DOI:
10.1039/C3PY00433C
(Paper)
Polym. Chem., 2013,
4, 4182-4191
Conjugated poly(azomethine)s via simple one-step polycondensation chemistry: synthesis, thermal and optoelectronic properties
Received
3rd April 2013
, Accepted 7th May 2013
First published on 13th May 2013
Abstract
Three conjugated triphenylamine-based poly(azomethine)s were prepared via well-known polycondensation chemistry using cheap and readily available starting materials and the results were contrasted with rrP3HT. Three functionalized diaminetriphenylamines (TPA(X), X = –H, –OMe, –CN) were polymerized in a simple one-step process with 2,3-dihydrothieno[3,4-b][1,4]dioxine-5,7-dicarbaldehyde (ThOx), with water being the only side product. The resulting polymers (TPA(X)ThOx, X = –H, –OMe, –CN) were characterized by GPC, IR and NMR, and show a good thermal stability. The opto-electronic properties could be tuned by changing the functionalization (X = –H, –OMe, –CN) on the triphenylamine moiety. Photovoltaic devices based on TPA(X)ThOx/PCBM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) showed power conversion efficiencies in the range of 0.02–0.04%. TRMC measurements showed that the presence of PCBM as an electron acceptor facilitates the formation of free mobile charges after excitation of the polymer. The low device efficiencies are attributed to a low hole-mobility of the polymer in combination with poor active layer morphology.
:2) showed power conversion efficiencies in the range of 0.02–0.04%. TRMC measurements showed that the presence of PCBM as an electron acceptor facilitates the formation of free mobile charges after excitation of the polymer. The low device efficiencies are attributed to a low hole-mobility of the polymer in combination with poor active layer morphology.
Introduction
In this paper we will present our efforts in exploring azomethine-based condensation polymers for photovoltaic applications. The development of conjugated polymers for use in organic photovoltaics (OPVs) has progressed rapidly in the last few decades. OPVs provide an attractive alternative to inorganic silicon-based solar cells because of their ease of processing and potential low cost. In the last few years, a wide range of (record breaking) small bandgap hole-transporting polymers have been developed, mostly synthesized via Suzuki and Wittig type reactions. These chemistries, however, make use of expensive metal catalysts, require inert reaction conditions, and need substantial product purification to obtain the polymers in the required purity, making large-scale application limited if not altogether impossible. An interesting alternative to these traditional conjugated polymers could come from polycondensation polymers. Poly(azomethine)s, or Schiff bases (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–), have already been applied, to some extent successfully, as hole transporting materials in organic solar cells by this group and others.1–3 The azomethine bond is isoelectronic with its vinyl analogue, however, its synthesis is more advantageous as water is the only by-product and (expensive) catalysts are not required.
C–), have already been applied, to some extent successfully, as hole transporting materials in organic solar cells by this group and others.1–3 The azomethine bond is isoelectronic with its vinyl analogue, however, its synthesis is more advantageous as water is the only by-product and (expensive) catalysts are not required.
Previous work from our group showed the first successful application of a series of conjugated poly(azomethine)s (PAMs) as photovoltaic materials in organic bulk heterojunction solar cells.1 These polymers were based on triphenylamine and (heterocyclic) aromatic dialdehydes. By varying the aromatic dialdehyde, the bandgap of the polymer could be tuned. The absence of alkyl side-chains and the all-aromatic structure of the polymer backbone ensured excellent thermal properties (Tg > 250 °C and T5%d > 400 °C). In addition, conjugated PAMs are known to be stable under ambient humid conditions and are also robust under common reducing reagents such as NaBH3CN, NaBH4, high pressure H2/Pd and even diisobutylaluminium hydride.4 To further explore the photovoltaic potential of PAMs and improve the photovoltaic performance compared to the previously reported series of PAMs, a new series of functionalized triphenylamine based poly(azomethine)s was designed. The TPA unit was functionalized to investigate the influence of substituent effects on the optoelectronic properties of the polymer (Scheme 1). An electron donating methoxy group was introduced in order to push electron density towards the polymer backbone thus reducing its bandgap with respect to the unsubstituted polymer. The introduction of a cyano group is expected to result in an increase in the bandgap compared to the unsubstituted polymer by reducing the electron density in the backbone. A cyclic ether was introduced on the thiophene moiety to further improve the solubility and absorption of the polymers compared to those reported previously.1 To reduce possible geminate recombination at unreacted functional end-groups all polymers were end-capped with mono-functionalized 4-aminotriphenylamine. The three different TPA(X)ThOx oligomers were synthesized with a number average molecular weight (Mn) of 5000 g mol−1. In order to investigate whether polymer molecular weight plays a role with respect to processing and device performance, we prepared TPA(OMe)ThOx with a Mn of 1000 and 9000 g mol−1.
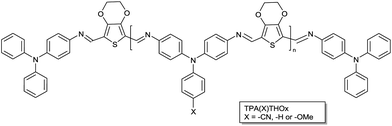 |
| | Scheme 1 Chemical structures of the triphenylamine based poly(azomethine)s (TPA(X)ThOx) with X = –H, –OMe, –CN). | |
Materials and methods
All chemicals were purchased from commercial sources and used as received unless stated otherwise. Lithium chloride and 2,3-dihydrothieno[3,4-b][1,4]dioxine-5,7-dicarbaldehyde were dried in an oven at 150 °C before use. rrP3HT reference material was purchased from Rieke metals (Mn = 50–70k). The structures were confirmed by 1H-NMR (Bruker WM-400, 400 MHz) and 13C-NMR (Bruker WM-400, 100 MHz). All samples were dissolved in deuterated solvents and the recorded spectra were referenced to the solvent (CDCl3: 1H 7.26 and 13C 77.0 ppm) relative to TMS. Mass spectra were recorded with a Shimadzu QP2010S and infrared spectra were obtained with a Perkin Elmer Spectrum 100. UV-vis spectra were collected using a Perkin Elmer Lambda 35 UV-vis spectrometer and photoluminescence spectra were collected by using a PTI Quantamaster spectrofluorimeter.
Electrochemical measurements
Cyclic voltammetry (CV) was measured using a Metrohm Potentiostat (PGSTAT302N) with platinum working and counter electrodes and a Ag/Ag+ reference electrode. Experiments were performed in oxygen-free chloroform solution of the polymer with 0.1 M tetrabutylammonium hexafluorophosphate as an electrolyte and measured at a scan rate of 10 mV s−1.
Molecular weight measurements
Gel permeation chromatography (GPC) measurements were performed on a Shimadzu Prominence GPC system equipped with a Shodex LF-804 column. N-Methylpyrrolidone (NMP) with 5 mM of LiBr was used as eluent at a flow rate of 0.5 mL min−1 at 60 °C. Data analyses were performed with LabSolutions software from the refractive index detector data. Quantification was made based on polystyrene standard calibration.
Thermal and calorimetric analysis
Thermogravimetric analysis (TGA) was performed on a Perkin Elmer Pyris diamond TG/DTA under a nitrogen atmosphere with a scan rate of 10 °C min−1. Differential scanning calorimetry was performed on a Perkin Elmer Sapphire DSC at a heating rate of 10 °C min−1.
Device preparation
Solar cells were prepared by covering pre-cleaned ITO patterned glass substrates with PEDOT:PSS (Clevios P, VP Al4083). The active layer, consisting of TPA(X)ThOx and PCBM (1:2 by weight), was spin-cast from a chloroform solution (15 mg mL−1). A 1 nm thick layer of lithium fluoride (LiF) followed by a 100 nm thick aluminum layer were deposited by thermal evaporation under vacuum. Current–voltage characteristics were recorded in a glovebox using a Keithley 2400 source meter under simulated solar light (1000 W m−2) from a tungsten–halogen lamp filtered by a Schott GG385 UV filter and a Hoya LB120 daylight filter was used to illuminate the devices. The layer thicknesses were measured on a Veeco Dektak 150 profilometer.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed using a FEI Tecnai TF20 electron microscope operated at 200 kV. The films were spin-coated on PEDOT:PSS and transferred to a copper grid.
Time-resolved microwave conductivity
The time-resolved microwave conductivity (TRMC) technique is employed to investigate the formation of mobile charge carriers and their decay with time. By using this technique, the change in conductance of the film on photoexcitation is recorded on a nanosecond time scale without applying external electrodes. The photoactive layers are placed in a microwave sample holder (cavity) with a response time of 18 ns. Samples were photoexcited with a 3 ns laser pulse from an optical parametric oscillator pumped at 355 nm with the third harmonic of a Q-switched Nd:YAG laser (Vibrant II, Opotek). Photogeneration of mobile charge carriers in the sample leads to an increase of the conductance, ΔG(t), and consequently to an enhanced absorption of microwave power by the sample. The time-dependent change of the conductance is obtained from the normalized change in microwave power (ΔP(t)/P) reflected from the cavity according to,| | 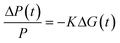 | (1) |
The geometrical dimensions of the cavity and dielectric properties of the media in the microwave cavity determine the sensitivity factor, K. From the maximum change in the conductance (ΔGmax) and the incident light intensity (I0), the parameter φΣμ denoting the product of the charge carrier generation yield (φ) per absorbed photon and the sum of the electron and hole mobilities (Σμ) can be calculated using,
| | 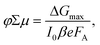 | (2) |
where
β is the ratio between the broad and narrow inner dimensions of the waveguide and
e is the elementary charge.
FA is the optical attenuation value calculated using
eqn (3). For more experimental details regarding
TRMC, see
ref. 5 and 6. The optical attenuation value (
FA) of the photoactive layer corresponds to the fraction of incident
photons that is absorbed by the sample. This was obtained according to,
| | 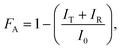 | (3) |
where
IT and
IR are transmission and reflection, respectively, recorded by a Perkin-Elmer Lambda 900 Vis/NIR
spectrophotometer equipped with an integrating sphere.
Synthesis
4,4′-Diamine-4′′-methoxytriphenylamine (2a).
A flame-dried 100 mL round-bottom flask equipped with a magnetic stir bar, a reflux condenser and a nitrogen inlet was charged with 20 mL dry DMSO, K2CO3 (17.89 g, 0.13 mol) and p-anisidine (2.93 g, 0.24 mol). This mixture was stirred under nitrogen for 10 min after which 1-fluoro-4-nitrobenzene (6.8 g, 0.48 mol) was added. The yellow reaction mixture was heated to 150 °C for 48 h under nitrogen. The reaction mixture was cooled to room temperature and precipitated in 200 mL cold water. The solids were collected by filtration, washed with water and dried under vacuum at 45 °C for 5 h. The product was purified by filtering over a short silica column using dichloromethane and recrystallization using glacial acetic acid. Yield: 3.8 g, 45%; TLC (3:1 hexane–ethyl acetate), tR 0.4 (one spot).
4,4′-Dinitro-4′′-methoxytriphenylamine (1a, 2.5 g, 6.9 mmol) was dissolved in THF–EtOH (100%) (50 mL/15 mL) and palladium on carbon (10%, 0.4 g) in EtOH (1 mL) was added to the reaction mixture. After degassing with argon for 15 minutes, the mixture was shaken for 19 h under a 2.4 bar hydrogen atmosphere before filtering over celite and a 0.45 μm filter. Removing the solvent yielded 2.03 g (6.7 mmol, 97%) of the title compound as a white solid. TLC (98% dichloromethane, 1.5% methanol, 0.5% NH4OH) tR 0.5 (one spot); FTIR υmax/cm−1: 3336, 2960, 1626, 1496, 1460, 1328, 1262, 1231, 1030, 824; 1H-NMR (CDCl3, 400 MHz) δ: 6.93 (d, J = 8.8 Hz, 2H); 6.87 (d, J = 8.0 Hz, 4H); 6.76 (d, J = 9.2 Hz, 2H); 6.59 (d, J = 8.4 Hz, 4H); 3.77 (s, 3H); 3.45 (br, 4H); 13C-NMR (CDCl3, 400 MHz) δ: 154.2; 142.5; 141.2; 140.5; 125.2; 123.7; 116.0; 114.3; 55.5; MS m/z (relative intensity): 306.10 (21.4%), 305.10 (100), 290.10 (46.8), 152.60 (27.1).
4,4′-Diamine-4′′-cyanotriphenylamine (2b).
4,4′-Dinitro-4′′-cyanotriphenylamine was obtained by a similar procedure as 4,4′-dinitro-4′′-methoxytriphenylamine, starting from 4-aminobenzonitrile (2.95 g, 25 mmol) and 4-fluoro-1-nitrobenzene (7.76 g, 55 mmol), and resulting in yellow/orange crystals (7.2 g, 80%).
4,4′-Dinitro-4′′-cyanotriphenylamine (1b, 1.0 g, 2.8 mmol) was reduced similarly as described above, resulting in the title compound as a white solid (0.80 g, 96%). TLC (95% dichloromethane, 4.5% methanol, 0.5% NH4OH) tR 0.5 (one spot); 1H-NMR (CDCl3; 400 MHz) δ: 7.31 (d, J = 9.2 Hz, 2H); 6.97 (d, J = 8.4 Hz, 4H); 6.74 (d, J = 8.8 Hz, 2H); 6.65 (d, J = 8.4 Hz, 4H); 3.68 (s br, 4H).
4,4′-Diaminetriphenylamine (2c).
4,4′-Dinitrotriphenylamine was obtained by a similar procedure as 4,4′-dinitro-4′′-methoxytriphenylamine, starting from aniline (10.0 g, 0.11 mol) and 4-fluoro-1-nitrobenzene (30.5 g, 0.22 mol), and resulting in orange crystals (24.18 g, 66%).
4,4′-Dinitrotriphenylamine (1c, 1.0 g, 3.0 mmol) was reduced similarly as described above, resulting in the title compound as a white solid (0.75 g, 91%). TLC (95% dichloromethane, 4.5% methanol, 0.5% NH4OH) tR 0.6 (one spot); 1H-NMR (CDCl3, 400 MHz) δ: 7.17 (t, J = 12 Hz, 2H); 6.97 (d, J = 8 Hz, 4H); 6.93 (d, J = 8 Hz, 2H); 6.85 (t, J = 8 Hz, 1H); 6.65 (d, J = 9 Hz, 4H); 3.53 (br, 4H).
4-Aminotriphenylamine (4).
4-Nitrotriphenylamine was obtained by a similar procedure as 4,4′-dinitro-4′′-methoxytriphenylamine, starting from diphenylamine (76 g, 0.45 mol) and fluoronitrobenzene (70 mL, 0.66 mol), and resulting in orange crystals (75 g, 57%).
4-Nitrotriphenylamine (3.5 g, 12 mmol) was reduced similarly as described above, resulting in the title compound as a white solid (3.0 g, 96%), TLC (97% dichloromethane, 2.5% methanol, 0.5% NH4OH) tR 0.6 (one spot); 1H-NMR (CDCl3, 400 MHz) δ: 7.19 (t, J = 7.8 Hz, 4H); 7.03 (d, J = 8 Hz, 4H); 6.95 (d, J = 8 Hz, 2H); 6.91 (t, J = 8 Hz, 2H); 6.61 (d, J = 9 Hz, 2H); 3.53 (br, 2H).
Representative example: polymerisation of TPA(OMe)ThOx 5K
Lithium chloride (0.5 g) was placed in a dry round-bottom flask under an argon atmosphere. 2,3-Dihydrothieno[3,4-b][1,4]dioxine-5,7-dicarbaldehyde (350 mg, 1.77 mmol) dissolved in dry NMP (7.5 mL) was added to the flask followed by a solution of 4,4′-diamino-4′′-methoxytriphenylamine (2a, 500 mg; 1.64 mmol) in dry NMP (7.5 mL). The reaction mixture turns deep red by adding a catalytic amount of p-toluene sulfonic acid and heating to 150 °C. After 24 h an excess of 4-aminotriphenylamine (4) in dry NMP (5 mL) was added to end-cap the polymer. After 48 h the viscous deep red reaction mixture was allowed to cool to room temperature and precipitated in methanol–water (50:50). The polymer was collected by filtration and washed with methanol–water (50:50).
The 1000 g mol−1 (1K) oligomer was washed with methanol and the 9000 g mol−1 (9K) oligomer was washed with refluxing acetone to remove the low molecular weight components and the unreacted end-cap. The final products were dried in a vacuum oven, resulting in deep red solids. FTIR υmax/cm−1: 3400 br. (Ar–H), 2926 (CH), 1601 (CN), 1493 (CC), 1239 (OMe), 956 (CN); 1H-NMR (CDCl3, 400 MHz) δ ppm: 8.63 (N–CH–); 7.26–6.65 (Ar.); 4.37 (O–CH2–); 3.82 (–OMe).
TPA(H)ThOx
Red polymer. FTIR υmax/cm−1: 3400 broad, 1594, 2926, 1488, 1443, 1269, 1084, 956, 835, 751; 1H-NMR (CDCl3, 400 MHz) δ: 8.64 (N–CH–); 7.27–6.65 (Ar.); 4.37 (O–CH2–).
TPA(CN)ThOx
Yellow polymer. FTIR υmax/cm−1: 3400 broad, 2925, 2214, 1595, 1487, 1442, 1265, 1082, 955, 827; 1H-NMR (CDCl3, 400 MHz) δ: 8.63 (N–CH–); 7.46–6.67 (Ar.); 4.38 (O–CH2–).
Results and discussion
Synthesis
Three different functionalized triphenylamine monomers, 4,4′-diaminotriphenylamine (TPA(–H)), 4,4′-diamino-4′′-methoxytriphenylamine (TPA(–OMe)), and 4,4′-diamino-4′′-cyanotriphenylamine (TPA(–CN)), were synthesized by the condensation of p-fluoronitrobenzene with the appropriate p-substituted aniline followed by subsequent reduction of the obtained dinitrotriphenylamines with hydrogen in the presence of palladium on carbon (Scheme 2). A one-step polycondensation reaction between diaminotriphenylamines (2) and 2,3-dihydrothieno[3,4-b][1,4]dioxine-5,7-dicarbaldehyde (3, ThOx) in the presence of p-toluene sulfonic acid and LiCl yielded poly(azomethine)s, which were subsequently end-capped with 4-aminotriphenylamine (4) to afford the polymers TPA(H)ThOx, TPA(OMe)ThOx, and TPA(CN)ThOx in good overall yields. The Carothers equation was used to obtain a Mn of approximately 5000 g mol−1 for all three polymers. TPA(–OMe)ThOx was also synthesized with a Mn of 1000 and 9000 g mol−1. The structure and composition of the oligomers were confirmed using GPC, 1H-NMR and FTIR. All compounds were analyzed with NMR, IR and mass spectroscopy. The molecular weights were analyzed by gel permeation chromatography (GPC) calibrated with polystyrene standards and are listed in Table 1.
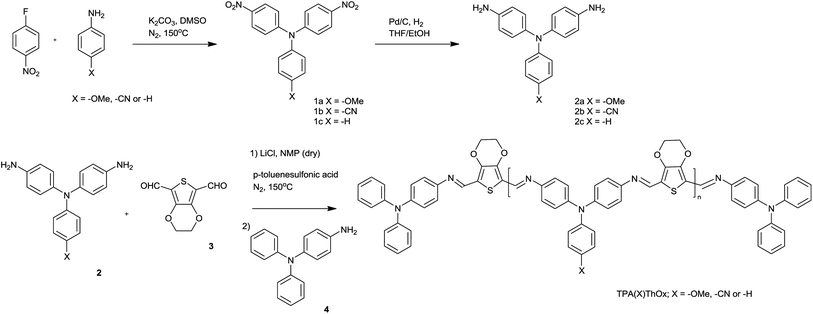 |
| | Scheme 2 Synthesis of the triphenylamine-based poly(azomethine)s (TPA(X)ThOx) with the different substituents on the triphenylamine unit. The polymers were subsequently end-capped with 4-aminotriphenylamine. | |
Table 1 Molecular weight of TPA(X)ThOx poly(azomethine)s determined by GPC
| Polymer |
Target Mn (g mol−1) |
M
n (g mol−1) |
M
w (g mol−1) |
PDI |
|
Could not be determined because of limited solubility.
|
| TPA(OMe)ThOx |
1000 |
1600 |
3400 |
2.1 |
| TPA(OMe)ThOx |
5000 |
5700 |
9700 |
1.7 |
| TPA(OMe)ThOx |
9000 |
—a |
—a |
—a |
| TPA(H)ThOx |
5000 |
5300 |
8400 |
1.6 |
| TPA(CN)ThOx |
5000 |
5500 |
14100 |
2.5 |
Thermal properties
The thermal properties of the 5K polymers were studied by thermogravimetric analysis (TGA) under an inert nitrogen atmosphere with a heating rate of 10 °C min−1 (Fig. 1). TPA(CN)ThOx shows the lowest thermal stability with a T5%d at 235 °C. TPA(OMe)ThOx as well as the unsubstituted TPA(H)ThOx show a higher thermal stability with T5%d at 291 and 321 °C respectively. Although the thermal stabilities of this new series are significantly lower than those of the previously reported poly(azomethine)s, the polymers still show a high thermal stability, compared to traditional OPV polymers. Mass spectrometry indicates that the weight loss below 350 °C can mainly be attributed to the degradation of the cyclic ether functionalized thiophene moiety. Although the substituents on the triphenylamine moieties have a significant influence on the thermal stability of the polymer, the (functionalized) triphenylamine itself is stable up to at least 400 °C.7
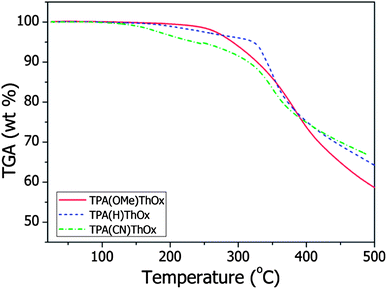 |
| | Fig. 1 TGA thermograms of the 5000 g mol−1 TPA(X)ThOx polyazomethine series recorded under a nitrogen atmosphere with a heating rate of 10 °C min−1. | |
Glass transition temperatures could not be observed with differential scanning calorimetry (DSC). The absence of a glass transition (Tg) is most likely due to the relative low degradation temperatures. The Tg for unsubstituted thiophene–triphenylamine-based azomethines is typically in the range of 250 to 300 °C.1
Optical properties
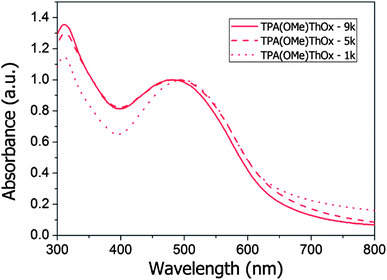
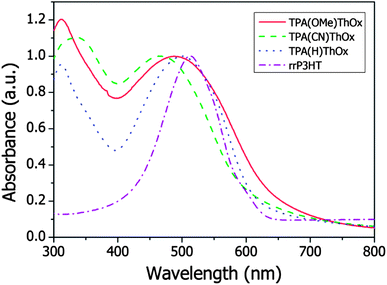
The absorption spectra of the three polymers as well as that of rrP3HT are measured both in solution and in the film and are shown in Fig. 2. The three TPA(X)ThOx poly(azomethine)s all show two clear absorption peaks in the measured absorption spectrum. As expected, functionalization of the TPA moiety with an electron withdrawing group resulted in a blue shift in the maximum of the absorption spectrum of TPA(CN)ThOx compared to that of the unfunctionalized TPA(H)ThOx. The introduction of an electron donating methoxy group on the other hand did not result in the expected red shift, however, it broadens the absorption spectrum compared to that of TPA(H)ThOX. The extinction coefficients of the TPA(X)ThOx poly(azomethine)s were found to be within the same order of magnitude and are comparable to that of rrP3HT. Molecular weight, however, did not significantly influence the absorption spectrum of TPA(OMe)ThOx (Fig. 3), as absorption spectra of polymers are known not to change significantly after approximately five repeating units.
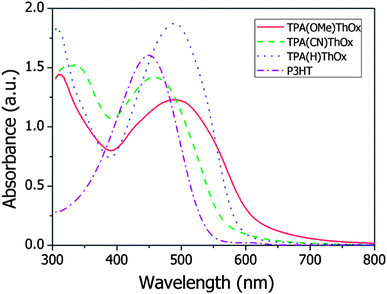 |
| | Fig. 2 UV-vis absorption spectrum of the 5000 g mol−1 TPA(X)ThOx polyazomethine series in solution at a concentration of 30 mg L−1 in pyridine. rrP3HT at a concentration of 30 mg L−1 in chloroform is included for reference purposes. | |
The absorption spectra of the polymers in the solid state showed a red shift of only a few nanometers (<15 nm) compared to those measured in solution (Fig. 4). The bandgap of the polymer was determined by using the onset of the absorption spectrum (Table 2). The bandgap of TPA(OMe)ThOx is found to be 1.9 eV, which is similar to rrP3HT. TPA(H)ThOx and TPA(CN)ThOx show, as expected from the theory, a slightly larger bandgap of 2.0 and 2.1 eV, respectively.
Table 2 Summary of the optoelectronic properties of TPA(X)ThOx
| Polymer |
UV-vis solutiona |
UV-vis filmb |
E
g
/eV |
E
onset/V |
E
max/V |
HOMOd/eV |
LUMOe/eV |
|
λ
max1/nm |
λ
max2/nm |
λ
max1/nm |
λ
max2/nm |
λonset/nm |
|
Measured in a quartz cuvette in chloroform.
Spin-cast on a quartz substrate from pyridine.
Estimated from the onset of the absorption spectrum of the film, 1240/λonset.
Determined by cyclic voltammetry, assuming Fc is 4.8 eV below vacuum.
Estimated by subtracting the band-gaps from the HOMO energy levels.
Electrochemical data from ref. 8.
|
| TPA(OMe)ThOx |
310 |
484 |
313 |
487 |
645 |
1.9 |
0.37 |
0.99 |
−4.8 |
−2.9 |
| TPA(H)ThOx |
318 |
491 |
310 |
500 |
606 |
2.0 |
0.35 |
1.11 |
−4.8 |
−2.8 |
| TPA(CN)ThOx |
324 |
454 |
335 |
467 |
598 |
2.1 |
0.38 |
1.28 |
−4.8 |
−2.7 |
| rrP3HT |
— |
449 |
— |
510 |
640 |
1.9 |
0.4f |
— |
−4.9f |
−3.0f |
Cyclic voltammetry
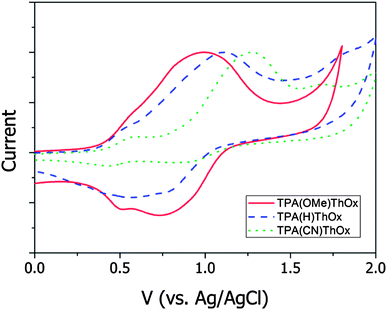
The redox behavior of TPA(X)ThOx poly(azomethine)s was studied by cyclic voltammetry (Fig. 5). A small oxidation wave with an onset of around +0.37 V vs. Ag/Ag+ can be observed in all three voltammograms. This oxidation is assumed to originate from the thiophene part of the polymer, as this oxidation is seen for all three polymers at the same potential and does not arise from the solvent or experimental conditions. The second larger oxidation wave shows a shift correlating with the electronic effect of the substituent on the TPA moiety; the electron donating methoxy group leads to a lower oxidation potential for TPA(OMe)ThOx, while the electron withdrawing cyano group results in a higher oxidation potential for TPA(CN)ThOx compared to that of the unsubstituted TPA(H)ThOx.
TPA(OMe)ThOx and TPA(CN)ThOx, both bearing a substituent at the para-position on the triphenylamine moiety, showed reversible oxidations, whereas TPA(H)ThOx, lacking a substituent at the para-position, showed a more irreversible behaviour (Fig. 6). The second oxidation peak, associated with the TPA moiety, disappeared on repeated cycling, while a new peak with slightly lower oxidation potential appeared, indicating the formation of a new oxidative species most likely formed by cationic radicals on the TPA moieties, which are known to dimerize at the para-position.9–11 The change in intensity of the observed signal during the measurement is attributed to the deposition of material on the electrodes, resulting in an increased signal.
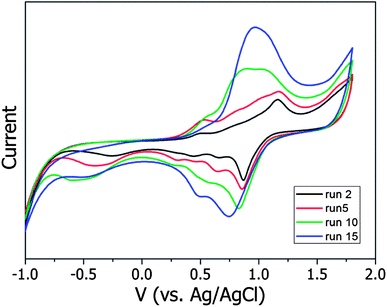 |
| | Fig. 6 Evolution of the cyclic voltammograms over 15 scans of TPA(H)ThOx in chloroform containing 0.1 M TBAPF6 measured with a scan rate of 10 mV s−1. | |
The positions of the HOMO levels of the polymers were calculated from the onsets (+0.37 V, −4.8 eV) of the oxidation relative to Ag/Ag+ and are shown in Table 2. The HOMO level determined for TPA(X)ThOx poly(azomethine)s is very similar to P3HT, which is found at −4.9 eV according to the literature.8 From combining the determined HOMO energy levels with the optical bandgaps, which were calculated from the UV-vis absorption onsets, the energy of the LUMO levels were estimated (Fig. 7).
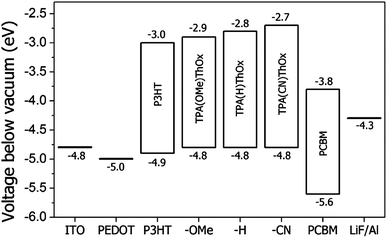 |
| | Fig. 7 Energy diagram of the different TPA(X)ThOx poly(azomethine)s and rrP3HT as a reference material, PCBM and electrodes. | |
Computational study
To gain insight into the electronic structure of the TPA(OMe)ThOx polymer, density functional theory (DFT) geometry optimizations (B3LYP, 6-31G(d,p)) were performed on oligomeric model structures; a tetramer (∼1K oligomer) of TPA(OMe)ThOx (Fig. 8a) and compared with our previously published TPA-thiophene polyazomethine (TPA(H)Th), which lacks both the methoxy group on the TPA unit and the cyclic ether on the thiophene moiety (Fig. 8b). The values for the HOMO energy levels of the polymers are extracted from fitting the calculated HOMO energy of the different length oligomers versus N, the minimum number of double bonds to go from one extremity of the oligomer to another, with an Kuhn-type expression found in ref. 12 The calculated energy difference between both structures resembles the energy difference found with cyclic voltammetry (ΔEexp = 0.5 eV versus ΔEth = 0.32 eV). The addition of the cyclic ether and the methoxy group destabilizes the HOMO of TPA(OMe)ThOx somewhat with respect to that of TPA(H)Th, which appears to be due to the electron donating character of both substituents. Nevertheless, the HOMO appears to be fairly delocalized over a large part of the optimized tetramers, suggesting that both (planar) model compounds are conjugated and the effect of the methoxy group and the cyclic ether is only minimal.
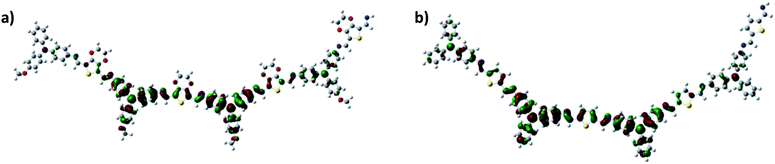 |
| | Fig. 8 DFT-calculated HOMO of the optimized tetramer structures of (a) TPA(OMe)ThOx and (b) TPA(H)Th. | |
Device fabrication
To evaluate the photovoltaic performance of the TPA(X)ThOx oligomer series, bulk heterojunction solar cells were fabricated by spin-coating a blend of polymer:[60]PCBM on ITO coated glass, on which a layer of electron blocking PEDOT:PSS was spin-coated, followed by a thermally evaporated LiF:Al top electrode. Processing conditions were screened in order to optimize the device parameters. The best performing devices were spin-coated from a chloroform solution with a 1:2 TPA(X)ThOx:PCBM ratio, at a concentration of 15 mg mL−1. The relatively low concentration of the spin-coating solutions is a result of the limited solubility of the oligomers. Higher concentrations resulted in residual undissolved material causing defects in the film. A spin speed of 2000 rpm resulted in an active layer thickness of ∼100 nm. Annealing at different temperatures had no significant influence on the device performance. TPA(H)ThOx, without a substituent on the triphenylamine moiety, showed slightly better performance compared to the other two TPA(X)ThOx oligomers. Although devices obtained from the 1K TPA(OMe)ThOx oligomer can perform better than those from the longer chain 5K oligomer, the overall efficiency is still very poor compared to state-of-the-art devices (I–V curve is shown in Fig. 9). We were not able to obtain reproducible devices from the 9K TPA(OMe)ThOx polymer because of the limited solubility of the polymer. Many of the fabricated devices showed leakage currents, which can be explained by defects in the film, most likely caused by the limited solubility of this polymer. The results of the device testing are summarized in Table 3.
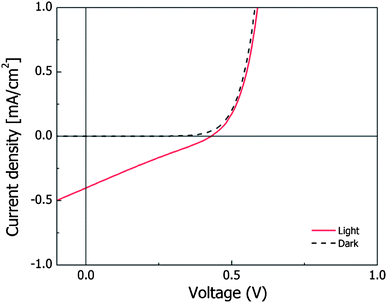 |
| | Fig. 9
I–V curve of 1K TPA(OMe)ThOx in the dark and under illumination. | |
Table 3 Device characteristics of polymer:[60]PCBM devices prepared from a 1:2 chloroform solution under simulated AM1 conditions
| Polymer |
V
oc (V) |
I
sc (mA cm−2) |
FF |
η (%) |
|
No reproducible devices could be obtained because of the limited solubility of the polymer.
|
| TPA(OMe)ThOx, 5K |
0.26 |
0.27 |
0.27 |
0.02 |
| TPA(H)ThOx, 5K |
0.27 |
0.45 |
0.37 |
0.04 |
| TPA(CN)ThOx, 5K |
0.26 |
0.29 |
0.26 |
0.02 |
| TPA(OMe)ThOx, 1K |
0.43 |
0.40 |
0.25 |
0.04 |
| TPA(OMe)ThOx, 9K |
—a |
—a |
—a |
—a |
The 1K TPA(OMe)ThOx polymer was further investigated to gain insight into the origin of the unexpected low performance of these polymers. The methoxy-substituted polymer was selected for it has the lowest bandgap and its synthesis is most convenient for scale up.
Photoluminescence
The photoluminescence (PL) quenching, to determine charge transfer characteristics, of the 1K TPA(OMe)ThOx polymer by [60]PCBM was investigated both in solution and in film. When TPA(OMe)ThOx was excited at its absorption maximum at 480 nm, a photoluminescence peak around 600 nm was observed (Fig. 10). However the photoluminescence efficiency was found to be around two orders of magnitude lower than P3HT, which is considered as a weakly fluorescent material itself. Other studies on conjugated poly(azomethine)s also show weak fluorescence for these types of polymers, indicating fast non-radiative decay channels.13–16 Skene and co-workers showed that suppressed fluorescence of azomethines can be the result of the deactivation of the excited state by internal conversion processes involving bond rotation.15 The fast deactivation of the excited state could be in competition with the charge transfer decreasing the amount of free charges that would be formed.
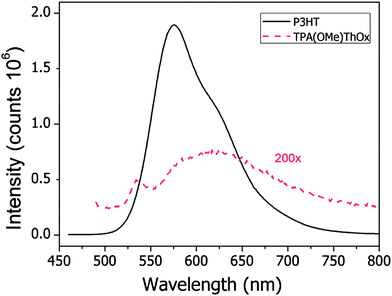 |
| | Fig. 10 Room temperature PL spectra of 1K TPA(OMe)ThOx and P3HT at a concentration of 13 mg L−1 in chloroform. TPA(OMe)ThOx was excited at 480 nm and P3HT at 450 nm. | |
Fluorescence from the film showed a small red-shift of approximately 20 nm compared to that measured in solution, which can be explained by a better ordering of the polymer chains in the solid-state. Nevertheless also in the solid-state, the photoluminescence of TPA(OMe)ThOx is very weak compared to that of P3HT. Due to the low fluorescence efficiency, quenching by [60]PCBM could not be observed, either in solution or in the film.
Time resolved microwave conductivity
In order to study dissociation of excited states into free mobile charges, we record microwave photoconductance transients for the TPA(OMe)ThOx polymer and its blend with the electron acceptor PCBM (Fig. 11). Upon photoexcitation of these thin films with a 3.5 ns laser pulse the transient shows a clear rise followed by a slow decay on a time scale of hundreds of nanoseconds. Due to the 18 ns response time of the microwave cavity the maximum of the photoconductance signal is observed at about 20 ns. It can be clearly observed that the amplitude of the photoconductance for the blend is increased by a factor of 25 as compared to that for the polymer alone. This demonstrates that the excited state dissociates into free mobile charge carriers, due to electron transfer from the polymer to PCBM. From the maximum change in conductance (ΔGmax) the product of the quantum yield per absorbed photon (φ) and the sum of the charge carrier mobilities (Σμ) was calculated using eqn (2). The φΣμ value at lower fluences remains constant, yielding a value of 3 × 10−4 cm2 V−1 s−1. However, at higher fluences a reduction of the φΣμ values is observed that has been previously attributed to higher order recombination processes.17 The φΣμ value observed for this blend is approximately two orders of magnitude smaller compared to that of P3HT:PCBM blends (Fig. 12).18 This difference can be both due to a smaller yield of charge carriers and/or lower mobility.
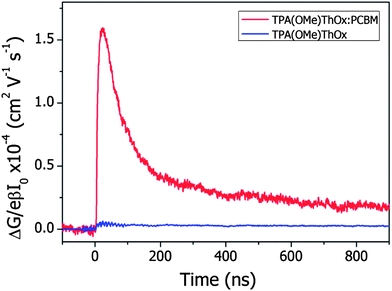 |
| | Fig. 11 Photoconductance transients obtained for the neat TPA(OMe)ThOx polymer and a TPA(OMe)ThOx:PCBM blend. Both thin films were prepared by spin-coating from chloroform. Traces were recorded at a 480 nm excitation wavelength using a fluence of 8.9 × 1014 photons per cm2. | |
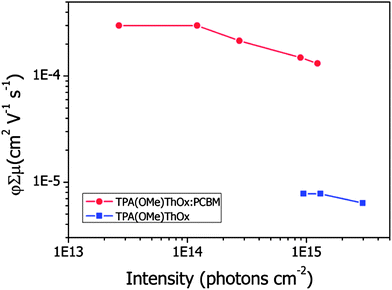 |
| | Fig. 12 Product of the quantum yield per absorbed photon (φ) and the sum of the charge carrier mobilities (Σμ) versus incident intensity normalized to the optical absorption of a TPA(OMe)ThOx polymer and a TPA(OMe)ThOx:PCBM blend. | |
Mobility study
Hole-only devices were prepared for the 1K TPA(OMe)ThOx polymer by evaporating gold, instead of a LiF/Al, top electrode on top of the active layer. The current–voltage characteristics (Fig. 13) can be fit assuming space-charge-limited currents with mobility exhibiting a Poole-Frenkel-type field dependence. The modeled mobilities are very low, for example, with a mobility of approximately 10−8 cm2 V−1 s−1 at a field of 3 × 105 V cm−1. Under working conditions this low hole mobility will almost certainly lead to unbalanced charge transport, explaining for a large part of the poor efficiency obtained with photovoltaic devices comprising this polymer.
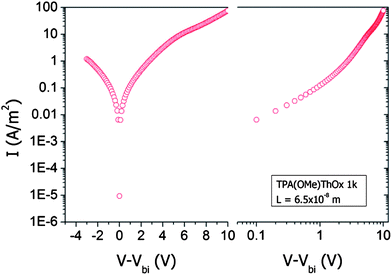 |
| | Fig. 13 Linear–log and log–log plot to determine the field independent mobility of TPA(OMe)ThOx by fitting the equation for space-charge limit current to the I–V characteristic of a hole only device in the high field region. | |
Transmission electron microscopy
The morphology of the active layer was studied by transmission electron microscopy (TEM). A P3HT sample was used for reference purposes and the morphology observed was very similar to what has been published for rrP3HT/PCBM (Fig. 14B). The films from a blend of 1K TPA(OMe)ThOx with PCBM (1:1 and 1:2) are amorphous and very homogeneous, as can be seen in Fig. 14A. No morphological features were visible on the image, indicating a completely homogeneous film. The polymer might dissolve the PCBM, resulting in a homogeneous mixed active layer. It is known from the literature that polymers with aromatic rings and atoms significantly larger than carbon, like sulphur, are good solvents for C60.19 Good mixing of the polymer with PCBM prevents phase separation, which leads to a poor interpenetrating network, which is an explanation for the low mobility as well as the poor device efficiency.
 |
| | Fig. 14 TEM image of (A) TPA(OMe)ThOx 1K:PCBM (ratio 1:2) and (B) rrP3HT:PCBM (1:1). In the rrP3HT sample, features with a length scale of ∼10 to 20 nm can been seen, while the TPA(OMe)ThOx sample shows no morphological features and appears to be completely homogeneous. | |
Conclusion
In conclusion, three new poly(azomethine)s with different substituents on the triphenylamine unit have been synthesized via a simple one-step polycondensation reaction. Although the functionalizations with respect to the previously reported poly(azomethine)s were expected to increase the solubility of these new polymers, we found that their solubility was actually very low. Because of the aromatic units and the absence of functional groups, the polymers showed good thermal stability but no glass transition (Tg). By changing the substituent on the triphenylamine moiety the bandgap of the polymer was influenced. The electronic properties of the polymers were studied by cyclic voltammetry and are found to be similar to those of rrP3HT. Photovoltaic devices were fabricated comprising the three TPA(X)ThOx polymers in combination with [60]PCBM. Power conversion efficiencies of around ∼0.02% were found, with the highest efficiency of 0.04% for the further optimized devices comprising the 1K TPA(OMe)ThOx polymer. The low efficiencies were explained by the low hole mobility of the polymer in combination with a poor morphology of the active layer. In addition, the weak fluorescence of the pristine polymer indicates the fast internal decay of the excited state, which will be in competition with dissociation into free charges. TRMC measurements showed that free mobile charges are produced by photoexcitation, albeit less efficiently than for rrP3HT-based devices.
Acknowledgements
This research forms part of the research programme of the Dutch Polymer Institute (DPI), project #717. The work of D. H. K. M. forms part of the research program of the Dutch Polymer Institute (DPI), project #681. Y. O. is a FNRS Post-doctoral Research Fellow. We would like to acknowledge Prof. René Janssen, Martijn Wienk and Dhritiman Gupta at the TU-Eindhoven for their assistance in preparing the photovoltaic devices.
Notes and references
- J. C. Hindson, B. Ulgut, R. H. Friend, N. C. Greenham, A. Kotlewski and T. J. Dingemans, J. Mater. Chem., 2010, 20, 937 RSC.
- G. D. Sharma, S. G. Sandogaker and M. S. Roy, Thin Solid Films, 1996, 278, 129 CrossRef CAS.
- A. Iwan, M. Palewicz, A. Chuchmala, L. Gorecki, A. Sikora, B. Mazurek and G. Pasciak, Synth. Met., 2012, 162, 143 CrossRef CAS.
- S. A. Pérez Guarìn, M. Bourgeaux, S. Dufresne and W. G. Skene, J. Org. Chem., 2007, 72, 2631 CrossRef.
- J. E. Kroeze, T. J. Savenije, M. J. W. Vermeulen and J. M. Warman, J. Phys. Chem. B, 2003, 107, 7696 CrossRef CAS.
- A. Huijser, T. J. Savenije, J. E. Kroeze and L. D. A. Siebbeles, J. Phys. Chem. B, 2005, 109, 20166 CrossRef CAS.
- H.-Y. Lin and G.-S. Liou, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 285 CrossRef CAS.
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272 CrossRef CAS.
- K. Y. Chiu, T. X. Su, J. H. Li, T. H. Lin, G. S. Liou and S. H. Cheng, J. Electroanal. Chem., 2005, 575, 95 CrossRef CAS.
- A. Ito, H. Ino, K. Tanaka, K. Kanemoto and T. Kato, J. Org. Chem., 2002, 67, 491 CrossRef CAS.
- H. J. Yen and G. S. Liou, Org. Electron., 2010, 11, 299 CrossRef CAS.
- J. Gierschner, J. Cornil and H.-J. Egelhaaf, Adv. Mater., 2007, 19, 173 CrossRef CAS.
- F.-C. Tsai, C.-C. Chang, C.-L. Liu, W.-C. Chen and S. A. Jenekhe, Macromolecules, 2005, 38, 1958 CrossRef CAS.
- M. Bourgeaux and W. G. Skene, Macromolecules, 2007, 40, 1792 CrossRef CAS.
- S. Dufresne and W. G. Skene, J. Phys. Org. Chem., 2012, 25, 211 CrossRef CAS.
- A. J. Ferguson, N. Kopidakis, S. E. Shaheen and G. Rumbles, J. Phys. Chem. C, 2008, 112, 9865 CAS.
- H. Niu, J. Cai, P. Zhao, C. Wang, X. Bai and W. Wang, Dyes Pigm., 2013, 96, 158 CrossRef CAS.
- W. J. Grzegorczyk, T. J. Savenije, T. E. Dykstra, J. Piris, J. M. Schins and L. D. A. Siebbeles, J. Phys. Chem. C, 2010, 114, 5182 CAS.
- C. M. Hansen and A. L. Smith, Carbon, 2004, 42, 1591 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a