literature.12
Polymer solutions (1 mg mL−1) in 20 mM phosphate buffer, 150 mM NaCl, at pH 5 were adjusted to pH 7.4 by step-wise addition of 0.1 M NaOH solution. The resultant colloidal dispersions were analysed by DLS to determine the mean size ± standard deviation (SD). ζ-Potential measurements were performed after diluting samples 10-fold in high purity water.
DNA-loaded polymersome preparation
A solution of dsDNA (100 μM) in 10 mM Tris–HCl, 50 mM NaCl, 1 mM EDTA at pH 7.8 was added to the polymer formulation (1 mg mL−1) to obtain 10:1 N/P and 1:1 N/P ratio (27 and 265 μL of dsDNA solution to 1 mL of polymer formulation respectively). Polymersomes were then prepared as described above.
Dynamic light scattering studies
Polymeric vesicles (1 mg mL−1) were prepared using 10 mM PBS, 150 mM NaCl at pH 7.4. Suspensions of polymers at varying pH were obtained by addition of aliquots (5 μL) of 0.1 M NaOH or 0.1 M HCl.Cell culture
KB cells (human cervical carcinoma) were grown at 37 °C, in 5% CO2 atmosphere, using folic acid free DMEM medium supplemented with 10% FBS, 2 mM L-glutamine, 100 IU per mL penicillin, 100 μg per mL streptomycin and 0.25 μg per mL of amphotericin B (Sigma-Aldrich). MCF7 (human breast adenocarcinoma) were grown at 37 °C, in 5% CO2 atmosphere, using RPMI-1640 medium supplemented with 10% FBS, 100 IU per mL penicillin, 100 μg per mL streptomycin and 0.25 μg per mL of amphotericin B.Cell up-take studies
Cells were seeded in a 12 well plate at a density of 5 × 105 cells per well. dsDNA–rhodamine loaded polymer assemblies obtained with 90:5:5 w/w of polymers 9/10/11 and control 90:10 w/w polymers 9/10 (500 μL of 1 mg mL−1) were added to the cells in the wells, and incubated at 37 °C for 2 hours in the dark to prevent photo-bleaching of labelled DNA. A control solution containing PBS (500 μL, 20 mM phosphate, 150 mM NaCl, pH 7.4) was also added to specific wells. After incubation, polymer dispersions were discharged and wells were washed with PBS. Cells were detached by treatment with 1% (w/v) trypsin in PBS. Cell suspensions from each well were transferred in microtubes and recovered by centrifugation at 1000 rpm for 5 minutes. The pellets were treated with 6 μL of 0.1% v/v Triton X-100 and lysates were analysed by fluorimetry (λex 526 nm, λem 555 nm).
Results and discussion
Synthesis of block co-polymers and supramolecular assembly
In this work a novel family of N-alkyl imidazole monomers was designed to produce the pH-responsive domains of the intended drug nanocarriers. Imidazole-containing molecules are common in biology and are known to possess tuneable acid/base behavior. The amino acid histidine, for example, exhibits an extremely wide range of pKa values for its imidazole side-chain, ranging from 2.3 to 9.2, dependent on its specific location and proximity to other residues within proteins.17 In addition, N-alkyl imidazole moieties are present in a number of clinically prescribed drugs, ranging from antifungal lanosterol 14 α-demethylase inhibitors – e.g. ketoconazole, miconazole, and clotrimazole – to nitroimidazole antibiotics such as metronidazole and tinidazole.18 With a pKa in the 6.5–7.5 range N-alkyl imidazoles appeared to be ideal precursors for the synthesis of pH-responsive drug nanocarriers since the monomer unit should, when present in a block co-polymer, alter the aggregation state of polymers across this pH range. In turn this should result in conformational changes of the polymers when passing from systemic circulation (pH 7.4), to more acidic conditions such as those found in hypoxic tumour tissue (pH 6.5–7.0) or endosomal/lysosomal acidic conditions (from 6.5 to 4.5) following cellular uptake.19 Thus the pH response might be transduced from local entrapment of drugs in containers at ambient pH to the controlled release of therapeutically relevant loaded molecules under more acidic conditions.In this study two N-alkyl imidazole monomers were prepared (Scheme 1). Briefly, imidazole was first treated with NaH, the resulting imidazolium Na salt was N-alkylated with α-ω O-acetyl chloroalcohols (1a and b) in DMSO at 100 °C for 3 hours, followed by hydrolysis in 10% aqueous NaOH to give the alcohol intermediates (2a and b). Subsequent treatment with methacroyloyl chloride in dichloromethane in the absence of additional acid scavengers then afforded the required 4-(1H-imidazol-1-yl)butyl methacrylate (3a) and 6-(1H-imidazol-1-yl)hexyl-methacrylate (3b) monomers as hydrochloride salts.
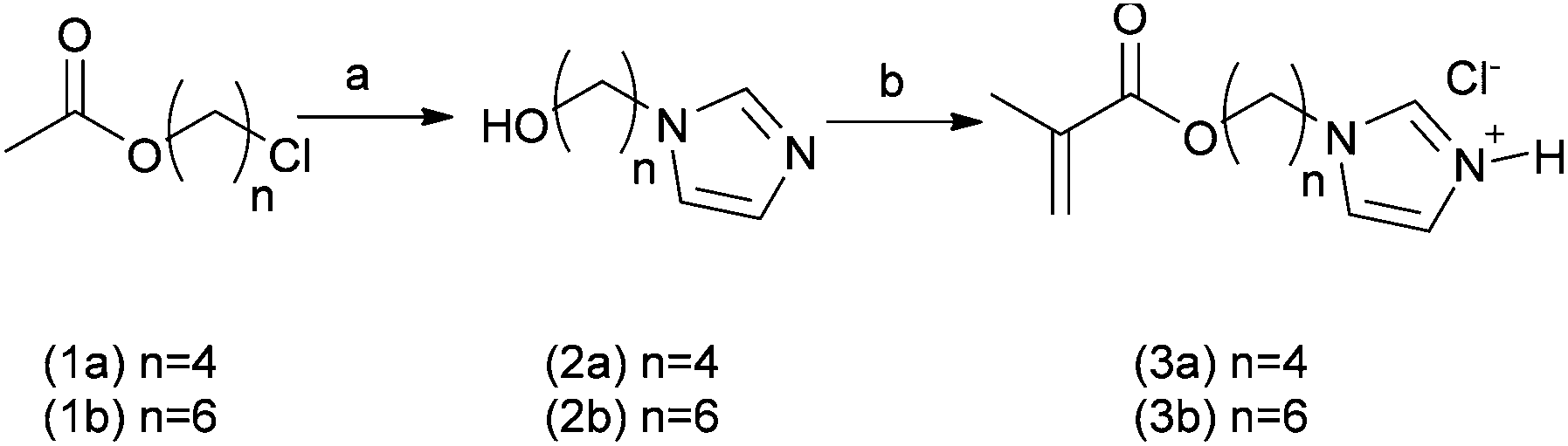 | ||
| Scheme 1 Reagents and conditions: (a) (i) NaH, imidazole, DMSO, 100 °C, 3 h; (ii) NaOHaq 10% w/w, 2 h; (b) methacryloyl chloride, −20 °C to ambient temperature, dichloromethane, 14 h. | ||
The use of standard acid scavengers – Et3N and DIPEA – for the latter step was found to reduce dramatically both yields (<10%) and shelf life of 3a and b monomers. However, as hydrochloride salts, they were found to be stable at −20 °C for several months. Structurally 3a and 3b possess 4 and 6 methylene spacers respectively, connecting the pH-responsive imidazole moiety to the methacrylic unit. An analogous N-alkyl monomer with a shorter C2 linker was also prepared, but was later found to produce very insoluble polymers. Through this methodology, linkers of various length were introduced as a means for modulating both the hydrophobicity and apparent pKa of the pH-responsive block copolymer domains. Since these polymers were designed to undergo self-assembly, the pH sensitive pendant groups were intended to promote hydrophobic interactions among the polymer chains through the “comb-like” structure of the alkyl linkers and guide their dissociation in acidic environments as a consequence of imidazole protonation.
A small library of well-defined PEG-b-p(3a and b) diblock co-polymers was then prepared by RAFT polymerisation of N-alkyl imidazole monomers 3a or 3b in the presence of either methoxyPEG1900 (4) or t-Boc–NH–PEG3500 (5) dithiobenzoates as the macro-transfer agents (Scheme 2).
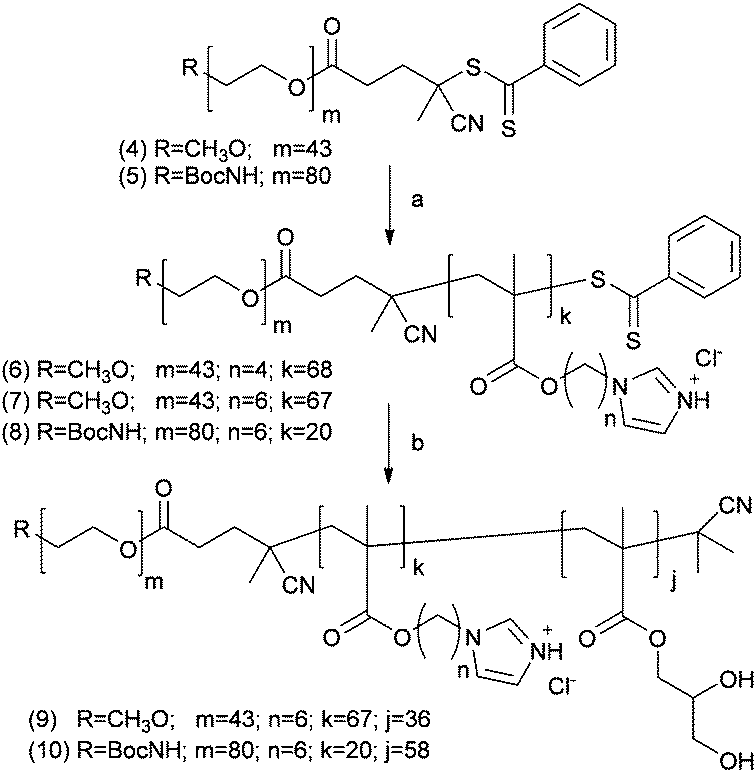 | ||
| Scheme 2 Reagents and conditions: (a) 4-(1H-imidazol-1-yl)butyl methacrylate (3a) or 6-(1H-imidazol-1-yl)hexyl-methacrylate (3b), AIBN, DMF, 65 °C; (b) (i) glycerol methacrylate, AIBN, DMF, 65 °C; (ii) AIBN (20 eq.), DMF, 80 °C. | ||
The t-Boc–NH-terminated PEG was used to obtain a sub-family of block co-polymers with a protected amino functionality at the polymer chain-end as a chemical handle amenable for subsequent functional group modification. Finally, chain extension with glycerol methacrylate (GMA) by RAFT afforded the required ABC amphiphilic triblock PEG-b-p(3b)-b-p(GMA) co-polymers.
The dithiobenzoate CTA end-functionalities were then removed with an excess of AIBN at 80 °C in DMSO for 3 h using the procedure developed by Perrier et al.16 We reasoned that the resultant polymers, with differentiated end-groups as well as A, B, and C blocks alternating in hydrophilic/hydrophobic nature and block length,20 would form stable vesicular assemblies at neutral pH but would disassemble at lower pH. In turn, it was expected that these vesicles could encapsulate drug payloads prior to pH-triggered controlled release. For this purpose, the self-assembly behaviour of mPEG1900-b-(3a)68 (6) and mPEG1900-b-(3b)67 (7) diblock co-polymers differing by only the length of the alkyl chain connecting the imidazole to the polymer backbone was investigated. Through these experiments we aimed to evaluate the effect of the alkyl linker length on the apparent pKa of the polymer, its association tendency and the response to pH alterations. Prior literature indicated that a number of pH-responsive diblock AB co-polymers have displayed switchable polymersome formation and disassembly over the pH ranges reported to be present across the cytosol (pH 7.4) to late endosomes pH (5–6).12,21 We thus adopted analogous methods of polymersome preparation over biologically relevant pH ranges even though the monomers and responsive blocks were different in our case. Specifically, with the intention of generating narrow size distributions of the particles, the self-assembly of polymers into polymersomes was carried out by the ‘pH-switch’ method.21,22 Diblock co-polymer 6 or 7 was dissolved in water at pH 3.0 then the pH was very slowly increased either by addition of NaOHaq under constant stirring or by dialysis against buffered aqueous media at increasing pH. In these experiments polymer 7, with a C6 linear aliphatic spacer linking the imidazole moieties to the polymer backbone, was found to produce aggregates that precipitate rapidly without forming well-defined particles. The related polymer 6, which contained less hydrophobic C4 linkers between the imidazole units and the methacrylate backbone, initially formed organised self-assembled structures of >1000 nm average size, but these proved to be very unstable over time and formed higher-ordered large aggregates (Fig. S3, ESI†).
The self-assembly behaviour of ABC amphiphilic triblock PEG-b-p(3b)-b-p(GMA) co-polymers 9 and 10 as a function of the pH was then investigated. Fig. 2 shows the 1H NMR spectrum of mPEG1900-b-(3b)67-b-(GMA)36 polymer 9, in D2O with NaCl.
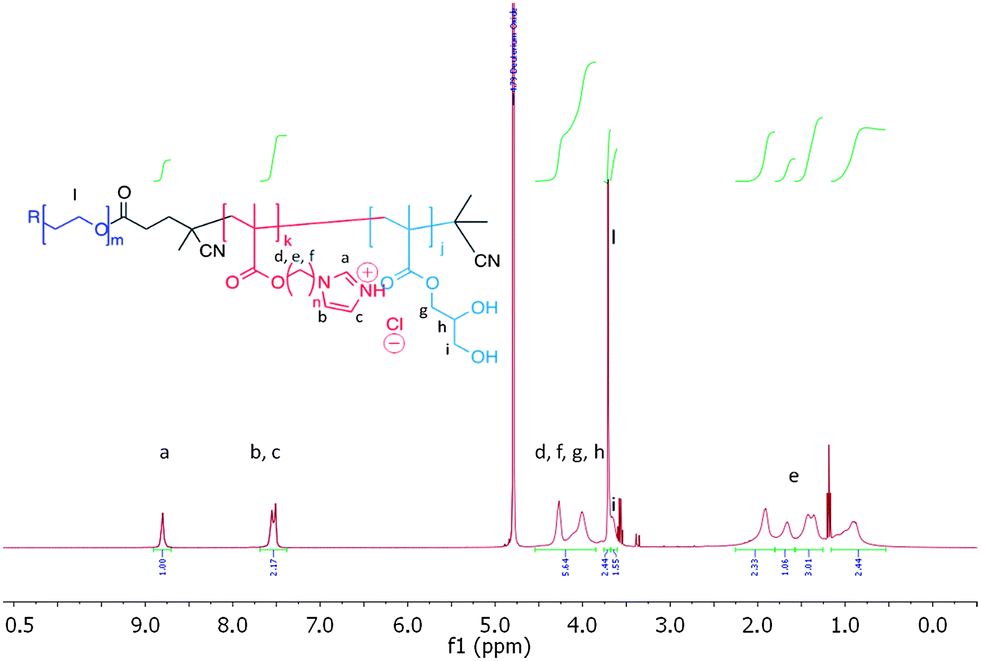 | ||
| Fig. 2 1H NMR spectrum of mPEG1900-b-(3b)67-b-(GMA)36 polymer 9, 1.0 mg mL−1 in D2O containing 150 mM NaCl. Traces of Et2O from polymer precipitation are visible in the spectrum. | ||
Repeating units of 3b were clearly visible, indicating efficient solvation of the p(3b) block under these conditions. However, 1H NMR analysis of 9 at different pH values revealed that, above pH 5.8, the area of the imidazole aromatic signals rapidly decreased, and disappeared completely at pH 7.0. This suggested that polymer 9 assembled into supramolecular structures wherein the p(3b) domain was poorly solvated and therefore not visible in the 1H NMR spectra. This is consistent with a mechanism by which at pH > 5.8 the protonated imidazole units were progressively converted into more hydrophobic free-base imidazole moieties, resulting in self-assembled structures (Fig. 3). Dynamic light-scattering (DLS) of the NMR samples confirmed this hypothesis, showing that by increasing the polymer solution pH from 3 to 5.6 unimeric polymer chains of 9 started to aggregate, generating particles with a maximum size of over 1 μm at pH ∼ 6.5–6.8. The size sharply decreased under more basic conditions, reaching a consistent size of ∼70 nm in size at pH ≥ 8.0. DLS studies with polymer 9 in salt-free conditions followed a similar trend, although the smaller aggregates (∼70 nm) were observed in the 6.5–7.0 range with this polymer rather than at higher pH values. Potentiometric titration of 9 suggested an apparent overall pKa of ∼5.9, although this value should be interpreted with care as self-assembly of polymer chains and association of imadazolic units into the core could potentially make a proportion of the acid/base functionalities inaccessible for titration over the timescale of the experiment. Turbidimetric assays carried out by gradually increasing the pH of a 1.0 mg mL−1 solution of 9 unimer at pH 3.0 in 150 mM NaClaq showed a cloud point at pH 5.7. Thus the pH value at which the polymer aggregated as shown by turbidimetry was in accord with the solution structure changes and conformational switching obtained by 1H NMR and DLS (see ESI†).
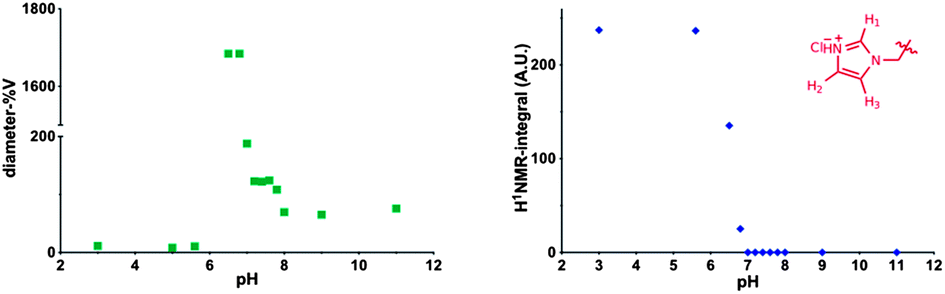 | ||
| Fig. 3 Left DLS analysis of mPEG1900-b-(3b)67-b-(GMA)36 polymer (9), 1 mg mL−1 at various pH values in D2O, 150 mM NaCl. Right integral of H1 proton in the pH-responsive p[6-(1H-imidazol-1-yl)hexyl-methacrylate (3b)] block, expressed in arbitrary units, A.U. Disappearance of the signal at pH > 6.8 is indicative of the formation of incorporation of deprotonated imidazolyl block into a poorly solvated hydrophobic core. | ||
The critical aggregation concentration (CAC) of copolymer 9 estimated by spectrofluorimetry using pyrene as a probe was found to be 21 μg mL−1.
Under the same conditions t-Boc–NH–PEG3500-b-(3b)20-b-(GMA)58 polymer (10), featuring a shorter hydrophobic p(3b) central block, formed smaller aggregates, reaching a size of 13 nm at pH 8.0 (Fig. S3, ESI†).
Encapsulation and release of oligonucleotides
The demonstrated ability of the newly-synthesised block co-polymers to form assemblies reversibly over pH ranges was encouraging for drug encapsulation and release experiments. The functional chain ends of these materials offered an additional advantage. Amphiphilic macromolecules bearing specific ligands at the hydrophilic polymer chain end have been used as selective targeted nanocarriers. For example, Zhang and co-workers engineered Tet1-functionalized PEG-b-PCL polymersomes as drug delivery vehicles to the inner ear by targeting trisialoganglioside clostridial toxin (GT1b) receptors,23 while Upadhay et al. utilised poly(γ-benzyl L-glutamate)-b-hyaluronan polymersomes to achieve enhanced intracellular uptake of doxorubicin in a murine model of Ehrlich Ascites Tumor (EAT) through CD44 receptor-mediated endocytosis.24 However, in certain cases, efficient recognition/endocytic processes do not require high densities of ligands at the surface of drug carriers,25,26 and this can be advantageous where a particular ligand is complex to obtain and/or expensive.26 Furthermore, an excess of ligands can negatively affect the ‘stealth’ properties conferred by hydrophilic polymer chains at the surface of nanoparticles. Ligand- and ligand-free block co-polymers can therefore be combined in a suitable ratio to achieve the desired ligand surface density. Kokkoli and co-workers used this approach to formulate a mixture of unmodified poly(1,2-butadiene)-b-poly(ethylene oxide) with its azido-terminated analogue. The azide functionalities at the surface of the resulting polymersomes were then reacted with PR_b – a ligand for α5β1 integrin targeting – to give targeted nanocarriers that were able to selectively deliver Orai3-specific siRNA to T47D breast cancer cells.27,28Accordingly, for encapsulation and cell uptake experiments we combined polymers 9 and 10 as the bulk and chemically modifiable components of the polymersome bilayer respectively. In a preliminary screening, formulations of 9/10 (1 mg mL−1) with ratios 99:1, 95:5 and 90:10 w/w were found to possess mean diameters of 139 ± 6, 128 ± 4 and 77 ± 2 nm, respectively. Incubation at 37 °C for 5 h showed that the 90:10 w/w formulation was very stable, with virtually no change in particle size over time, whereas the 99:1 and 95:5 w/w samples showed a 2-fold increase in size. Thus the 90:10 w/w formulation was used for subsequent oligonucleotide entrapment and release studies. A short dsDNA 19-mer (see ESI†) was used as a model oligonucleotide, as it was expected to display very similar physico-chemical properties as siRNA but did not require formulation under rigorously RNAse-free conditions. Polymers 9 and 10 at a weight ratio of 9:1 were dissolved in buffer at pH 5, then dsDNA (1:1 N/P) was added and the pH was raised to 7.4 and any unentrapped oligonucleotide was removed by dialysis against PBS pH 7.4. DLS analysis revealed the formation of aggregates with a mean diameter of 230 nm, whilst in the absence of DNA smaller assemblies (60 nm mean diameter) were detected (Fig. 4).
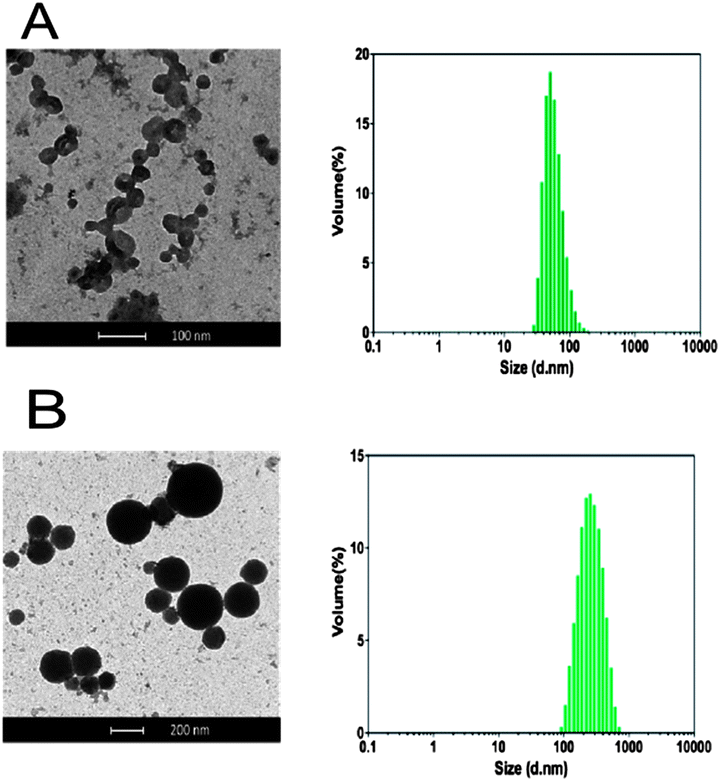 | ||
| Fig. 4 (A) Vesicles obtained from 2 mg per mL of polymers 9/10 9:1 w/w in 20 mM phosphate, 150 mM NaCl at pH 7.4 in the absence, and (B) in the presence of dsDNA (19-mer GAGATGTAAGGCCAGGCCG and its complementary strand), 1:1 N/P, loading capacity = 14% mol/mol. | ||
Additional experiments showed higher incorporation of DNA within polymer nanoparticles when the starting materials were formulated at pH < pKa of the polyimidazole block, when the latter is mostly protonated. However, when the polymer–DNA nanoparticles were brought to pH 7.4, the overall charge on the particles was found to be close to zero (Fig. S11, ESI†), indicating that there was no surface segregation of the polymer cationic blocks or dsDNA, and the stability of the particles in aqueous suspension was therefore attributable to a hydrophilic but non-charged outer polymer corona. After the induced dissociation of the aggregates by decreasing the pH to 5, quantification of entrapped dsDNA showed a loading capacity (LC) of 14% mol/mol (mol dsDNA/mol polymer chains), corresponding to 7% w/w.
These results might suggest that DNA was likely not encapsulated solely in a discrete ‘water pool’ inside a polymersome, but was at least partially complexed with the polymer by electrostatic interactions.
TEM results provided supporting evidence of particle formation and also indicated a clear difference in nanoparticle structure. Dense, almost spherical, objects were observed in the polymer–DNA formulations, whereas hollow-looking vesicles were present in the samples containing polymers alone (Fig. 4). Although TEM analysis itself can neither support nor eliminate the possibility of a polyplex assembly, the observed differences in volume and shape between the two samples may suggest that additional electrostatic interaction may have played a role in forming stable DNA–polymer assemblies.
Release of the 19-mer oligomeric dsDNA from 90:10 w/w polymer 9/10 nanocarrier was then investigated in buffer solutions at 37 °C at pH 7.4 and 5.0,29 to mimic the acid–base conditions found in systemic circulation and in late endosomal/lysosomal intracellular compartments, respectively. As apparent from Fig. 5, release of the dsDNA was markedly pH-dependent, with 85% of the original encapsulated/complexed nucleic acid released at pH 5.0 after 8 h, whilst only 15% of entrapped dsDNA was released by the nanocarriers at pH 7.4 over the same time.
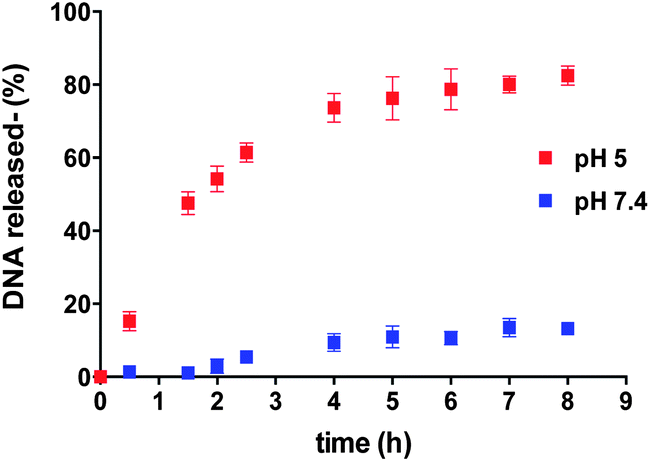 | ||
Fig. 5 Kinetics of release of dsDNA from polymersomes formed by 9:1 w/w mixture of polymers 9 and 10 at pH 5 ( ) and 7.4 ( ) and 7.4 ( ), at 37 °C. ), at 37 °C. | ||
The dsDNA release experiments were an important test of the polymer design criteria, as they implied that the imidazole-containing blocks were at a physico-chemically critical state over the key cytosol–endosome pH range. The fact that dsDNA could be reversibly associated with the polymer implied that not enough imidazole units in the block were protonated for the polymer as a whole to bind strongly to dsDNA at pH 5.0, yet there were nevertheless sufficient numbers of positive charges over the pH-responsive blocks to repel each other at the lower pH and prevent self-association into polymersomes.
Having established the primary utility of the polymers as nucleic acid carriers, the cytocompatibility of the materials and the functionality of the polymer chain ends were explored. Initial experiments showed that polymer 9 was well-tolerated by a range of cell-lines including KB human cervical carcinoma, MCF-7 breast cancer cells and B16-F10 mouse melanoma cell (data not shown). For formulation studies we therefore used polymer 9 with an end-modified version of polymer 10 in ratios such that polymersomes bearing a targeting agent at their surface were obtained.
The chain ends of the t-Boc-protected polymer 10 were thus converted to a ligand-functionalized polymer, α-folate–PEG3500-b-(3b)20-b-(GMA)58 (11) by acid hydrolysis followed by coupling of the resulting terminal primary amine with folic acid N-hydroxysuccinimide activated ester (Scheme 3). 11 was thus used to prepare folate-modified dispersions. Previous stability experiments indicated that a 90:5:5 w/w ratio of polymers 9/10/11 led to the smallest and better defined polymersomes (Fig. S8, ESI†).
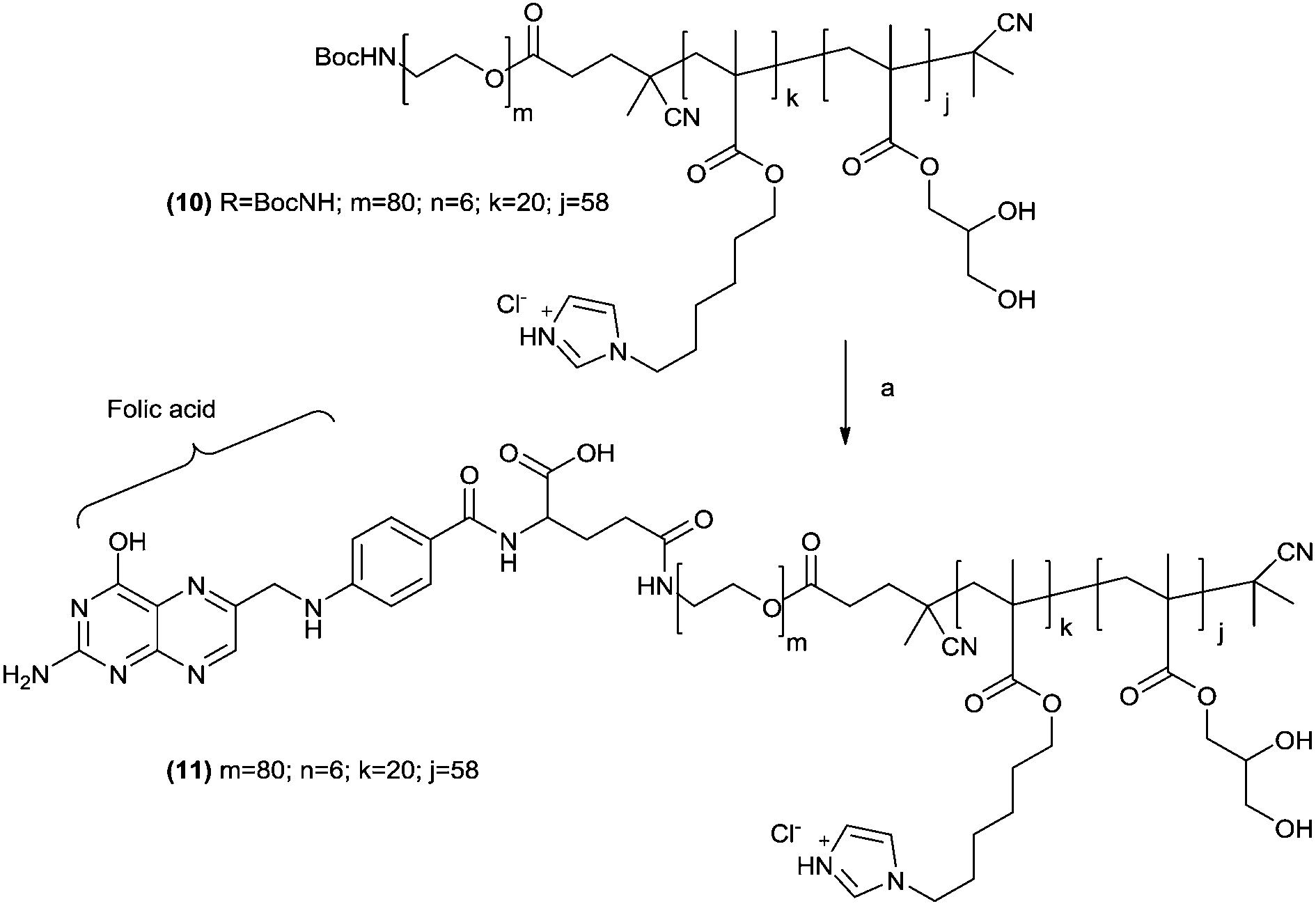 | ||
| Scheme 3
t-Boc removal and folic acid conjugation to polymer 10. Reagents and conditions: (a) (i) TFA–DCM 1:1 v/v, ambient temperature, 2 h; (b) folic acid–NHS ester, DMSO, ambient temperature, 14 h. | ||
Cellular uptake
Polymer formulations with, and without, the folate-terminal functionality were incubated with KB and MCF-7 cells to investigate feasibility for cell delivery by passive or receptor mediated internalization mechanisms.30–32 The former cancer cell line over-expresses the folate receptor, while the latter does not. The polymer nanoparticles for these assays were prepared in the presence of rhodamine-6G labelled dsDNA (with the same sequence as that used in the encapsulation assays) to facilitate cross-comparison. The particle sizes (DLS) for these polymer/dsDNA materials were similar to those obtained in the previous experiments.Quantification of polymer nanoparticles uptake was via fluorescence intensities derived from the rhodamine channel for rhodamine-6G–DNA emission. As apparent from Fig. 6, marked differences in cell association occurred dependent on whether the polymer–DNA particles were ligand-functionalised and if the cells expressed the folate receptor. In general, uptake of the nanoparticles was higher in KB cells compared to MCF-7 cells, and the highest uptake overall was obtained for the polymer formulations containing 5% of the folate-tipped polymer 11. The fact that the polymer nanoparticles were of similar sizes and ζ-potential across the set of folate-tipped and non-folate tipped materials, when formulated with dsDNA, strongly suggests that the uptake pathways were unlikely to have been a function of the nanoparticle geometries. Thus, while we did not conduct specific inhibition studies with free folic acid to saturate any folate receptors on the KB cells, the ability of these folate receptor positive cells to internalise the folate-tipped polymers nevertheless was supportive of a specific uptake pathway for the folate–polymer formulations. These experiments also highlighted the ability of mixed polymer formulations with relatively low ligand densities at their surfaces being able to enter cells via a receptor-mediated process.
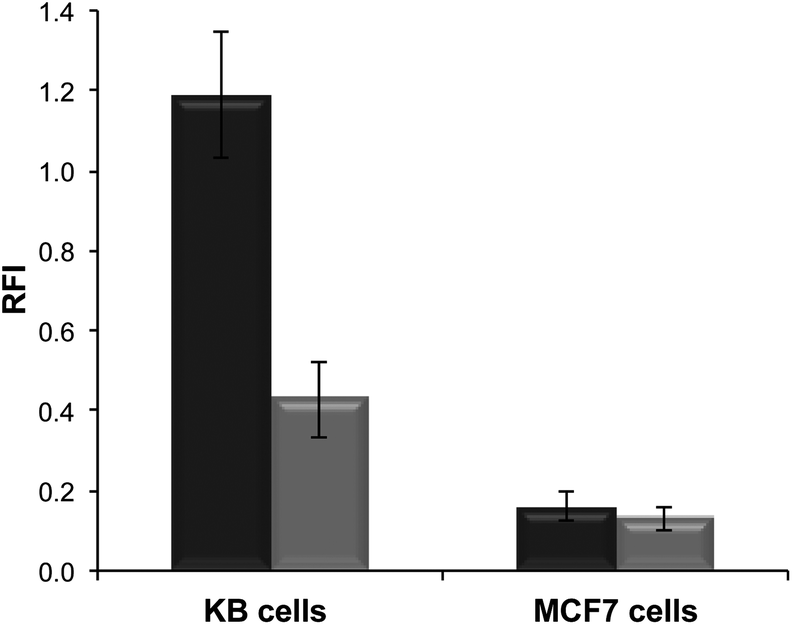 | ||
Fig. 6 Relative Fluorescence Intensity (RFI) of cells incubated with polymersomes loaded with rhodamine 6G-labeled DNA. Targeted nanoparticles were prepared from polymer 9 with 10 w/w polymer 10 ( ) or 5:5 w/w polymer 10 and polymer 11 ( ) or 5:5 w/w polymer 10 and polymer 11 ( ). Cell lines employed for this study were KB cells (human cervical carcinoma), which over-express the folate receptor, and MCF7 cells (human breast adenocarcinoma), which do not over-express the folate receptor. ). Cell lines employed for this study were KB cells (human cervical carcinoma), which over-express the folate receptor, and MCF7 cells (human breast adenocarcinoma), which do not over-express the folate receptor. | ||
When considered in context, the materials we have described exhibit some key properties that are favorable for pharmaceutical applications. The ability to encapsulate biopolymers such as DNA is clearly advantageous for emerging medical technologies, since many new therapeutic entities (proteins, siRNA) are biological in origin and need a delivery system to be used in practice. In addition, the ability to fine-tune polymer structures across physiological pH ranges by mixed responsive monomer block combinations may allow control over tissue and cell localization. We observed very significant size changes for some of the self-assembled polymer structures over pH ranges varying from 5.5–6.5, 6.8–7.0 and 7.2–7.4, and these might in turn result in nanocarrier entrapment and retention in tumours through pH-dependent swelling,33 followed by intracellular uptake and drug release. Recent papers from a number of groups have shown intriguing shapes through polymer responses to pH,34 and particle shape itself is being increasingly investigated as a means to influence biodistribution and targeting.35–37 Finally the facile route to ligand-functionalized formulations by mixing 2 or more co-polymers with different end-groups potentially enables multi-modal targeting, whereby much more specific cell- or sub-cellular organelle delivery might be addressed.
As a corollary, it should be noted that while the imidazole ring on the responsive block components is present in natural molecules (e.g. purine, histamine, histidine) and is therefore considered to be cytocompatible and non-toxic, there are many examples of polymers with positive charges (e.g. polylysine) which can be toxic.38 Furthermore, as noted earlier, imidazole containing antifungal agents include potent inhibitors of the CYTP450 enzymes lanosterol α demethylase (CYP3A4) and Δ-22 desaturase (CYP2C19), leading to blocking of the ergosterol pathway and subsequent membrane destabilization and toxicity.39,40 Breakdown in vivo of the polymers used in this study would likely lead to PEG, polymethacrylic acid and imidazolic residues which may be tolerable for acute therapies but unacceptable for longer-term treatments. Nevertheless, the design rules obtained for responsive materials from this study are useful to inform further studies wherein specific components of each responsive block and/or polymeric amphiphile are re-engineered or synthesised from more pharmacologically acceptable materials.
Conclusions
In conclusion, this study has demonstrated the successful synthesis of a range of new monomers with a variety of physico-chemical features (hydrophobicity, pKa, alkyl linker length) and the preparation of pH-responsive block co-polymers by RAFT techniques. The ability of these polymers to change conformation over specific biologically relevant pH ranges has been shown by NMR and DLS studies and the pH-switching behavior has been used to encapsulate DNA and release the nucleic acid in a triggered fashion. Finally, selected polymers and their formulations with DNA have been shown to be non-toxic, colloidally stable at ambient pH and temperature, and able to transport DNA via a receptor-mediated uptake pathway into specific cell lines. We are currently conducting full biological evaluations of these materials and will report the results in a future manuscript.Acknowledgements
We thank the University of Nottingham, EPSRC (Grants EP/H006915/1 and EP/H005625/1) and EU (NanoSciERA research programme) for funding and Christine Grainger-Boultby for technical assistance.Notes and references
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS.
- J. Sanchis, F. Canal, R. Lucas and M. J. Vicent, Nanomedicine, 2010, 5, 915–935 CrossRef CAS.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- J. P. Magnusson, A. O. Saeed, F. Fernandez-Trillo and C. Alexander, Polym. Chem., 2011, 2, 48–59 RSC.
- R. Federico, A. Cona, P. Caliceti and F. M. Veronese, J. Controlled Release, 2006, 115, 168–174 CrossRef CAS.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- J. A. Mindell, in Annual Review of Physiology, ed. D. Julius and D. E. Clapham, 2012, vol. 74, pp. 69–86 Search PubMed.
- J. W. Wojtkowiak, D. Verduzco, K. J. Schramm and R. J. Gillies, Mol. Pharmaceutics, 2011, 8, 2032–2038 CrossRef CAS.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharmaceutics, 2010, 7, 1913–1920 CrossRef CAS.
- E. S. Lee, H. J. Shin, K. Na and Y. H. Bae, J. Controlled Release, 2003, 90, 363–374 CrossRef CAS.
- J. Gaitzsch, I. Canton, D. Appelhans, G. Battaglia and B. Voit, Biomacromolecules, 2012, 13, 4188–4195 CAS.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS.
- A. Kishimura, S. Liamsuwan, H. Matsuda, W. F. Dong, K. Osada, Y. Yamasaki and K. Kataoka, Soft Matter, 2009, 5, 529–532 RSC.
- W. Agut, A. Brulet, C. Schatz, D. Taton and S. Lecommandoux, Langmuir, 2010, 26, 10546–10554 CrossRef CAS.
- R. Nasanit, P. Iqbal, M. Soliman, N. Spencer, S. Allen, M. C. Davies, S. S. Briggs, L. W. Seymour, J. A. Preece and C. Alexander, Mol. BioSyst., 2008, 4, 741–745 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- M. W. Jones, G. Mantovani, C. A. Blindauer, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 7406–7413 CrossRef CAS.
- L. De Luca, Curr. Med. Chem., 2006, 13, 1–23 CAS.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- A. Blanazs, M. Massignani, G. Battaglia, S. P. Armes and A. J. Ryan, Adv. Funct. Mater., 2009, 19, 2906–2914 CrossRef CAS.
- Y. Zhang, W. Zhang, A. H. Johnston, T. A. Newman, I. Pyykko and J. Zou, Int. J. Nanomed., 2012, 7, 1015–1022 CrossRef CAS.
- K. K. Upadhyay, A. K. Mishra, K. Chuttani, A. Kaul, C. Schatz, J.-F. Le Meins, A. Misra and S. Lecommandoux, Nanomedicine, 2012, 8, 71–80 CrossRef CAS.
- G. Cavallaro, L. Mariano, S. Salmaso, P. Caliceti and G. Gaetano, Int. J. Pharm., 2006, 307, 258–269 CrossRef CAS.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS.
- T. O. Pangburn, K. Georgiou, F. S. Bates and E. Kokkoli, Langmuir, 2012, 28, 12816–12830 CrossRef CAS.
- M. A. Petersen, L. Yin, E. Kokkoli and M. A. Hillmyer, Polym. Chem., 2010, 1, 1281–1290 RSC.
- In the experiment at pH 5.0 the sample was first prepared at pH 7.0 at ambient temperature, then the pH adjusted to the desired value with diluted HCl. Strictly speaking under these conditions at pH 5.0 the sample is not buffered, the pH of the solution was monitored during the experiment and no significant changes were observed.
- G. Khraund, J. Dubois and N. Lavignac, J. Pharm. Pharmacol., 2009, 61, 71 Search PubMed.
- F. P. Seib, A. T. Jones and R. Duncan, J. Drug Targeting, 2006, 14, 375–390 CrossRef CAS.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- P. Vaupel, D. K. Kelleher and O. Thews, Exp. Oncol., 2000, 22, 15–25 Search PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- J.-W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS.
- S. Mitragotri and J. Lahann, Nat. Mater., 2009, 8, 15–23 CrossRef CAS.
- A. C. Hunter and S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 1203–1209 CrossRef CAS.
- H. Matsui, Y. Sakanashi, T. M. Oyama, Y. Oyama, S.-i. Yokota, S. Ishida, Y. Okano, T. B. Oyama and Y. Nishimura, Toxicology, 2008, 248, 142–150 CrossRef CAS.
- L. Di and E. H. Kerns, Drug-like properties: concepts, structure design and methods, Academic Press, Elsevier, London, UK, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Titration and turbidimetry assays, DNA loading and release studies, and TEM analysis. See DOI: 10.1039/c3py00744h |
| This journal is © The Royal Society of Chemistry 2014 |