 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of photo-isomerisation on host–guest interactions in poly(azocalix[4]arene)s†
Szymon
Wiktorowicz
,
Heikki
Tenhu
and
Vladimir
Aseyev
*
Department of Chemistry, Laboratory of Polymer Chemistry, University of Helsinki, A.I.Virtasen aukio 1, PB 55, FIN-00014, HY, Finland. E-mail: Vladimir.Aseyev@helsinki.fi; Fax: +358-9-19150330; Tel: +358 9 191 50333
First published on 13th March 2013
Abstract
Poly(azocalix[4]arene)s with varying length of lower rim substituents (n-butyl and n-dodecyl) have been evaluated in terms of photo-isomerisation behaviour and its influence on the complex formation with a low molar mass pyridinium-based guest. Irradiation induces slight changes in the cone conformation of the calix[4]arene units. This in turn affects the degree of host–guest interaction, which is decreased when the polymers are in the cis-rich state. The complexation can thus be controlled by adjusting the photostationary state of the poly(azocalix[4]arene)s.
Introduction
Ever since the structure proof of calixarenes was obtained in 1978 by Gutsche1 and in 1979 by Andreetti,2 calixarenes have attracted a considerable amount of attention from the scientific community. Over the course of time and synthetic progress, they have demonstrated unique and interesting properties that have led to a consistent pursuit of new derivatives, with the main focus being placed on the smallest member of the calixarene family – the calix[4]arene. This cyclic tetramer comprising four phenolic rings bridged together by methylene units can undergo a conformational inversion in solution, thus enabling four different conformers3 – cone, partial cone, 1,2-alternate and 1,3-alternate. By choosing an appropriate solvent, temperature and considering the upper rim substituents (para to the hydroxyl group) of the compound, one can induce the persistence of a specific conformation. In 1982, Donald J. Cram4 introduced the concept of restricting the interconversion through modification of the hydroxyl groups (lower rim). This proved to be an unprecedented milestone in the syntheses of a vast array of structures demonstrating unique shape derived properties. From then on, calix[4]arenes have been used as platforms for amphiphilisation,5 photochromic hosts,6 anchoring groups7 and investigated for film forming abilities,8 metal ion recognition,9,10etc.Naturally, the crossover to other areas of chemistry was inevitable and the preparation of polymeric systems containing calixarenes soon followed. Several approaches have been used, but the most common at present is the substitution of the lower rim with initiating groups11 for atom transfer radical polymerization.12,13 Another approach would be to utilize the upper rim for derivatization – the polymers can then e.g. be ‘clicked’ onto terminal alkyne groups in a Huisgen 1,3-dipolar cycloaddition reaction. Calix[4]arenes have also been embedded into the main chain,14,15 or as a side group.16 Interesting block copolymers of tetrathiophene and calixarenes have been recently reported.17 However, the preparation of polymers comprising exclusively of calix[4]arenes, bound through the upper rim, has not been a feasible task, with majority of attempts resulting in the formation of short oligomers.18,19
In the last decade numerous azocalix[4]arene derivatives20 have been synthesized, with their primary usage in detection and extraction of specific metal ions.21 Dimers of azocalix[4]arenes22 have been synthesized with varying bridge lengths, as well as directly linked bisazocalix[4]arenes.23 In the latter case, however, no UV-induced photo-isomerisation of the azo-moiety was observed.
We have recently communicated on the successful syntheses of poly(azocalix[4]arene)s,24 which comprise exclusively of calix[4]arenes, locked in the cone conformation and joined by azo-bridges. These polymers were shown to undergo reversible photo-isomerisation between the trans and the cis form.
In the current study, poly(azocalix[4]arene)s (Fig. 1) with n-butyl and n-dodecyl chains in the lower rim are subjected to irradiation experiments to determine the possible extent of the photo-isomerisation and its effect on conformational properties of the calix[4]arene units. Titration of the butoxy-substituted polymers with pyridinium-based guest reveals a different degree of interaction before and after irradiation, as well as in the case when polymers with bulkier dodecyl chains in the lower rim are used. The unique photo-induced differences in complex formation are then used to visualize a controllable release mechanism of the guests upon irradiation with 365 nm.
![Structure of poly(azocalix[4]arene)s.](/image/article/2013/PY/c3py00194f/c3py00194f-f1.gif) | ||
| Fig. 1 Structure of poly(azocalix[4]arene)s. | ||
Experimental section
Materials and characterization
All chemicals were of reagent grade quality as obtained from commercial sources and were used without further purification. Solvents were freshly distilled and stored over molecular sieves. Anhydrous inhibitor-free tetrahydrofuran (THF) used for UV-Vis and light scattering (LS) measurements was purchased from Sigma-Aldrich. Vitride, (sodium bis(2-methoxyethoxy)aluminum hydride), (Red-Al) was purchased from Sigma Aldrich as a ≥ 65 wt% solution in toluene. Reactions were carried out under nitrogen atmosphere unless stated otherwise. Compounds 1,25 2–426 (ESI, Scheme S1†) were synthesized according to known literature procedures. Detailed syntheses of cone 5,17-dinitrocalix[4]arenes with n-butyl and n-dodecyl chains (Compounds 6, Scheme S1†) as well as general procedures for coupling of 5,17-dinitrocalix[4]arenes to poly(azocalix[4]arene)s were described in an earlier communication.24 Synthesis and irradiation studies of model compounds, MC4, MC12 and MCazo, are presented in the ESI.†Fractionation of poly(azocalix[4]arene)s with aliphatic side chains
Upon quenching and extraction of crude reaction mixtures, the polymeric products were dissolved in tetrahydrofuran (THF c = 25 g L−1) and left stirring overnight to ensure complete solubilization. 2-Propanol was then slowly added to the solution until the cloud point was reached and the suspensions were stirred for 20 hours. The resulting precipitate was collected by filtration and the residual solution was evaporated to dryness. The residue was re-dissolved in THF and 2-propanol was once more added until the cloud point and the procedure was repeated several times until no formation of precipitate could be observed after addition of a significant amount of 2-propanol. In the case of the finer suspensions, centrifugation was used to separate the fractions from the liquid phase. All collected fractions were analyzed with SEC in THF against PS standards.Instrumentation
The NMR spectra were recorded with a 200 MHz Varian Gemini 2000 NMR or with a 500 MHz Bruker Avance III spectrometer using deuterated chloroform (CDCl3) or dimethyl sulfoxide (DMSO) as solvents (polymer concentrations varying depending on the experiment). Tetramethylsilane was the reference in all measurements and chemical shifts are presented in parts per million from the internal TMS standard.For determining the molar mass of the crude products and precipitated polymeric fractions size exclusion chromatography (SEC) was performed using a Waters instrument equipped with Waters Styragel HR6, HR4 and HR2 columns (7.8 × 300 mm each), with Waters 2487 UV (set to 230 nm or 365 nm) and Waters 2410 RI detectors. Tetrahydrofuran with 1% toluene was used as the eluent, with a flow rate of 0.8 mL min−1. Conventional calibrations were performed using poly(styrene) standards (Scientific Polymer Products Inc.).
MALDI-ToF mass spectra were obtained on a Bruker Microflex, which was equipped with 337 nm N2 laser in the reflector mode for determination of the molecular weight of all primary compounds. Sample preparation: 2,5-dihydroxybenzoic acid (DHB) in THF was used as the matrix (20 μL of 40 g L−1 solution), sodium trifluoroacetate in THF (0.5 μL of 10 mg mL−1 solution) as the cationizing agent and the analyte samples were dissolved in THF (5 μL of 10 mg mL−1).
UV-Vis spectra were recorded with a Shimadzu 250 1 PC spectrometer. Irradiation of samples was done using an internal xenon lamp of a Fluoromax-4 Spectrofluorometer from HoribaJobin Yvon (365 nm and 450 nm, exit slit: 3 nm, detection mode blocked) over a varying period of time specific to the conducted experiment. Calibration of the lamp was done prior to each measurement. Solutions were prepared in anhydrous tetrahydrofuran (unless stated otherwise) and concentrations varied depending on the experiment (0.025 mg mL−1 to 20 mg mL−1). When the concentration of irradiated samples exceeded the absorbance measurement capability of the spectrometer, samples were diluted for UV-Vis absorbance determination.
Light scattering measurements were conducted with a Brookhaven Instruments goniometer BIC-200SM, a BIC-TurboCorr digital auto/crosscorrelator and a BIC-CrossCorr detector equipped with two BIC-DS1 detectors. A BIC Mini-L30 diode laser operating at a wavelength of 637 nm and a power of 30 mW was used as a light source. Pseudo-cross-correlation functions of the scattered light intensity were collected at a 90° scattering angle with the self-beating scheme. For each sample, 4–5 correlation functions recorded during 40 min were averaged and then analyzed with an inverse Laplace transform program CONTIN. The temperature of the sample was controlled with a Lauda RC 6C thermostat. All measurements were performed on samples in THF at 20 °C.
Results and discussion
The key factors behind the design of the poly(azocalix[4]arene)s include the locking of the cone conformation of the monomers: 5,17-dinitrocalix[4]arenes with aliphatic chains to ensure the geometrical persistence of the calix[4]arene moiety and bridging of the units with an azo-linkage that is capable of photo-isomerisation. As a result, any significant change in the conformation of the polymeric backbone is governed by the structural transition of the azo-linkage (trans–cis and cis–trans). It is however noteworthy to mention that the calix[4]arene moieties still remain somewhat flexible, however no conformational inversion (cone, partial cone, 1,2-alternate or 1,3-alternate) can occur. The choice of the length of the aliphatic chains in the lower rim was dictated by the urge to study the effect of the substituents on the coupling reaction, as well as photo-isomerisation behaviour.Fractionation of the polymers
In a previous report,24 we have described the optimal conditions for the synthesis of poly(azocalix[4]arene)s. As is the case with all step coupling procedures, the size distribution of the polymeric chains obtained from these reactions was broad. In order to determine any possible differences derived from the chain length, it was necessary to consider numerous ways of extracting polymeric fractions with low polydispersity and different degrees of polymerisation. Our first trials with membrane dialysis in THF or chloroform solutions were unsuccessful due to permeation of the larger molecular weight species through the membrane. Precipitation of the polymers with methanol from their chloroform solutions typically resulted in an uncontrolled fractionation, leading to a mixture of polymer chains with a size distribution pattern comparable to the crude reaction mixtures but without the smallest of the oligomeric species (n < 4; n is the number of calix[4]arene units in the chain). The best results were obtained from careful precipitation of the polymers from their tetrahydrofuran solutions upon addition of 2-propanol. The onset of clouding of the solution was a direct indication of when the addition had to be stopped. By repeating the procedure on the residue after a careful collection of the precipitate, we could isolate fractions of the polymers with polydispersity index between 1.2 (for oligomeric species) and 1.6 (for the highest molecular weight polymers). Typical eluograms for crude and fractionated poly(azocalix[4]arene)s with n-dodecyl chains are presented in Fig. 2a.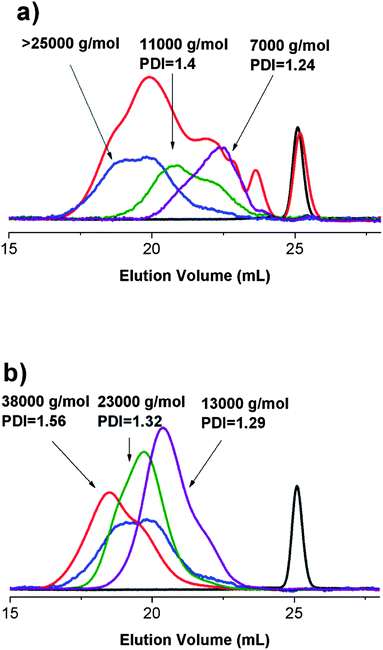 | ||
Fig. 2 (a) Fractions collected from the crude reaction mixture. Monomer, DNCalixC12 ( ), crude reaction, AZ2C12 ( ), crude reaction, AZ2C12 ( ), fraction 1 ( ), fraction 1 ( ), 2 ( ), 2 ( ), 3 ( ), 3 ( ); (b) fractions collected from reprecipitation of fraction 1. Monomer, DNCalixC12 ( ); (b) fractions collected from reprecipitation of fraction 1. Monomer, DNCalixC12 ( ), fraction 1 ( ), fraction 1 ( ), precipitate 1 ( ), precipitate 1 ( ), 2 ( ), 2 ( ), 3 ( ), 3 ( ) from fraction 1. ) from fraction 1. | ||
As is seen from Fig. 2, the largest molecular weight fraction from the crude reaction mixture, fraction 1 (>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) had a broad size distribution. By re-dissolving this fraction and repeating the precipitation protocol we could collect 3 more fractions of a lower polydispersity (Fig. 2b). The same procedure worked well for poly(azocalix[4]arene)s with n-butyl chains (ESI, Fig. S2†) and all collected fractions could be characterized with SEC. For the purpose of this study, we have chosen 6 representative fractions for further characterization, the molecular weight characteristics of which are presented in Table 1.
000 g mol−1) had a broad size distribution. By re-dissolving this fraction and repeating the precipitation protocol we could collect 3 more fractions of a lower polydispersity (Fig. 2b). The same procedure worked well for poly(azocalix[4]arene)s with n-butyl chains (ESI, Fig. S2†) and all collected fractions could be characterized with SEC. For the purpose of this study, we have chosen 6 representative fractions for further characterization, the molecular weight characteristics of which are presented in Table 1.
| Fraction name | Substituent chains | M n [g mol−1] | M w [g mol−1] | PDI | DP (Mn) | DP (Mw) |
|---|---|---|---|---|---|---|
| A13C4 | C4 | 9000 | 12000 |
1.29 | 13 | 18 |
| A13C12 | C12 | 14000 |
20000 |
1.36 | 13 | 18 |
| A19C4 | C4 | 13000 |
15000 |
1.24 | 19 | 22 |
| A25C12 | C12 | 28000 |
40000 |
1.42 | 25 | 36 |
| A37C4 | C4 | 25000 |
39000 |
1.56 | 37 | 57 |
| A35C12 | C12 | 38000 |
59000 |
1.56 | 35 | 54 |
Photo-isomerisation studies
In order to determine the differences in photo-isomerisation of the azo bridges with respect to unbound azobenzene analogues, a detailed study on the behavior of the model compounds, MC4 and MC12, upon irradiation needed to be performed (ESI†). Similar experiments on a series of concentrations were done for the polymer fractions A13C4, A13C12, A37C4 and A35C12 (ESI, Fig. S6–S9†). An optimal concentration was then set for the model compounds, MC4 (0.06 g L−1) and MC12 (0.1 g L−1), and the n-butoxy (c = 0.1 g L−1) and n-dodecyloxy (c = 0.18 g L−1) polymers. This is due to the fact that within the same volume the model compounds contain double the chromophore amount as compared to the calix[4]arene units in the polymer.UV-Vis absorption measurements of the samples (ESI, Fig. S10†) indicate that the polymers exhibit a shift in the position of the absorbance maximum,27 which is located at 365 nm, in comparison to the maximum of the model compounds, which is at 360 nm. This is not surprising, as all meta positions (3,3′ and 5,5′) of the azobenzene incorporated into the polymer are substituted with the methylene bridge of the calix[4]arene. The spectra were then compared by plotting the ratio of the maximum absorption at the given time to the maximum absorption prior to irradiation (Amax(t)/Amax(0)) for all samples (Fig. 3).
![Example of trans-to-cis photo-isomerisation: comparison of the absorption decays for (a) n-butyl substituted model compound, MC4, and poly(azocalix[4]arenes) fractions A13C4 and A37C4 in THF. (b) n-Dodecyl substituted model compound, MC12, and poly(azocalix[4]arenes) fractions A13C12 and A35C12 in THF upon irradiation of the samples with 365 nm light (polymer) and with 360 nm light (model compounds). For model compounds and the polymer cazo = 0.184 mM.](/image/article/2013/PY/c3py00194f/c3py00194f-f3.gif) | ||
| Fig. 3 Example of trans-to-cis photo-isomerisation: comparison of the absorption decays for (a) n-butyl substituted model compound, MC4, and poly(azocalix[4]arenes) fractions A13C4 and A37C4 in THF. (b) n-Dodecyl substituted model compound, MC12, and poly(azocalix[4]arenes) fractions A13C12 and A35C12 in THF upon irradiation of the samples with 365 nm light (polymer) and with 360 nm light (model compounds). For model compounds and the polymer cazo = 0.184 mM. | ||
At first glance it is feasible to deduce that the trans-to-cis photo-isomerisation is significantly slower for the polymers with respect to the reference compounds. The photostationary state (PSS) of the model compounds, MC4 and MC12, is reached after approximately 40 minutes of irradiation, at which the polymers still contain over 35% trans bonds. This state is achieved for the model compounds after only 10 minutes of irradiation. We also observe that the PSS of the model compounds is at a much lower value (0.06), indicating that most of the species are in fact in the cis form. All poly(azocalix[4]arene)s reach the photostationary state in this concentration regime after 80 minutes of irradiation, and the values of the absorption maxima ratios suggest the trans content to be around 20%. Despite elongating the irradiation time, no significant decrease of this value could be observed. This is not surprising, as the chain-like structure of the poly(azocalix[4]arene)s may induce steric restrictions in the photo-isomerisation process.
In a subsequent step, all irradiated samples were subjected to irradiation with 450 nm, to promote reverse cis-to-trans photo-isomerisation (Fig. 4). The model compounds photo-isomerize completely, regaining their absorption maximum value from before irradiation within 30 minutes.
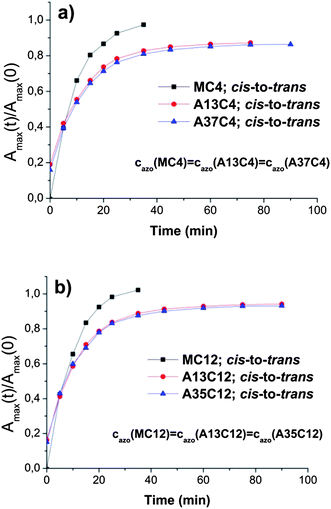 | ||
| Fig. 4 Example of cis-to-trans photo-isomerisation. Change in the absorption maxima ratios (Amax(t)/Amax(0)) with time upon irradiation with 450 nm wavelength. (a) MC4, A13C4 and A37C4 (n-butyl derivatives); (b) MC12, A13C12 and A35C12 (n-dodecyl derivatives). For model compounds and the polymer cazo = 0.184 mM. | ||
The isomerisation in the case of the polymers is somewhat slower and the value of the absorption maximum prior to irradiation is not completely regained within the duration of the experiment (85–95% trans in 90 min). This may suggest that upon isomerisation back to the trans form the polymers still possess areas of distorted conformation (cis), which are less prone to immediate photo-isomerisation (e.g. sterically hindered) and relax back to the preferred trans form during a longer period of time (ESI, Fig. S11†).
A similar experiment was performed to determine how temperature influences the relaxation of the predominantly cis polymers, obtained upon irradiation with 365 nm wavelength, to the trans species. All samples were irradiated to PSS and divided into 2 separate batches, one of which was kept in the dark at 20 °C, and the other was transferred to an oven at 50 °C. Monitoring of the absorption maxima values with time was done for both batches to determine how elevated temperature influences the thermal relaxation of model compounds and the polymers (Fig. 5).
![Change in the UV-Vis absorption maxima upon thermal relaxation for the model compound, MC4 (c = 0.06 g L−1 in THF): after irradiation with 360 nm, maintained at 20 °C () at 50 °C () and without irradiation () and for poly(azocalix[4]arene) fractions A13C4 after irradiation with 365 nm at 20 °C () at 50 °C () and A37C4 at 20 °C (), at 50 °C () (c = 0.1 g L−1 in THF). For model compounds and the polymer cazo = 0.184 mM.](/image/article/2013/PY/c3py00194f/c3py00194f-f5.gif) | ||
Fig. 5 Change in the UV-Vis absorption maxima upon thermal relaxation for the model compound, MC4 (c = 0.06 g L−1 in THF): after irradiation with 360 nm, maintained at 20 °C ( ) at 50 °C ( ) at 50 °C ( ) and without irradiation ( ) and without irradiation ( ) and for poly(azocalix[4]arene) fractions A13C4 after irradiation with 365 nm at 20 °C ( ) and for poly(azocalix[4]arene) fractions A13C4 after irradiation with 365 nm at 20 °C ( ) at 50 °C ( ) at 50 °C ( ) and A37C4 at 20 °C ( ) and A37C4 at 20 °C ( ), at 50 °C ( ), at 50 °C ( ) (c = 0.1 g L−1 in THF). For model compounds and the polymer cazo = 0.184 mM. ) (c = 0.1 g L−1 in THF). For model compounds and the polymer cazo = 0.184 mM. | ||
The model compound, MC4, regains the maximum absorption state prior to irradiation after approximately 200 minutes at 50 °C. When left in the dark at 20 °C, the relaxation occurs at a much lower rate and only 40% of the trans content is achieved in the same time interval. In the case of the polymer fractions, the relaxation process at both temperatures is slower than for the model compound. After 200 minutes at 50 °C, the trans content of the polymer solutions reaches a plateau at 78% (A37C4) and 86% (A13C4) suggesting that the abrupt thermal relaxation of the polymers leads to the occurrence of sterically hindered azo-bonds that cannot participate in the cis-to-trans transition to the same extent as the reference non-linked compounds. Given enough time (1–2 days), however, all poly(azocalix[4]arene) samples regain the pre-irradiation maximum absorption values. Thus we have seen that all the UV-Vis experiments show differences in the isomerisation behaviour between the poly(azocalix[4]arene)s and the model compounds.
Conformation and size
To further understand the transition and how the molecular conformation is affected upon irradiation and the thermal relaxation, 1H NMR spectra were taken in deuterated chloroform and the samples were subsequently irradiated to the PSS. A representative spectrum of the model compound, MC4 (Fig. 6), indicates that the observation of the structural change induced by the UV-absorption at 360 nm is possible.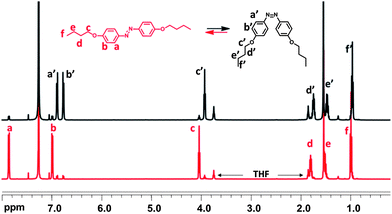 | ||
| Fig. 6 1H NMR spectrum of MC4 in CDCl3 before (red) and after (black) irradiation. | ||
In the trans form, the signals of the protons in the ortho (a) and meta (b) positions to the azo bridge appear at 7.85 and 6.98 ppm while signals from the protons of the n-butyl chain can be seen at 4.04 (c), 1.81 (d), 1.54 (e) and 0.99 (f), respectively. Irradiation of the samples to the PSS induces a structural transition and the cis species are in majority. The signals from the protons in the ortho position (a′) to the azo bridge are shifted upfield to 6.88 ppm, while the meta-position protons (b′) experience a smaller chemical shift upfield to 6.76 ppm. Also a slight change in the signals for the protons of the n-butyl chains can be observed, which now appear at 3.93 (c′), 1.74 (d′), 1.48 (e′) and 0.96 (f′) ppm. In both cases, prior to and after irradiation, trace signals from different isomers can be observed.
Subsequently, analysis of the 1H NMR spectra of the polymers (Fig. 7, fraction A35C12) was done in a similar manner. Before irradiation, the polymers exist in a predominantly trans form, which is supported by the occurrence of an intense peak at 7.92 ppm (a – protons adjacent to the azo-linkage). The remaining protons of the 2 unsubstituted aromatic rings of the calix[4]arene moiety give a signal at 6.24 ppm (b) and the methylene bridge protons (c and c*) appear at 4.56 and 3.36 ppm. The protons of the n-dodecyl chains give signals at 4.20 (d – para in aromatic rings bearing the azo bridge), 3.77 (e – para in aromatic rings without the azo bridge), 1.88 (f), 1.32 (g) and 0.94 (h) ppm. The large upfield shift observed for protons of the unsubstituted aromatic rings (6.24 ppm) would correspond to a pinched cone conformation, in which these groups are in close proximity to each other.28
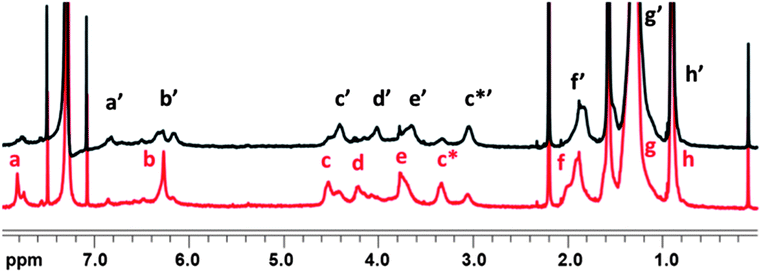 | ||
| Fig. 7 1H NMR spectrum of A35C12 in CDCl3 before (red) and after (black) irradiation. | ||
Upon irradiation, certain differences can be observed in the 1H NMR spectra. Firstly, the sharp peak from the protons (a) adjacent to the azo bridge in the aromatic rings decreases in intensity and a new peak can be observed at 6.92 ppm (a′). This corresponds well to the observed shift upon isomerisation to the cis form of the model compounds. The peak of the other aromatic protons splits upon irradiation into 2 signals, which may suggest a slight change in the conformation of the calix[4]arene units as a result of their flexibility. This is further supported by an upfield shift observed for the methylene bridge protons (c′ and c*′), which now appear at 4.40 and 3.04 ppm (as opposed to 4.56 and 3.36 ppm, c and c*). Thus the separation of the methylene signals changes from 1.20 (Δδc–c*) to 1.36 (Δδc′–c*′) ppm. Also the separation of the OCH2 proton signals changes from 0.35 to 0.43 ppm. The remainder of the signals (f′–h′) for the n-dodecyl chain protons also shows upfield shifts.
Simultaneous decrease in the chemical shift difference for the OCH2 hydrogens and increase in the difference for methylene bridge protons may indicate a flattening of the already pinched cone conformation (C2v symmetry) of the calix[4]arene building block (Fig. 8) upon irradiation to the cis state.
![Graphic depiction of flattening of the pinched cone conformation of calix[4]arene.](/image/article/2013/PY/c3py00194f/c3py00194f-f8.gif) | ||
| Fig. 8 Graphic depiction of flattening of the pinched cone conformation of calix[4]arene. | ||
1H NMR spectra were also measured at varying time intervals (0.5, 1, 12, 24 and 48 h) after irradiation at 365 nm. It can be clearly seen, that given enough time (48 h), all the polymers regain their original structure (ESI, Fig. S13–S16†).
We also wanted to see whether the size of the polymers is affected by the trans-to-cis photo-isomerisation, as was observed for rigid main-chain azopolymers reported by Hecht et al.29 Measurements were done using size exclusion chromatography (ESI, Fig. S18†). Samples of A37C4 and A12C4 in THF, before and after irradiation, were analyzed for differences in elution behavior. Monitoring of the experiment was done with a UV detector set to 365 nm to determine whether the irradiated samples, upon passing through the column still possess a low trans content.
Only minimal changes in the elution volume (0.15 mL for A37C4 and 0.25 mL for A12C4, measured from the peak maximum) could be observed for both, predominantly cis polymeric samples in comparison to the relaxed trans polymers. The large difference in absorption at 365 nm proves that the samples retained their cis conformation upon passing through the column.
Light scattering measurements (ESI, Fig. S19†) were done using a 637 nm laser, to avoid any overlap with absorbance. The size distributions (the mean hydrodynamic diameter, dh = 20 nm) were in accord with the SEC results and showed no significant change in size of macromolecules before and after irradiation. This is not surprising considering the conformational flexibility of the calix[4]arene units and the presence of trans distortive bonds (20%) when the polymers have been irradiated to the cis-rich PSS.
Complex formation
The polymers under investigation comprise of calix[4]arenes in the main chain, which are known cavitands in supramolecular chemistry. When in the cone conformation, calix[4]arenes possess a cyclic array of π-rich aromatic rings capable of interaction with low molar mass guests and cations. In recent years, several studies on the complexation of calix[4]arene derivatives with pyridinium-based guests have been reported.30 The strength of the cation–π interaction can be evaluated through 1H NMR titration of the host compound with the pyridinium-based guest.In our experiments, hexadecylpyridinium chloride, C16Py+, was used as the guest compound for titration of poly(azocalix[4]arene) fractions, trans A19C4 and A25C12 in deuterated chloroform. 1H NMR spectra were recorded upon each addition and pyridinium ring hydrogen signals (Fig. 9; Hα, Hβ, Hγ) were monitored for difference in the chemical shift positions in the presence of the A19C4 polymer fraction. Upon addition of the guest compound, C16Py+, the pyridinium aromatic group signals exhibit a clear difference in the chemical shift, the largest of these being for the 4-position, Hγ (8.28 ppm upon the first addition, in comparison to 8.50 ppm for solution of C16Py+ in the absence of the host). This indicates that an interaction between the host and the guest compound is possible even without the presence of free hydroxyl groups in the lower rim.
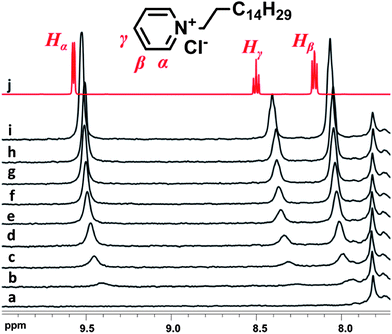 | ||
| Fig. 9 Magnification of the 1H NMR spectra (7.7–10 ppm) of the pyridinium-based guest, C16Py+ upon titration of the host A19C4 in CDCl3: (a) A19C4 before addition, (b–i) subsequent equimolar additions, and (j) C16Py+ in the absence of host. | ||
However, 2D NOESY experiments failed to provide any relevant data suggesting a complex formation in which the guest is spatially fixed with respect to the host. This may be attributed to the short lifetime of the interaction compared to the NMR timescale. Furthermore, NMR diffusion measurements were done to determine whether any difference can be observed upon addition of the guest (DG = 6.8 × 10−10 m2 s−1) to the host (DH = 1.54 × 10−10 m2 s−1). The values obtained from the titration measurements show that the diffusion coefficient of the supposed complex remains unchanged (average of Dcomp = 1.58 × 10−10 m2 s−1). These results led us to believe that the host–guest interaction is a short-lived one and no permanent complex is formed but a dynamic one, in which the pyridinium guest ‘senses’ the presence of the poly(azocalix[4]arene) host.
The data obtained from the 1H NMR measurements do however show the differences when comparing with the results from A19C4 and A25C12 fractions (Fig. 10a). Not surprisingly, in the case of the poly(azocalix[4]arene)s with n-dodecyl chains the difference in the chemical shifts of the guest is smaller than for the polymers with n-butyl chains, suggesting that the interaction is suppressed because of steric reasons. Bulky n-dodecyl chains shield the calix[4]arene units more effectively than the short n-butyl equivalents. Furthermore, we wanted to study whether irradiation of the poly(azocalix[4]arene) solutions would lead to a difference in the complex formation.
![(a) Comparison of the chemical shifts monitored for Hγ of the pyridinium guest in the absence () and presence of A19C4 () and A25C12 () poly(azocalix[4]arene) fractions in CDCl3. (b) Chemical shifts monitored for Hγ of the pyridinium guest in the absence () and presence of the A19C4 poly(azocalix[4]arene) fractions in the trans () and cis form () in CDCl3.](/image/article/2013/PY/c3py00194f/c3py00194f-f10.gif) | ||
Fig. 10 (a) Comparison of the chemical shifts monitored for Hγ of the pyridinium guest in the absence ( ) and presence of A19C4 ( ) and presence of A19C4 ( ) and A25C12 ( ) and A25C12 ( ) poly(azocalix[4]arene) fractions in CDCl3. (b) Chemical shifts monitored for Hγ of the pyridinium guest in the absence ( ) poly(azocalix[4]arene) fractions in CDCl3. (b) Chemical shifts monitored for Hγ of the pyridinium guest in the absence ( ) and presence of the A19C4 poly(azocalix[4]arene) fractions in the trans ( ) and presence of the A19C4 poly(azocalix[4]arene) fractions in the trans ( ) and cis form ( ) and cis form ( ) in CDCl3. ) in CDCl3. | ||
Interestingly, we observed that titration of the trans n-butyl substituted poly(azocalix[4]arene) with the pyridinium guest leads to larger shifts in the 1H NMR spectra (Fig. 10b) than for the irradiated cis counterparts. This may once again indicate that the pinched cone conformation adopted by the calix[4]arene units in the cis form of the polymers is more flattened in comparison to the trans species and hence the calix[4]arene becomes less prone to the interaction with the pyridinium guests.
To provide a reference point for the results of the complex formation experiments with the poly(azocalix[4]arene) hosts, another model compound was prepared (ESI, Fig. S1†). Irradiation of the compound to cis-PSS was done to determine (Fig. S21†) whether change in conformation could influence the complexation behaviour similarly as in the case of the polymers.
Surprisingly, the model compound did not exhibit any dynamic complex formation with the pyridinium-based guest in either the trans (ESI, Fig. S22†) or the cis (ESI, Fig. S23†) forms. This is supported by the lack of change in the 1H NMR chemical shifts of protons of the pyridinium guest upon consecutive additions to a solution of the host. Only increase in overall signal intensity was observed. The absence of interaction suggests that the cavity is inaccessible, which may be a result of aggregation of the free calix[4]arene moieties. This suggests that the host–guest interaction observed in the case of the polymers is a unique feature of the systems under study.
Moreover, a study to realize whether photo-isomerisation of the polymers could be induced in the presence of the pyridinium-based guest was done. The polymer in the preferred trans form was titrated with the guest and the addition was stopped upon reaching the value of 9.20 ppm of the Hα proton signal (Fig. 11). The solutions were subsequently irradiated with 365 nm wavelength to induce the cis form of the polymers. A clear change in the chemical shift for the protons of the pyridinium guest was observed (δHα = 9.31 ppm), indicating that fewer guests interact with the polymeric host. The solutions were then irradiated with 450 nm to promote return of the poly(azocalix[4]arene)s to the trans conformation. A 1H NMR spectrum was recorded prior to completion of the transition to monitor the change in the pyridinium guest signals. An intermediate value of 9.25 ppm was obtained when the polymers contained 70% trans bonds. The value returned to 9.20 ppm when the irradiation process was complete. The same results could be achieved when the cis-to-trans transition was induced thermally.
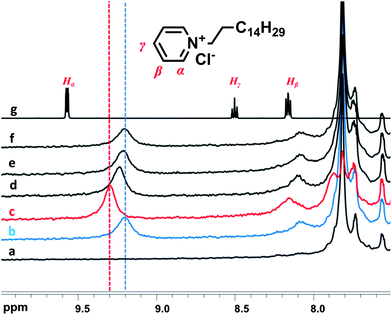 | ||
| Fig. 11 Magnification of the 1H NMR spectra (7.5–10 ppm) of the pyridinium-based guest, C16Py+ (c = 1.0 mmol L−1) upon titration of the host A19C4 (c(calix) = 3.6 mmol L−1) in CDCl3: (a) trans A19C4 before addition, (b) trans A19C4 in the presence of guest, (c) cis A19C4 after irradiation with 365 nm (d) intermediate state A19C4 – 450 nm irradiation (e) after irradiation with 450 nm (f) upon thermal relaxation, (g) C16Py+ in the absence of host. | ||
The obtained results showed that by adjusting the degree of cis–trans-form content of the polymers different extent of dynamic complex formation could be achieved (Fig. 12). This offers an exciting possibility to control the strength of the interaction of the guests with the polymeric host template by means of irradiation and/or thermal relaxation.
![Illustration of difference in conformation and complex formation upon irradiation of poly(azocalix[4]arene)s with specific wavelengths.](/image/article/2013/PY/c3py00194f/c3py00194f-f12.gif) | ||
| Fig. 12 Illustration of difference in conformation and complex formation upon irradiation of poly(azocalix[4]arene)s with specific wavelengths. | ||
Conclusions
Poly(azocalix[4]arene)s with n-butyl and n-dodecyl chains in the lower rim of the calix[4]arene units have been prepared and subsequent careful fractionation of the crude reaction products yielded polymer fractions with moderate polydispersities, two of which were chosen for detailed analysis. Irradiation studies showed that photo-isomerisation of the low molar mass model compounds is faster and more complete than that of the azobenzene moieties in the poly(azocalix[4]arene) backbone. The characteristics of the photostationary state (PSS) observed for all polymer samples upon the trans-to-cis transition clearly indicate that the polymers still contain some trans bonds despite the majorly induced cis form. This suggests that for some units within the polymer chain the transition is sterically hindered. The same is true for the reverse cis-to-trans photo-isomerisation upon irradiation with a 450 nm light source, as well as for the cis-to-trans thermal relaxation. NMR studies indicated structural and conformational changes upon irradiation and the reversibility of the transition upon thermal relaxation. The lack of significant changes in the size of the polymers in the trans and cis form, as observed with SEC and DLS experiments, may be attributed to the fact that the calix[4]arene units are still flexible enough to slightly deviate from the locked cone conformation.Secondly, complexation studies were done using NMR to determine a possible interaction of pyridinium-based guests with the poly(azocalix[4]arene) host. The formation of a dynamic complex between the species could be observed. When comparing the polymers with n-butyl and n-dodecyl side chains, it has been shown that the interaction is suppressed when long sterically hindering chains are present in the lower rim of the polymer. When the n-butyl substituted polymers in the cis form are titrated with the pyridinium guest, the host–guest interaction is decreased, which may be attributed to a less favorable flattened pinched cone conformation of the calix[4]arene units in the chain after irradiation. Interestingly, no interaction with the guest could be observed for a model, low molar mass calix[4]arene compound bearing two diametrically positioned azo-groups in the upper rim.
A subsequent complexation experiment explored the differences in the interaction of the guest with the poly(azocalix[4]arene) host under different irradiation conditions. In the trans state the interaction with the guest is most pronounced. Upon photo-assisted transition to the cis form, the interaction becomes weaker. Upon relaxation back to the trans form, the interaction becomes stronger. The degree of interaction with the guest may thus be controlled by means of irradiation.
The obtained results lead us to believe that combining the properties derived from calix[4]arenes, with photo-switchable bridges and macromolecular architecture, may result in the preparation of numerous interesting polymeric templates for hosting of low molar mass guests with an in-built tunable release mechanism upon irradiation.
Acknowledgements
We thank the Academy of Finland (project numbers 127329 and 260486) for financial support and Dr Sami Hietala for assistance with NMR measurements.Notes and references
- C. D. Gutsche and R. Muthukrishnan, J. Org. Chem., 1978, 43, 4905 CrossRef CAS.
- G. D. Andreetti, R. Ungaro and A. Pochini, J. Chem. Soc., Chem. Commun., 1979, 1005 RSC.
- C. D. Gutsche and L. J. Bauer, J. Am. Chem. Soc., 1985, 107, 6052 CrossRef CAS.
- J. R. Moran, S. Karbach and D. J. Cram, J. Am. Chem. Soc., 1982, 104, 5826 CrossRef CAS.
- E. van Dienst, W. I. Bakker, J. F. I. Engbersen and D. N. Reinhouldt, Pure Appl. Chem., 1993, 65(3), 387 CrossRef CAS.
- S. Bew, R. Brimage, N. L'Hermite and S. Sharma, Org. Lett., 2007, 9(19), 3713 CrossRef CAS.
- T. H. Kim, S. H. Kim, L. V. Tan, Y. Dong, H. Kim and J. Kim, Talanta, 2008, 74, 1654 CrossRef CAS.
- T. Fujimoto, C. Shimizu, O. Hayashida and Y. Aoyama, J. Am. Chem. Soc., 1997, 119, 6676 CrossRef CAS.
- Y. Ishikawa, T. Kunitake, T. Otsuka and S. Shinkai, J. Chem. Soc., Chem. Commun., 1989, 736 RSC.
- A. Casnatti and R. Ungaro, in Calixarenes in Action, L. Mandolini and R. Ungaro, Imperial College Press, London, 2000, p. 62 Search PubMed.
- J. Ueda, M. Kamigaito and M. Sawamoto, Macromolecules, 1998, 31, 6762 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 1998, 31, 7218 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 2000, 33, 7261 CrossRef CAS.
- Y. Yang and T. Swager, Macromolecules, 2006, 39, 2013 CrossRef CAS.
- Y. Yang and T. S. wager, Macromolecules, 2007, 40, 7437 CrossRef CAS.
- P. D. Barata, A. I. Costa, L. F. Ferreira and J. V. Prata, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(22), 5040 CrossRef CAS.
- (a) H.-h. Yu, B. Xu and T. Swager, J. Am. Chem. Soc., 2003, 125, 1142 CrossRef CAS; (b) H.-h. Yu, A. E. Pullen, M. G. Büschel and T. Swager, Angew. Chem., Int. Ed., 2004, 43, 3700 CrossRef CAS.
- H. Kudo, K. Shigematsu, K. Mitani, T. Nishikubo, N. C. Kasuga, H. Uekusa and Y. Ohashi, Macromolecules, 2008, 41, 2030 CrossRef CAS.
- A. Dondoni, C. Ghiglione, A. Mara and M. Scoponi, J. Org. Chem., 1998, 63, 9535 CrossRef CAS.
- (a) F. Oueslati, I. Dumazet-Bonnamour and R. Lamartine, Tetrahedron Lett., 2001, 42, 8177 CrossRef CAS; (b) H. Halouani, I. Dumazet-Bonnamour, C. Duchamp, C. Bavoux, N. Ehlinger, M. Perrin and R. Lamartine, Eur. J. Org. Chem., 2002, 4202 CrossRef CAS; (c) H. Deligöz and N. Ercan, Tetrahedron, 2002, 58, 2881 CrossRef; (d) E.-H. Ryu and Y. Zhao, J. Org. Chem., 2006, 71, 9491 CrossRef CAS.
- (a) Q. Ma, H. Ma, M. Su, Z. Wang, L. Nie and S. Liang, Anal. Chim. Acta, 2001, 439, 73 CrossRef CAS; (b) H. Chawla, S. Singh, S. Sahu and S. Upreti, Tetrahedron, 2006, 62, 7854 CrossRef CAS; (c) L. Lu, S. Zhu, X. Liu, Z. Xie and X. Yan, Anal. Chim. Acta, 2005, 535, 183 CrossRef CAS.
- T. Tilki, İ. Şener, F. Karci, A. Gülce and H. Deligöz, Tetrahedron, 2005, 61, 9624 CrossRef CAS.
- A. Arduini, A. Pochini and A. Secchi, Eur. J. Org. Chem., 2000, 2325 CrossRef CAS.
- S. Wiktorowicz, V. Aseyev and H. Tenhu, Polym. Chem., 2012, 3, 1126 RSC.
- C. D. Gutsche and M. Iqbal, Org. Synth., 1989, 68, 234 Search PubMed.
- J.-O. Dalbavie, J.-B. Regnouf-de-Vains, R. Lamartine, S. Lecocq and M. Perrin, Eur. J. Org. Chem., 2000, 683 CAS.
- C. Ruslim and K. Ichimura, J. Mater. Chem., 2000, 10, 2704 RSC.
- O. Struck, W. Verboom, W. J. J. Smeets, A. L. Spek and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 1, 1997, 2, 223 Search PubMed.
- D. Bléger, T. Liebig, R. Thiermann, M. Maskos, J. P. Rabe and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 12559 CrossRef.
- (a) S. Ishihara and S. Takeoka, Tetrahedron Lett., 2006, 47, 181 CrossRef CAS; (b) F. Ciesa, A. Plech, C. Mattioli, L. Pescatori, A. Arduini, A. Pochini, F. Rossi and A. Secchi, J. Phys. Chem. C, 2010, 114, 13601 CrossRef CAS; (c) L. Pescatori, A. Arduini, A. Pochini, A. Secchi, C. Massera and F. Ugozzoli, Org. Biomol. Chem., 2009, 7, 3698 RSC; (d) P. Pulkkinen, S. Wiktorowicz, V. Aseyev and H. Tenhu, RSC Adv., 2013, 3, 733 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEC eluograms and UV-Vis spectra for other polymeric fractions, normalisation of UV-Vis experiments, 1H NMR data for other photo-isomerisation and complexation experiments, LS measurements, synthesis, photo-isomerisation and complexation data of model compounds for complexation studies. See DOI: 10.1039/c3py00194f |
| This journal is © The Royal Society of Chemistry 2013 |