 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic acids can crosslink poly(ionic liquid)s into mesoporous polyelectrolyte complexes†
Qiang
Zhao
,
Sebastian
Soll
,
Markus
Antonietti
and
Jiayin
Yuan
*
Max-Planck-Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, Am Muehlenberg 1, D-14476 Golm, Germany. E-mail: jiayin.yuan@mpikg.mpg.de; Fax: +49-(0)331-567-9502; Tel: +49-(0)331-567-9552
First published on 18th February 2013
Abstract
A template-free method was introduced to prepare mesoporous poly(ionic liquid) complexes by employing a variety of multivalent carboxylic acids to electrostatically crosslink imidazolium-based poly(ionic liquid)s in diethyl ether. The as-synthesized porous networks exhibit good structural stability and large specific surface area up to 290 m2 g−1. Their performance in dye removal from ethanol was demonstrated to be superior to activated carbon and mesoporous silica.
Polymerized ionic liquids or poly(ionic liquid)s (PILs), which are usually synthesized by polymerization of ionic liquid (IL) monomers, constitute a subclass of polyelectrolytes that combine some IL properties with the common features of polymers.1–7 As such, PILs hold some unique structural merits and are advantageous in a multitude of materials applications, such as gas separation/absorption, carbon preparation, energy conversion, catalysis, and many more.8–19 Porous PILs possess increased surface area and can accelerate the interfacial mass and energy exchange, thus are important in some fields, for instance catalysis or fast stimuli-responsive materials.20–22 In light of this, mesoporous PILs with pore sizes between 2 and 50 nm are promising candidates, as they simultaneously provide adequate mass transfer and large surface area. It has been proven recently that mesoporous PILs absorbed faster and more CO2 than their bulk.23 Very recently, we found that ionic complexation between cationic PILs and deprotonated poly(acrylic acid) could create a micro/mesoporous matrix without using a template, a phenomenon that, to the best of our knowledge, has not been introduced yet in any polyelectrolyte system.24 In general, mesoporous polyelectrolytes with high surface area are less reported,12,25–29 and majorly prepared from a hard templating or post-modification method. Taking into account that the ionic complexation is one of the most fundamental and characteristic properties of polyelectrolyte materials,30–36 the PIL-based ionic complex architectures are of great scientific curiosity. In addition they may bring in new functional materials to the polyelectrolyte family.
In this communication, we prepared mesoporous polyelectrolyte networks through the ionic complexation between imidazolium-based cationic PILs and organic oligoacids in ammonia-containing diethyl ether. Although this approach is differentiated from the previous one simply by replacing poly(carboxylic acid)s with small organic acids, thus simplifying the synthetic procedure, the incorporation of natural acids is a striking benefit, making the synthesis a step forward towards flexible functionalization, following the framework of green chemistry at the same time.37
Scheme 1 illustrates the chemical structures of a PIL poly(3-cyanomethyl-1-vinylimidazolium bis(trifluoro methanesulfonyl)imide) (PCMVImTf2N) and a multivalent acid molecule 1,2,4,5-benzenetetracarboxylic acid (BCA), which were employed as a model pair to prepare the mesoporous PIL-based networks. In detail, PIL and BCA mixed at different molar ratios were dissolved in DMSO solvent, forming a homogeneous blend solution. Under this condition BCA molecules stayed in a neutral state, therefore no or only weak ionic complexation occurred at this stage. The blend solution was added dropwise into diethyl ether containing 0.3 wt% of ammonia under sonication conditions. Upon addition the four COOH groups on BCA molecules were immediately deprotonated by excessive NH3 into carboxylate anions (COO−NH4+), triggering the in situ ionic crosslinking of the cationic PIL. This process was facilitated here by homogeneous pre-mixing of both components in DMSO at a molecular level before the precipitation step. Owing to the multi-charge character, a single BCA molecule can effectively crosslink different PIL chains (at least two), producing insoluble networks that were easily collected as precipitates by centrifugation. This material is termed PIL complexes (PILC) with regard to its electrostatic complexation character.
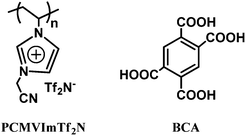 | ||
| Scheme 1 Chemical structures of the PIL polymer PCMVImTf2N and 1,2,4,5-benzenetetracarboxylic acid (BCA). | ||
The ionic complexation and the morphology of the as-synthesized PILC material made from PCMVImTf2N and BCA were first studied by Fourier transform infrared (FT-IR) spectroscopy and scanning electron microscopy (SEM). In the FT-IR spectra of intact BCA and the PILC in Fig. 1a, the marked absorption peaks at 1700 and 1550 cm−1 were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in COOH and COO− groups, respectively. The significant shrinkage of the COOH band and the appearance of an intense COO− band in the PILC product confirmed the deprotonation state of the organic acids.24 SEM examination (Fig. 1b) visualized that the PILC networks were composed of nanosized particles (25–50 nm) with a distinctive morphology. The secondary structure, the interlinked random nanoparticle packing might provide large transport channels favourable for many practical applications.
O stretching in COOH and COO− groups, respectively. The significant shrinkage of the COOH band and the appearance of an intense COO− band in the PILC product confirmed the deprotonation state of the organic acids.24 SEM examination (Fig. 1b) visualized that the PILC networks were composed of nanosized particles (25–50 nm) with a distinctive morphology. The secondary structure, the interlinked random nanoparticle packing might provide large transport channels favourable for many practical applications.
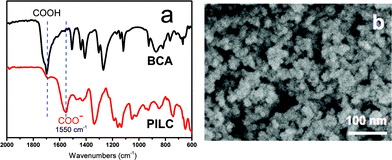 | ||
| Fig. 1 (a and b) FT-IR and SEM characterization of the PILC product. | ||
The pore characteristics of the PILC product were investigated by nitrogen sorption measurements (Fig. 2a). It presents a type-IV isotherm with a hysteresis loop in a P/P0 range between 0.7 and 0.9, suggesting that in addition to the features visible in SEM mesopores of 3–10 nm in diameter are the major species (Fig. S2†).38–40 The specific surface area calculated from the Brunauer–Emmett–Teller (BET) equation and the pore volume calculated from the Barrett–Joyner–Halenda (BJH) method were 250 m2 g−1 and 0.63 cm3 g−1, respectively.23,40 For such systems these values are in an unusually high range. This illustrates the big difference between our system and the classical polyelectrolyte complexes, which are essentially non-porous as such. Thanks to the strong ionic complexation between PIL and BCA, and appropriate polarizability of the PIL part, these mesopores are mechanically and physicochemically robust and change little under refluxing with some organic solvents, like acetone, THF or ethanol (Fig. S3†). In contrast, the simple physical blend of PIL and BCA is nonporous (Fig. 2a). In this context the pore structure seems to be related to the ionic complexation structures. Indeed, a positive correlation between the specific surface area of PILCs and the degree of ionic complexation (Scheme S2†) was observed in the PILC products (Fig. 2b). Varying the BCA/PIL (represented by COOH/imidazolium) feeding ratio, the complexation and the surface characteristics peak at a COOH/imidazolium molar ratio of 0.85. This value hints that a complete ionic complexation between the imidazolium cations in PILs and the 4 carboxylate anions in BCA for all molecules is sterically impossible (otherwise COOH/imidazolium = 1). The average coordination number is calculated to be 3.4. This is reasonable, because the imidazolium–carboxylate charge pair formed at an earlier stage might introduce spatial hindrance which prevents the later coming BCA molecules from closing their coordination shell along the same polymer chain.
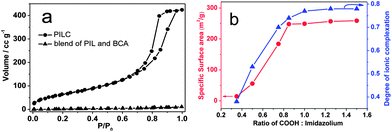 | ||
| Fig. 2 (a) Nitrogen sorption isotherms of the PILC and the PCMVImTf2N–BCA physical blend. (b) Plot of the specific surface area (red) and the degree of ionic complexation (blue) of the PILC products against various COOH/imidazolium molar ratios undertaken in different complexation experiment runs. | ||
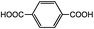
Compared to the common interpolyelectrolyte complex systems,30–34 introducing multivalent small molecules into the ionic complexation might afford some advantages such as cheap resources, defined geometries, an abundance of candidate choices, and a broad range of added orthogonal chemical functionalities, opening a new avenue to a rich family of mesoporous PILC materials. To demonstrate this potential, several more PILC networks were similarly prepared from PCMVImTf2N with different acids. Table 1 summarizes the chemical structure of the acids and the pore properties of the corresponding PILC products. Generally, PILC materials based on the benzene series (entry 1–4, Table 1), citric acid, trans-aconitic acid, and even oxalic acid (entry 5–7, Table 1) are all porous rigid solids with specific surface area values ranging from 230 to 290 m2 g−1. The relatively large variation in the pore volume (0.63 cm3 g−1 to 1.54 cm3 g−1) can be attributed to the different chemical structures and charge densities of the acid molecules, and the corresponding varied pore sizes of these PILC systems. In contrast, the monovalent benzoic acid (entry 8, Table 1) produced only a nonporous soft sticky material in a low yield.
| Entry | Acids | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|---|
| 1 |
|
263 | 1.54 |
| 2 |

|
277 | 0.77 |
| 3 |
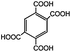
|
249 | 0.63 |
| 4 |
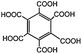
|
233 | 0.83 |
| 5 |
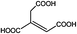
|
255 | 1.23 |
| 6 |

|
233 | 1.11 |
| 7 |
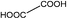
|
290 | 1.35 |
| 8 |
|
<10 | 0.01 |
As stated before, polyelectrolyte complex systems have been widely investigated so far and have exclusively low surface area and mesoporosity, if not prepared from a template method.30–34,41,42 A comparison of our system with previous ones highlights two unique features: (1) premixing of the two components at a molecular level prior to carrying out complexation, and (2) the neutralization-triggered in situ ionic crosslinking in a poor solvent. Under these conditions, a fast and efficient ionic complexation (min. diffusion for both components) takes place, fixing the non-equilibrium (“frozen state” due to the insolubility) polymer chain conformation into a porous state.24,43,44 A supportive proof is that the same PILCs (entry 1–7 in Table 1) were nonporous when prepared by dropping their mixture solution into water, ethanol, acetone or tetrahydrofuran, simply because these solvents can dissolve or heavily solubilize the PILs or the organic acids.
Being highly dipolar, polarizable, and in a mesoporous state, the PILC materials are expected to find potential applications in heterogeneous catalysis, absorption, separation, purification, etc.45,46 This can be illustrated here by using PILCs as absorbents for the efficient removal of organic dye molecules from organic solvents (note that this is a more difficult problem as compared to the removal of dyes from water). Such a function is frequently required for organic solvents recycling. Currently, approaches such as solvent resistant nanofiltration (SRNF) are investigated for this purpose because the traditional means like thermal distillation is too energy-demanding.47,48Fig. 3 shows the performance of the PILC material (SBET ∼ 250 m2 g−1, entry 3 in Table 1), activated carbon (SBET ∼ 800 m2 g−1) and mesoporous silica (SBET ∼ 400 m2 g−1) in absorbing methyl orange (MO) under a default set of conditions from ethanol. When the mesoporous PILC was used, the UV absorption intensity of MO molecules decreased by 90% and 95% after 5 and 60 minutes, respectively. In contrast, under the same conditions after 5 minutes only 70% decrease in the absorption intensity was observed by activated carbon and only 2% by mesoporous silica. We also tested anion exchange resins (Fig. S4†), and the absorbing process is much slower compared to the PILC. Hence, PILC's superior efficiency in capturing dye molecules seems to stem from a judicious combination of its polarity, mesopores, charge character as well as its fine dispersibility in ethanol. All of these enable a shortened diffusion distance and a strong interaction with MO molecules.
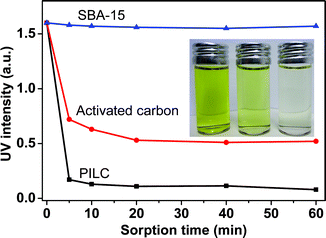 | ||
| Fig. 3 UV absorption (λ ∼ 435 nm) of methyl orange solution (0.02 M in ethanol) after being treated with 10 mg of PILC (black line), activated carbon (red line), and SBA-15 (blue line), respectively. The inset is a photograph of the methyl orange solution after 1 h treatment with SBA-15, activated carbon, and PILC from left to right. | ||
In conclusion, mesoporous poly(ionic liquid) complex networks with specific surface area up to 290 m2 g−1, large pore volume, and stable pore structures were prepared by employing electrostatic interaction between an imidazolium-based PIL and multivalent organic acids. This unique template-free strategy is exceptionally simple in operation and was shown to be generally valid for a variety of other multivalent carboxylic acids, including many natural acid molecules. As an applicative example, the mesoporous PILC products were tested and showed an enhanced dye absorption performance in ethanol, making them promising candidates for impurity removal even from organic solvents.
Acknowledgements
The authors thank the Max Planck Society for financial support. J.Y. enjoyed helpful discussions with Dr Jens Weber, and thanked Dr Christoph Wieland for the GPC measurements.Notes and references
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339–360 CrossRef CAS.
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- X. Sui, M. A. Hempenius and G. J. Vancso, J. Am. Chem. Soc., 2012, 134, 4023–4025 CrossRef CAS.
- Y. Xiong, J. Liu, Y. Wang, H. Wang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS.
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS.
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, Macromol. Rapid Commun., 2012, 33, 1149–1153 CrossRef CAS.
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2011, 5, 436–442 CrossRef CAS.
- F. Liu, L. Wang, Q. Sun, L. Zhu, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS.
- M. Fevre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J.-M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776–6784 CrossRef CAS.
- J. Yuan, S. Soll, M. Drechsler, A. H. E. Mueller and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 17556–17559 CrossRef CAS.
- G. A. Becht, M. Sofos, S. Seifert and M. A. Firestone, Macromolecules, 2011, 44, 1421–1428 CrossRef CAS.
- Y. Ye, J.-H. Choi, K. I. Winey and Y. A. Elabd, Macromolecules, 2012, 45, 7027–7035 CrossRef CAS.
- M. D. Green, J.-H. Choi, K. I. Winey and T. E. Long, Macromolecules, 2012, 45, 4749–4757 CrossRef CAS.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu, Y. Zhang and F. Yan, J. Mater. Chem., 2012, 22, 18018–18024 RSC.
- F. Schuler, B. Kerscher, F. Beckert, R. Thomann and R. Mulhaupt, Angew. Chem., Int. Ed., 2013, 52, 455–458 CrossRef.
- F. Yan and J. Texter, Angew. Chem., Int. Ed., 2007, 46, 2440–2443 CrossRef CAS.
- D. England, N. Tambe and J. Texter, ACS Macro Lett., 2012, 1, 310–314 CrossRef CAS.
- J. Texter, Macromol. Rapid Commun., 2012, 33, 1996–2014 CrossRef CAS.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- Q. Zhao, P. Zhang, M. Antonietti and J. Yuan, J. Am. Chem. Soc., 2012, 134, 11852–11855 CrossRef CAS.
- Y. J. Wang, A. M. Yu and F. Caruso, Angew. Chem., Int. Ed., 2005, 44, 2888–2892 CrossRef CAS.
- Q. Li, J. F. Quinn, Y. Wang and F. Caruso, Chem. Mater., 2006, 18, 5480–5485 CrossRef CAS.
- Y. Wang and F. Caruso, Chem. Mater., 2006, 18, 4089–4100 CrossRef CAS.
- A. Wilke and J. Weber, Macromol. Rapid Commun., 2012, 33, 785–790 CrossRef CAS.
- Q. Sun, X. Meng, X. Liu, X. Zhang, Y. Yang, Q. Yang and F.-S. Xiao, Chem. Commun., 2012, 48, 10505–10507 RSC.
- D. V. Pergushov, A. H. E. Mueller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- Q. Zhao, Q. F. An, Y. Ji, J. Qian and C. Gao, J. Membr. Sci., 2011, 379, 19–45 CrossRef CAS.
- Y. Li, T. K. Bronich, P. S. Chelushkin and A. V. Kabanov, Macromolecules, 2008, 41, 5863–5868 CrossRef CAS.
- S. A. Sukhishvili, E. Kharlampieva and V. Izumrudov, Macromolecules, 2006, 39, 8873–8881 CrossRef CAS.
- A. F. Thunemann, M. Muller, H. Dautzenberg, J. F. O. Joanny and H. Lowen, Polyelectrolyte complexes, in Polyelectrolytes with Defined Molecular Architecture II, ed. M. Schmidt, 2004, vol. 166, pp. 113–171 Search PubMed.
- G. Godeau, L. Navailles, F. Nallet, X. Lin, T. J. McIntosh and M. W. Grinstaff, Macromolecules, 2012, 45, 2509–2513 CrossRef CAS.
- M. Wathier and M. W. Grinstaff, Macromolecules, 2010, 43, 9529–9533 CrossRef CAS.
- M. Ali Aboudzadeh, M. Eugenia Munoz, A. Santamaria, M. Jose Fernandez-Berridi, L. Irusta and D. Mecerreyes, Macromolecules, 2012, 45, 7599–7606 CrossRef.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS.
- K. Okada, M. Nandi, J. Maruyama, T. Oka, T. Tsujimoto, K. Kondoh and H. Uyama, Chem. Commun., 2011, 47, 7422–7424 RSC.
- Y. Li, X. Wang and J. Sun, Chem. Soc. Rev., 2012, 41, 5998–6009 RSC.
- Y. Xiang, S. Lu and S. P. Jiang, Chem. Soc. Rev., 2012, 41, 7291–7321 RSC.
- E. K. Penott-Chang, D. V. Pergushov, A. B. Zezin and A. H. E. Mueller, Langmuir, 2010, 26, 7813–7818 CrossRef CAS.
- E. K. Penott-Chang, M. Ruppel, D. V. Pergushov, A. B. Zezin and A. H. E. Mueller, Polymer, 2011, 52, 4296–4302 CrossRef CAS.
- Q. Chen, M. Luo, P. Hammershoj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS.
- Q. Chen, Q. Wang, M. Luo, L.-J. Mao, C.-G. Yan, Z.-H. Li and B.-H. Han, Polymer, 2012, 53, 2032–2037 CrossRef CAS.
- P. Vandezande, L. E. M. Gevers and I. F. J. Vankelecom, Chem. Soc. Rev., 2008, 37, 365–405 RSC.
- X. Li, S. De Feyter, D. Chen, S. Aldea, P. Vandezande, F. Du Prez and I. F. J. Vankelecom, Chem. Mater., 2008, 20, 3876–3883 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039c3py00159h. |
| This journal is © The Royal Society of Chemistry 2013 |