 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The balance between intramolecular hydrogen bonding, polymer solubility and rigidity in single-chain polymeric nanoparticles†
Patrick J. M. Stals, Martijn A. J. Gillissen, Renaud Nicolaÿ, Anja R. A. Palmans* and E. W. Meijer*
Laboratory of Macromolecular and Organic Chemistry and Institute of Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl; e.w.meijer@tue.nl; Fax: +31 (0) 40 245 1036; Tel: +31 (0) 40 245 3105
First published on 13th February 2013
Abstract
A library of copolymers with pendant, protected ureido-pyrimidinone (UPy) groups was prepared applying controlled polymerization techniques. The polymer backbones were based on polyacrylate, polymethacrylate, polystyrene and polynorbornene and differ in stiffness, molecular weight and the linking moiety between the backbone and the UPy group. In all cases, the percentage of protected UPy groups was kept constant. The effect of solvent on the behaviour of the polymers before and after removal of the protecting groups was evaluated in, among others, chloroform and tetrahydrofuran (THF). After deprotection of the UPy protecting group, the UPys dimerize via four-fold H-bonding in THF, inducing a collapse into single-chain polymeric nanoparticles (SCPNs), as evidenced by a combination of 1H-NMR spectroscopy, size-exclusion chromatography and dynamic light scattering. In chloroform, on the other hand, dimerization of the UPy groups is present but interchain interactions occur as well, resulting in less-defined SCPNs. Remarkably, the flexibility of the polymer backbone, the polymer molecular weight and the nature of the linker unit all do not affect SCPN formation. In contrast, the interaction between solvent and the UPy moiety is a critical parameter for SCPN formation. For example, strong intramolecular dimerization of the UPys is observed in THF while interparticle interactions are suppressed. From this investigation we conclude that a wide variety of polymer backbones are suitable for polymer collapse via supramolecular interactions and thus allow for the formation of SCPNs but that the solvent choice is crucial to enhance intramolecular H-bonding and, at the same time, to suppress interparticle interactions.
Introduction
The field of supramolecular polymer chemistry has developed from the fusion of classic polymer science and supramolecular chemistry.1,2 Herein, the creation of functional, highly ordered architectures that combine the excellent material properties of a classic polymer with the reversible and dynamic nature of non-covalent interactions is pursued.1,3 In the past decade, a number of applications emerged from this interdisciplinary field, ranging from innovative scaffolds for biomedical applications,4 to electronic devices,5 and self-healing materials.6 The seminal work of Stadler7 and Rotello8 highlighted the potential of covalent polymers with pendant H-bonding groups, while advances in controlled polymerization techniques triggered renewed interest in the synthesis and application of such “sticky” polymers.9,10 Recently, the tuneable, highly directional and orthogonal interactions offered by the toolbox of supramolecular chemistry were evaluated to collapse individual chains of synthetic polymers into defined single-chain polymeric nanoparticles (SCPNs).11 Supramolecular motifs such as diamides,12 benzene-1,3,5-tricarboxamides (BTAs),13 ureido-pyrimidinones (UPys),14 BTA-bipyridines,15 cucurbit[8]uril,16 and a combination of thymine–diaminopyridine and six-point cyanuric acid–Hamilton wedge interactions17 are highly promising for this purpose, but also weak, reversible covalent interactions such as disulfide bridges,18 or hydrazone groups19 permit the preparation of dynamic nanoparticles.In a number of examples, secondary structures mimicking α-helices,13,15 β-sheets14a,b or both14c have been observed within the SCPNs, sometimes even when the solvent is water. In fact, the quest for SCPNs that display a compartmentalised, defined three-dimensional shape as a result of a tertiary structure, a feat currently only attainable by DNA and proteins, is a crucial theme. Understanding the rules governing the folding of SCPNs will allow the full potential of supramolecular polymer chemistry to be exploited and the folding of SCPNs will also serve as a simplified model system for understanding protein folding. We envision that these SCPNs will spur novel directions in areas such as catalysis and sensing.13b,c,15
While several reports convincingly show the collapse of individual polymer chains into particles of defined, nanometer-sized dimensions, the relation between the nature of the polymer backbone (stiffness, solubility), the H-bonding group and its collapse behaviour has not been addressed. Recent theoretical calculations by Markvoort et al. showed the critical importance of chain-flexibility on the collapse of single-polymer chains.20 A deeper understanding of this relationship is imperative for the design of future, functional SCPNs.
In this contribution, we systemically investigate the formation of SCPNs as a function of polymer backbone, polymer length, nature of the connecting linker and solvent. We select four of the most widely used polymer backbones in side-chain functionalised supramolecular polymers: poly n-butylacrylate and polymethyl methacrylate which have a relatively flexible backbone and the more stiff polystyrene and polynorbornene. In all cases, we select 2-ureido-pyrimidinone (UPy) units as self-assembling moieties because of their ability to dimerise via strong four-fold hydrogen bonds (Kdim = 6 × 107 mol L−1 in chloroform).21 The application of a UV-labile o-nitrobenzyl-protecting group to temporarily “cage” the UPy enhances the solubility of the polymer constructs and allows initiating of folding of the individual polymer chains on demand by UV irradiation (Fig. 1).14,22 With the help of size exclusion chromatography (SEC) and dynamic light scattering (DLS), we evaluate the importance of solvent, polymer molecular weight, spacer length between UPy and polymer and the stiffness of the polymer in the folding behaviour of the SCPNs. The results indicate that, of all the parameters we anticipated to be involved in SCPN folding, SCPN–solvent interactions are by far the most important one.
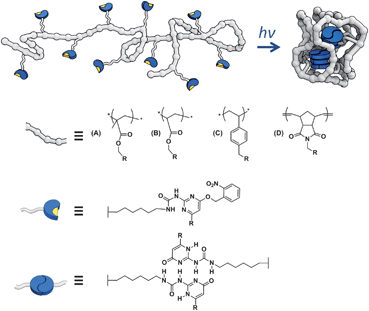 | ||
| Fig. 1 Schematic representation of the collapse of UPy containing polymers. | ||
Experimental section
Materials and methods
Commercial reagents were obtained from Acros or Sigma-Aldrich. Solvents were obtained from Biosolve. Deuterated solvents were obtained from Cambridge Isotope Laboratories. Styrene was distilled under reduced pressure prior to use, methyl methacrylate, 2-((trimethylsilyl)oxy)ethyl methacrylate and n-butylacrylate were passed through a short column filled with an inhibitor remover (Aldrich) or basic alumina. AIBN was recrystallized from methanol. Dry, degassed toluene, THF and dichloromethane were tapped off a solvent purification column. Chloroform was dried over molecular sieves and triethylamine was stored over KOH pellets. All other commercial chemicals were used as received. 4-Cyano-4-methyl-5-(phenylthio)-5-thioxopentanoic acid was kindly provided by SyMO-Chem (Eindhoven, the Netherlands). Aminomethylated polystyrene resin was obtained from NovaBioChem. N-(6-Methyl-4-oxo-1,4-dihydropyrimidin-2-yl)-1H-imidazole-1-carboxamide (1a),23N-(4-oxo-6-tridecyl-1,4-dihydropyrimidin-2-yl)-1H-imidazole-1-carboxamide (1b),24tert-butyldimethyl(4-vinylbenzyloxy) silane (9),25 (N-dodecyl)-5-norbornene-exo-2,3-dicarboximide,26N-acetyloxy-2,5-(pyrrolidinedione)-5-norbornene-exo-2,3-dicarbox-imide,27 4-vinylbenzyl-6-(tert-butoxycarbonylamino)hexanoate,25 and 3rd generation Grubbs catalyst (dichloro[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene](benzylidene)-bis-(pyridine)ruthenium(II))28 were prepared according to literature procedures. NMR spectroscopy was performed on a Varian Mercury Vx 400 MHz and/or a Varian 400 MR 400 MHz (400 MHz for 1H and 100 MHz for 13C). To improve the signal to noise ratio, typical measurements on polymer samples were carried out with 3000 scans and a delay time of 5 seconds. Deuterated solvents used are indicated in each case. 1H chemical shifts are reported in ppm downfield from tetramethylsilane (TMS), 13C chemical shifts in ppm downfield of TMS using the resonance of the deuterated solvent as internal standard. Abbreviations used are s: singlet, d: doublet, dd: double doublet, t: triplet, q: quartet, and m: multiplet. Matrix assisted laser desorption/ionization (MALDI) mass spectra were obtained on PerSeptive Biosystems Voyager DE-PRO or Bruker MALDI-TOF spectrometers using α-cyano-4-hydroxycinnamic acid (CHCA) and 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]-malononitrile (DCTB) as matrices. GC-MS measurements were performed on a Shimadzu GC-MS system equipped with an autosampler with a Zebron ZB-35 column of 30 m × 0.25 mm with a 0.25 μm coating and using the following temperature program: 80 °C|1 min → 330 °C, 30 °C min−1|3 min. Manual column chromatography was performed using Merck 60 Å pore size silica gel (particle size: 63–200 μm). Flash chromatography was performed using an automatic flash chromatography instrument Biotage Isolera One equipped with Biotage SNAP Cartridge KP-Sil silica cartridges. IR spectroscopy was performed on a Perkin-Elmer FTIR Spectrum 2 equipped with a Perkin-Elmer UATR Two accessory. Dynamic light scattering measurements were performed on a Malvern μV Zetasizer equipped with an 830 nm laser and a scattering angle of 90° at a temperature of 20 °C. Samples were prepared by filtering solutions through a 0.2 μm PTFE-filter (Whatman) in a fluorescence cell with a path length of 1 cm. DMF-SEC measurements were carried out in PL-GPC-50 plus from Polymer Laboratories (Agilent Technologies) with the refractive index detector working in DMF containing 10 mM LiBr at 50 °C at a constant flow rate of 1 mL min−1 on a Shodex GPC-KD-804 column (exclusion limit = 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da; 0.8 cm i.d. × 300 mL) which was calibrated with polyethyleneoxide (PEO) samples with a range from 282–77350 Da (Polymer Laboratories – Agilent Technologies). THF-SEC-measurements were performed on a Shimadzu-system with two Agilent Technology columns in series (PLgel 5 μm mixed C [200–2000000 Da] and PLgel 5 μm mixed D [200–40000 Da]) and equipped with a RI detector (Shimadzu RID-10A) and a PDA detector (Shimadzu SPD-M10A), with THF as eluent at a constant flow rate of 1.0 mL min−1. The system was calibrated with polystyrene (PS) samples with a range of 580–100000 Da (Polymer Laboratories). CHCl3-SEC-measurements were performed on a Shimadzu-system equipped with an Agilent Technology Resipore column [200–400000 Da] and equipped with a RI detector (Shimadzu RID-10A) and a PDA detector (Shimadzu SPD-M20A), with CHCl3 as eluent at a constant flow rate of 1.0 mL min−1. The system was calibrated with polystyrene (PS) samples with a range of 92–371000 Da. SEC-measurements were performed on purified polymers on a THF-system, unless otherwise noted. All measurements shown for CHCl3 and THF-measurements were those recorded with the PDA-detector. Photoirradiation experiments were performed in a Luzchem LZC-4V UV reactor equipped with 8 × 8 watt UV-A light bulbs (λmax = 350 nm). The photoirradiation reactions were performed in a quartz cuvette or NMR tube at room temperature.
000 Da; 0.8 cm i.d. × 300 mL) which was calibrated with polyethyleneoxide (PEO) samples with a range from 282–77350 Da (Polymer Laboratories – Agilent Technologies). THF-SEC-measurements were performed on a Shimadzu-system with two Agilent Technology columns in series (PLgel 5 μm mixed C [200–2000000 Da] and PLgel 5 μm mixed D [200–40000 Da]) and equipped with a RI detector (Shimadzu RID-10A) and a PDA detector (Shimadzu SPD-M10A), with THF as eluent at a constant flow rate of 1.0 mL min−1. The system was calibrated with polystyrene (PS) samples with a range of 580–100000 Da (Polymer Laboratories). CHCl3-SEC-measurements were performed on a Shimadzu-system equipped with an Agilent Technology Resipore column [200–400000 Da] and equipped with a RI detector (Shimadzu RID-10A) and a PDA detector (Shimadzu SPD-M20A), with CHCl3 as eluent at a constant flow rate of 1.0 mL min−1. The system was calibrated with polystyrene (PS) samples with a range of 92–371000 Da. SEC-measurements were performed on purified polymers on a THF-system, unless otherwise noted. All measurements shown for CHCl3 and THF-measurements were those recorded with the PDA-detector. Photoirradiation experiments were performed in a Luzchem LZC-4V UV reactor equipped with 8 × 8 watt UV-A light bulbs (λmax = 350 nm). The photoirradiation reactions were performed in a quartz cuvette or NMR tube at room temperature.Synthesis of UPy-building blocks
1H-NMR (CDCl3): δ = 9.12 (s, 1H, N–H), 8.13 (d, 1H, Ar–H), 7.65 (d, 2H, Ar–H), 7.50 (m, 1H, Ar–H), 7.03 (s, 1H, N–H), 6.28 (s, 1H, Ar–H), 5.73 (s, 2H, Ar–CH2–O), 3.38–3.08 (m, 4H, NH–CH2, OCN–CH2), 2.37 (s, 3H, Ar–CH3), 1.73–1.28 (m, 8H, CH2). 13C-NMR (CDCl3): δ = 169.5, 157.3, 154.3, 154.1, 147.6, 133.7, 132.4, 128.9, 128.7, 125.0, 100.2, 64.7, 42.9, 39.8, 31.2, 29.7, 26.4, 26.2, 23.8. MALDI-TOF-MS: m/z: 429.31 (M + H+).
Synthesis of additional monomers
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CO–CHCH2), 5.80 (dd, J = 10.4, 1.4 Hz, 1H, CC–H), 4.17 (t, 2H, CO–O–CH2), 3.63 (t, 2H, Si–O–CH2), 1.72 (q, 2H, CH2), 1.58 (q, 2H, CH2), 0.88 (s, 9H, Si–C–CH3), 0.04 (s, 6H, Si–CH3). 13C-NMR (CDCl3): δ = 166.3, 130.4, 128.6, 64.5, 62.6, 31.9, 29.2, 25.9, 25.2, −5.4. GC/MS tR = 4.508; m/z: 129.C–H), 5.54 (s, 1H, CC–H), 4.50 (s, 1H, N–H), 4.13 (t, 2H, O–CH2), 3.10 (t, 2H, N–CH2), 1.93 (s, 3H, CC–CH3), 1.72–1.26 (m, 17H, CH2). 13C-NMR (CDCl3): δ = 167.5, 156.0, 136.5, 125.2, 79.1, 64.6, 40.5, 30.0, 28.5, 28.4, 26.4, 25.7, 18.3.
C–H), 6.11 (dd, J = 17.3, 10.4 Hz, 1H, CO–CHCH2), 5.80 (dd, J = 10.4, 1.4 Hz, 1H, CC–H), 4.17 (t, 2H, CO–O–CH2), 3.63 (t, 2H, Si–O–CH2), 1.72 (q, 2H, CH2), 1.58 (q, 2H, CH2), 0.88 (s, 9H, Si–C–CH3), 0.04 (s, 6H, Si–CH3). 13C-NMR (CDCl3): δ = 166.3, 130.4, 128.6, 64.5, 62.6, 31.9, 29.2, 25.9, 25.2, −5.4. GC/MS tR = 4.508; m/z: 129.C–H), 5.54 (s, 1H, CC–H), 4.50 (s, 1H, N–H), 4.13 (t, 2H, O–CH2), 3.10 (t, 2H, N–CH2), 1.93 (s, 3H, CC–CH3), 1.72–1.26 (m, 17H, CH2). 13C-NMR (CDCl3): δ = 167.5, 156.0, 136.5, 125.2, 79.1, 64.6, 40.5, 30.0, 28.5, 28.4, 26.4, 25.7, 18.3.Synthesis of polymers
:95, the solvent decanted and the polymer dried under high vacuum at room temperature to a constant weight to obtain a very viscous, slightly yellow oil. 1H-NMR (CDCl3): δ = 4.02 (t, broad, CO–O–CH2), 3.62 (t, Si–O–CH2), 2.38–1.30 (m, CH, CH2), 0.92 (t, CH3), 0.88 (s, Si–C–CH3), 0.04 (s, Si–CH3). SEC: Mn = 34.8 kDa, PDI = 1.83.000, PDI = 1.08.P3b1H-NMR (CDCl3): δ = 7.20–6.24 (m, Ar–H), 4.97 (s, Ar–CH2–O–CO), 4.45 (s, N–H), 3.05 (s, broad, CO–NH–CH2), 2.32 (s, broad, O–CO–CH2), 2.16–1.20 (m, CH, CH2). SEC: Mn = 18100, PDI = 1.10.
CH), 4.52 (s, NCH2CO), 3.80–2.54 (aliphatic side chain and polymer backbone), 2.38–1.89 (aliphatic side chain and polymer backbone), 0.87 (t, CH3). SEC: Mn = 58.2 kDa, PDI = 1.33.P1a′ was obtained as a slightly yellow solid. 1H-NMR (CDCl3): 4.12 (s, broad, CO–O–CH2), 3.84 (s, broad, CH2–OH), 3.60 (s, broad, CO–O–CH3), 2.07–0.76 (m, CH2, CH3). SEC: Mn = 18.4 kDa, PDI = 1.10.
P1b′ was obtained as a slightly yellow solid. 1H-NMR: the spectroscopic data were identical to those of P1a′. SEC: Mn = 40.8 kDa, PDI = 1.33.
P2′ was obtained as a colourless sticky oil and was purified by a short plug of silica, followed by precipitation in MeOH–H2O, 15:85. 1H-NMR (CDCl3): δ = 4.03 (t, broad, CO–O–CH2), 3.65 (t, broad, HO–CH2), 2.38–1.32 (m, CH, CH2), 0.93 (t, CH3). SEC: Mn = 38.7 kDa, PDI = 1.96.
P3a′ was obtained as a white solid.251H-NMR (CDCl3): δ = 7.20–6.24 (m, Ar–H), 4.57 (s, broad, Ar–CH2), 2.19–1.15 (m, CH2). SEC: Mn = 18.4 kDa, PDI = 1.08.
P1c′ was obtained as a slightly yellow solid. 1H-NMR (CDCl3): δ = 7.78 (s, broad, NH3+), 3.95 (s, broad, CO–O–CH2), 3.60 (s, broad, CO–O–CH3), 3.09 (s, broad, N–CH2), 2.09–0.79 (m, CH2, CH3).
P3c′ was obtained as a white solid.251H-NMR (CDCl3): δ = 7.85 (s, broad, NH3+), 7.45–6.23 (m, Ar–H), 4.98 (s, broad, Ar–CH2), 3.05 (m, CH2–NH2), 2.37 (m, CH2–CO), 2.13–1.10 (m, CH2).
P5a: 1H-NMR (CDCl3): δ = 9.16 (s, broad, N–H), 8.14 (d, Ar–H), 7.65 (d, 2× Ar–H), 7.51 (m, Ar–H), 7.16 (s, N–H), 6.29 (s, broad, 1H, Ar–H), 5.88 (s, broad, N–H), 5.74 (s, Ar–CH2–O), 4.26 (s, broad, NH–CO–O–CH2), 4.12 (s, broad, CO–O–CH2), 3.60 (s, broad, CO–O–CH3), 3.33 (m, NH–CH2, UPy), 3.18 (s, broad, O–CO–NH–CH2), 2.37 (s, Ar–CH3), 2.07–0.76 (m, CH2, CH3). Conversion alcohols: 47%. SEC: Mn = 24.9 kDa, PDI = 1.12.
P5b: 1H-NMR (CDCl3): δ = 9.16 (s, broad, N–H), 8.11 (d, Ar–H), 7.71 (m, 2× Ar–H), 7.51 (m, Ar–H), 7.09 (s, N–H), 6.25 (s, broad, Ar–H), 5.83 (s, broad, N–H), 5.75 (s, 2H, Ar–CH2–O), 4.26 (s, broad, NH–CO–O–CH2), 4.11 (s, broad, CO–O–CH2), 3.60 (s, broad, CO–O–CH3), 3.33 (m, NH–CH2, UPy), 3.18 (s, broad, O–CO–NH–CH2), 2.58 (s, Ar–CH2), 2.18–0.79 (m, CH2, CH3). Conversion alcohols: 32%. SEC: Mn = 67.9 kDa, PDI = 1.43.
P5c: In the case of P5c, no DBTDL was added, instead 50 μL triethylamine was added and the mixture was stirred at room temperature. 1H-NMR (CDCl3): δ = 9.18 (s, broad, N–H), 8.12 (d, Ar–H), 7.66 (m, 2× Ar–H), 7.50 (m, Ar–H), 7.14 (s, N–H), 6.27 (s, Ar–H), 5.73 (s, 2H, Ar–CH2–O), 4.70 (s, broad, N–H), 3.94 (s, broad, CO–O–CH2), 3.60 (s, broad, CO–O–CH3), 3.32 (m, NH–CH2, UPy), 3.16 (s, broad, NH–CO–NH–CH2), 2.60 (s, Ar–CH2), 2.07–0.77 (m, CH2, CH3). SEC: Mn = 24.1 kDa, PDI = 1.16.
P6: 1H-NMR (CDCl3): δ = 9.11 (s, broad, N–H), 8.11 (d, Ar–H), 7.63 (m, 2× Ar–H), 7.50 (m, 1× Ar–H), 7.13 (s, N–H), 6.27 (s, Ar–H, UPy), 5.72 (s, Ar–CH2–O), 4.03 (s, broad, CO–O–CH2), 3.77 (s, broad, NH–CO–O–CH2), 3.32 (m, NH–CH2, UPy), 3.13 (s, broad, O–CO–NH–CH2), 2.36 (s, Ar–CH3), 2.34–1.32 (m, CH, CH2), 0.91 (t, CH3). Conversion alcohols: 70%. SEC: Mn = 48.7 kDa, PDI = 1.84.
P7a: 1H-NMR (CDCl3): δ = 9.16 (s, broad, N–H), 8.11 (d, Ar–H), 7.63 (m, 2× Ar–H), 7.58 (m, 1× Ar–H), 7.33–6.84 (m, Ar–H, styrene), 6.83–6.28 (m, Ar–H, styrene), 6.25 (s, Ar–H, UPy), 5.71 (s, Ar–CH2–O), 4.99 (s, broad, Ar–CH2–O–CO), 4.70 (s, broad, N–H), 3.33 (t, NH–CH2, UPy), 3.17 (t, O–CO–NH–CH2), 2.57 (t, Ar–CH2), 2.30 (t, O–CO–CH2), 2.22–1.09 (m, CH2, CH2), 0.87 (t, CH3). Conversion alcohols: 49%. SEC: Mn = 23.6 kDa, PDI = 1.17.
P7b: In the case of P7b, no DBTDL was added, instead 50 μL triethylamine was added and the mixture was stirred at room temperature. 1H-NMR (CDCl3): δ = 9.21 (s, broad, N–H), 8.06 (d, Ar–H), 7.64 (m, 2× Ar–H), 7.47 (m, 1× Ar–H), 7.37–6.82 (m, Ar–H, styrene), 6.82–6.32 (m, Ar–H, styrene), 6.27 (s, Ar–H, UPy), 5.68 (s, Ar–CH2–O), 4.92 (s, broad, Ar–CH2–O–CO), 4.49 (s, broad, N–H), 3.31 (t, NH–CH2, UPy), 3.09 (t, NH–CH2, urea), 2.57 (t, Ar–CH2), 2.30 (t, O–CO–CH2), 2.22–1.09 (m, CH2, CH2), 0.87 (t, CH3). SEC: Mn = 28.7 kDa, PDI = 1.17.
CH), 5.68 (s, Ar–CH2–O), 4.72 (s, broad, Ar–CH2–O–CO), 4.02 (s, NCH2CO), 3.52–2.56 (broad, aliphatic side chain and polymer backbone), 2.35 (broad, O–CO–CH2), 2.32–1.80 (broad, aliphatic side chain and polymer backbone), 1.68–1.40 (broad, aliphatic side chain and polymer backbone), 1.40–1.06 (m, CH2, CH2), 1.06–0.67 (m, CH3). SEC: Mn = 58.2 kDa, PDI = 1.33.Results and discussion
Design and synthetic strategy
Four types of polymers were selected that differ in backbone rigidity, namely polymethyl methacrylates (series A), poly n-butylacrylates (series B), polystyrenes (series C) and polynorbornenes (series D). All polymers comprise pendant self-assembling UPy motifs (Scheme 1). Two different methods to incorporate the self-assembling motif are possible: either via direct copolymerization of a UPy-based monomer or via post-functionalisation of the polymer with a suitable UPy-based synthon.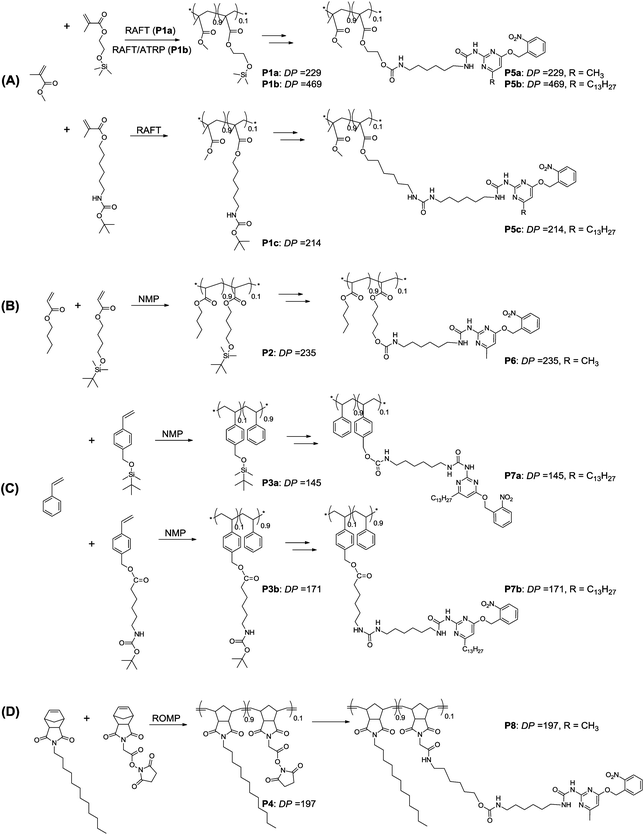 | ||
| Scheme 1 Synthesis of polymers discussed in this contribution, DP refers to average degree of polymerization. | ||
In light of a recent report on issues with copolymerizing UPy-based monomers, we opted for the post-functionalization strategy.14b Random copolymers with a degree of polymerization (DP) of around 200 and a loading of 10% of post-functionalizable groups were selected. Such DPs are readily attainable via a number of controlled polymerization techniques, while a 10% loading of self-assembling groups is a convenient compromise between polymer solubility and a sufficient number of interacting groups. Vinyl-based monomers can be polymerized using controlled radical polymerization techniques,29–32 wherein excellent control over the molecular weight, the polydispersity index (PDI) and end-groups can be achieved. On the other hand, ring-opening metathesis polymerization (ROMP) using the 3rd generation Grubbs catalyst offers the possibility to produce norbornene based polymers with narrow molecular weight distributions.33
To enable post-functionalization, we selected comonomers comprising silyl-protected alcohols, t-BOC-protected amines or N-succinimide esters. The presence of a protective group in the case of alcohol or amine containing monomers proved to be important to ensure good control during the polymerization reaction.25 The silyl and t-BOC protection groups were selected because they are deprotected under mild conditions. We designed novel o-nitrobenzyl protected UPy synthons to react with the pendant alcohol/amine (via reaction with isocyanate) or active ester groups (via reaction with amine) on the desired polymers (Scheme 2). We selected two different substituents (a: –CH3 and b: –C13H27) on the alkylidene-position of the UPy moiety to show the wide applicability of this novel synthon. The CH3-substituent is the substituent most commonly observed in UPy molecules while the C13H27-substituent is expected to enhance the solubility of the synthon.
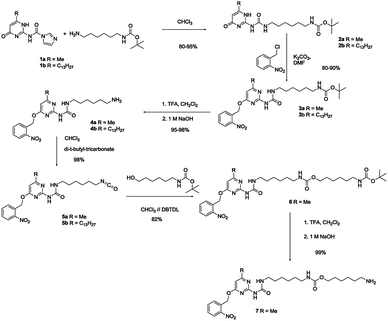 | ||
| Scheme 2 Synthesis of phUPy synthons 5 and 7. | ||
The “caging” of the UPy group is crucial since “free” UPys frequently induce solubility problems, hampering proper characterization of the polymers. In addition, the o-nitrobenzyl protective group is UV-labile and conveniently removed upon UV-irradiation.14,22 In this way, conditions can be selected (e.g. in dilute solutions) to induce intramolecular dimerization of the UPy motifs, resulting in folding of individual polymer chains into SCPNs.
In addition to differences in polymer flexibility when keeping the DP constant (P5a, P6, P7a and P8), polymers were designed that allow evaluation of the effect of polymer molecular weight on the folding behavior by varying the degree of polymerization (P5a and P5b). Moreover, the use of a different connectivity (urea versus urethane) to attach the UPy group to the polymer (P5a/c and P7a/b) will provide information on how additional H-bonding interactions affect the folding behavior. Finally, we selected 3 solvents to investigate the ability of all polymers to form SCPNs: chloroform, a weak H-bond donor, tetrahydrofuran (THF), a weak H-bond acceptor and polar dimethylformamide (DMF), a strong H-bond disruptor.
Synthesis of o-nitrobenzyl-protected UPy synthons
To graft an o-nitrobenzyl protected UPy (phUPy) to the desired amine/alcohol or activated ester pendant prepolymers, we prepared two different phUPy synthons, one comprising an isocyanate group (compound 5, Scheme 2) and one comprising an amine group (7, Scheme 2). The synthesis of phUPy building blocks 5 and 7 is outlined in Scheme 2 and starts from known CDI-activated isocytosines 1a,b (Scheme 2).23,24The CDI-activated isocytosines 1a,b were reacted with mono-BOC protected hexyldiamine, to afford UPys 2a,b in high yield (80–95%). Application of ortho-nitrobenzylchloride in DMF/K2CO3 afforded O-alkylation and protected UPys 3a,b were obtained in excellent yields (80–90%). Removal of the t-BOC with trifluoroacetic acid yielded free amines 4a,b. Reaction of the free amines with di-tert-butyltricarbonate34 afforded isocyanates 5a,b in near quantitative yield and excellent purity. Finally, isocyanate 5a was reacted further with tert-butyl (6-hydroxyhexyl)carbamate, and after deprotection of the t-BOC group, amine 7 was obtained in high yield and purity. Amine 7 incorporating an additional urethane bond was prepared to facilitate comparison between the vinyl polymer series and the norbornene polymer series (Scheme 1).
Synthesis of prepolymers P1–4
Prepolymers based on vinyl-based monomers (series A–C, Scheme 1) were prepared using RAFT,31 RAFT/ARGET ATRP,30 or NMP.32 Polymethyl methacrylate based copolymers P1a and P1c were prepared using RAFT polymerizations using 4-cyano-4-methyl-5-(phenylthio)-5-thioxopentanoic acid as the chain transfer agent (CTA) under standard conditions for RAFT polymerizations.35 Two different polymers were prepared: polymer P1c, the copolymer of methyl methacrylate with 10 mol% 6-((tert-butoxycarbonyl)amino)hexyl methacrylate, providing protected amines on the polymer scaffold and polymer P1a, the copolymer of methyl methacrylate with 10 mol% 2-((trimethylsilyl)oxy)ethyl methacrylate (HEMA-TMS), providing protected alcohols on the polymer scaffold. The SEC traces in THF of both polymers showed similar characteristics: Mn ∼ 21 kDa, with an excellent PDI (1.07) for P1a and Mn ∼ 28 kDa, with PDI = 1.20 for P1c. To compare the influence of molecular weight on the self-assembly, we also prepared copolymer P1b which like P1a contained HEMA-TMS and methyl methacrylate in a 9:1 molar ratio but with a molecular weight twice as high as P1a. To achieve this, a new controlled polymerization method for obtaining high molecular weight polymers recently reported by Matyjaszewski et al. was applied.36 This method, a combination of RAFT with ARGET ATRP, resulted in a copolymer with a high DP (∼470) and a low PDI (1.14).Polystyrene based polymers were prepared using NMP utilizing N-tert-butyl-N-(2-methyl-1-phenylpropyl)-O-(1-phenylethyl) hydroxylamine as the NMP-agent under previously described conditions.25 Two different polymers were prepared:25 polymer P3b, the copolymer of styrene with 5 mol% 4-vinylbenzyl 6-(tert-butoxycarbonylamino)hexanoate and polymer P3a, the copolymer of styrene with 12 mol% tert-butyldimethyl(4-vinylbenzyloxy) silane. This affords protected amines and alcohols, respectively, on the polymer scaffold (Table 1). The SEC traces of both polymers showed narrow polydispersities (Mn = 18 kDa, with PDI = 1.10 for polymer P3b and Mn = 19 kDa, with PDI = 1.08 for polymer P3a).
| Series | Polymer | Method | Feed ratio (mA/mB) | mA | Conversion (%) | Observed ratio (mA/mB) | Mn,thb (Da) | DPthb | Mn,expc (Da) | PDIc |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conversions and observed incorporation of the two monomers mA and functional monomer mB as determined by 1H-NMR.b Based on conversion.c All SEC-measurements were carried out on a THF system using a polystyrene calibration, after precipitation of the polymer. | ||||||||||
| A | P1a | RAFT | 90/10 | MMA | 80 | 90/10 | 25200 | 229 | 21200 | 1.07 |
| A | P1b | ATRP/RAFT | 90/10 | MMA | 59 | 89/11 | 51800 | 469 | 59400 | 1.14 |
| A | P1c | RAFT | 90/10 | MMA | 72 | 92/8 | 22800 | 214 | 28000 | 1.20 |
| B | P2 | NMP | 91/9 | nBA | 93 | 91/9 | 33400 | 235 | 34800 | 1.83 |
| C | P3a | NMP | 91/9 | St | 63 | 88/12 | 15900 | 145 | 19000 | 1.08 |
| C | P3b | NMP | 95/5 | St | 74 | 95/5 | 20200 | 171 | 18100 | 1.10 |
| D | P4 | ROMP | 90/10 | Norb. | >99 | 93/7 | 65200 | 197 | 58200 | 1.33 |
Using similar polymerisation conditions, poly((n-butyl)acrylate) based copolymer P2 was prepared by copolymerizing n-butylacrylate with 10 mol% 4-((tert-butyldimethylsilyl)oxy)butyl acrylate using N-tert-butyl-N-(2-methyl-1-phenylpropyl)-O-(1-phenylethyl)hydroxylamine as the NMP-agent. A copolymer with a molecular weight of 35 kDa and a DP of ∼235 was obtained. This copolymer showed a larger PDI (∼1.8, Table 1) than its styrene counterpart.
Polynorbornene based copolymer P4 was prepared via ring-opening metathesis polymerisation of (N-dodecyl)-5-norbornene-exo-2,3-dicarboximide with (N-acetyloxy-2,5-pyrrolidinedione)-5-norbornene-exo-2,3-dicarboximide in a molar ratio of 9:1, using a third generation Grubbs catalyst.28 A polymer with a molecular weight Mn of 58 kDa and a decent PDI (1.33) was obtained.
All prepolymers P1–P4 were characterised by SEC and 1H-NMR, the results are summarised in Table 1. In all cases, the observed polymer composition matches very well with the composition of the feed.
Synthesis of phUPy containing polymers P5–P8
Polymers P1–P3 were deprotected using either trifluoroacetic acid (P1c and P3b) to yield the corresponding free amines or tetrabutylammonium fluoride (TBAF) (P1a,b, P2 and P3a) to yield the corresponding free alcohols. Subsequently, phUPy building blocks 5a and 5b were coupled (see Scheme 2 for details) to the free alcohol and free amine containing polymers, affording polymers P5–P7. Reaction of the isocyanates to the free amines was quantitative, while reaction with the free alcohols proceeded with conversions of 32–70%. The activated NHS-ester in P4 was reacted with phUPy 7, affording polymer P8 in quantitative yield. PhUPy building blocks and polymers P5–P8 were fully characterised with SEC (Fig. S1–S7†), DLS (Fig. S8–S14†), 1H-NMR (Fig. S15–S19†) and infrared spectroscopy (Fig. S20–S24†). The relevant data are summarised in Table 2. As an example, the 1H-NMR of P7b in CDCl3 is shown in Fig. 2. The spectrum shows the presence of the phUPy-moiety (peaks a–g), while comparison of the integrals of peaks m with i, j and s indicates a quantitative reaction of the amines of prepolymer P3b.| Series | Polymer | Prepolymer | Obs. ratio (mA/mB)a | DPtha | # UPysc | Mn,expb (Da) | PDIb |
|---|---|---|---|---|---|---|---|
| a Calculation based on corresponding prepolymers.b All SEC-measurements were carried out on a THF system using a polystyrene calibration after isolation of the polymer.c Calculation based on conversion of alcohols/amines/activated ester. | |||||||
| A | P5a | P1a | 90/10 | 229 | 11 | 24900 | 1.12 |
| A | P5b | P1b | 89/11 | 469 | 17 | 67900 | 1.43 |
| A | P5c | P1c | 92/8 | 214 | 17 | 24100 | 1.16 |
| B | P6 | P2 | 91/9 | 235 | 15 | 48700 | 1.84 |
| C | P7a | P3a | 88/12 | 145 | 9 | 23600 | 1.17 |
| C | P7b | P3b | 95/5 | 171 | 9 | 28700 | 1.17 |
| D | P8 | P4 | 93/7 | 197 | 14 | 57000 | 1.26 |
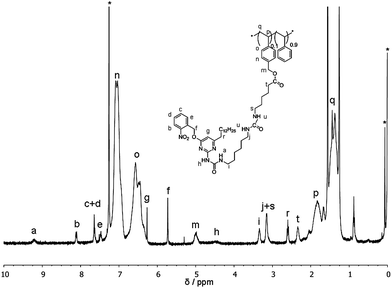 | ||
| Fig. 2 1H-NMR of P7b in CDCl3. | ||
Thus, the influence of the nature of the backbone on the polymer folding behavior can be evaluated by comparing representatives of the 4 different series, P5a, P6, P7 and P8, all showing similar DPs. In addition, by comparing P5a (DP = 230) with P5b (DP = 460), the influence of molecular weight on the polymer folding behavior can be assessed. Finally, the influence of the linker group (urea or urethane) can be evaluated for series A (flexible polymer, P5a and P5c) and series C (more rigid polymer, P7a and P7b).
Influence of polymer stiffness and solvent on intramolecular UPy dimerization
We previously reported that UPy moieties attached to a polymer can dimerize intramolecularly under sufficiently dilute conditions.14 The formation of the hydrogen bonds restricts the conformational freedom of the polymer chain and results in a chain collapse. This collapse was conveniently probed by SEC and AFM. Here, we induced deprotection of the protected UPys by illuminating a solution of the desired polymer (c = 1 mg mL−1) with UV light (UV-A, λmax = 350 nm) in a Luzchem photoreactor for 2 hours. We apply a combination of different techniques to assess the effect of o-nitrobenzyl deprotection. First, 1H-NMR provides information on the ability of the UPy groups to dimerize after deprotection. Size exclusion chromatography (SEC) and dynamic light scattering (DLS) were used to evaluate the size of the polymers before and after deprotection and quantify the degree of chain collapse and single chain character. Finally, we used atom force microscopy to visualise the single chain character of the formed SCPNs. All techniques were performed in three different solvents: chloroform, DMF and THF. Although solvent–polymer backbone interaction parameters are important variables, this study shows that the main solvent dependences here are related to the interaction of the supramolecular unit and the solvent (vide infra). In fact, the second virial coefficients for all polymer systems investigated here are similar for both THF and chloroform (5–9 × 10−4 mol cm3 g−2).37 In chloroform, UPys with an aliphatic substituent on the pyrimidinone ring usually dimerize via strong four-fold hydrogen bonds via the 4[1H]-pyrimidinone dimer.21b In more polar solvents UPy-dimerisation is weaker, as is evidenced by the presence of a large percentage of the weaker pyrimindin-4-ol dimer (THF) or its complete absence (DMSO/DMF).38 In all cases, the polymer solutions were evaluated before and after illumination with UV light. First, we discuss in detail the results of these studies on polymethacrylate P5a.Fig. 3 shows the 1H-NMR spectrum of P5a dissolved in CHCl3 (c = 2 mg mL−1) before and after deprotection. Before deprotection, the intramolecular H-bonded proton of the urea of the phUPy is clearly visible at 9.2 ppm, while no other H-bonded protons are visible. After deprotection three additional signals appeared around 10.3, 11.9 and 13.1 ppm. These are attributed to the UPy dimer in its 4[1H]-pyrimidinone tautomer. Compared to the “free” UPy dimer in CDCl3, the signals are significantly broadened, indicative of a reduced mobility of the UPy dimer. The latter is in line with our expectations for UPy dimers in a SCPN. In THF-d8, a similar pattern is observed before and after deprotection of the UPy group, indicating that also in this rather polar solvent at rather low concentrations 4[1H]-pyrimidinone dimers are formed. Remarkably the NH peaks are much sharper compared to those in THF. These results indicate that in THF the UPys are able to dimerize into the strong 4[1H]-pyrimidinone dimer when attached to a polymer chain and that the high local concentration of UPys inside a polymer chain helps dimerization into a 4[1H]-pyrimidinone dimer. In DMF-d7 after deprotection (Fig. S16†), two signals at 11.8 ppm and 9.3 ppm suggest the presence of the 6[1H]-pyrimidinone monomer (Fig. S16†),38 while smaller subsets at 13.0 and 10.3 ppm suggest the presence of a 4[1H]-pyrimidinone dimer.38
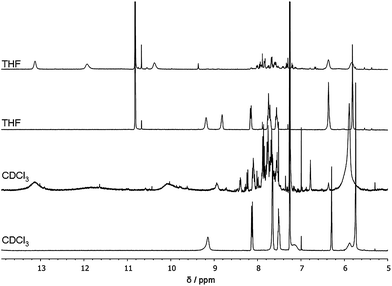 | ||
| Fig. 3 1H-NMR spectra of P5a in CDCl3 before (bottom) and after (second from bottom) deprotection and in THF-d8 before (second from top) and after (top) deprotection. The peak at 10.8 ppm in THF-d8 corresponds to an impurity in the deuterated solvent. | ||
SEC gives qualitative information on the coil to globule transition in polymers because it probes the hydrodynamic volume of macromolecules. In our previous work, we observed that removal of the protecting group results in an increase of the SEC retention time, indicating a reduction of the hydrodynamic volume of the polymer chain. However, since the measurements are typically conducted relative to a standard and interactions of the polymer with the column may occur, the results are not always straightforward to interpret. In contrast, dynamic light scattering gives a direct way of determining the size of the polymers in solution provided that intermolecular aggregation can be suppressed.
We performed SEC and DLS measurements on solutions of P5a in THF (c = 1 mg mL−1) before and after deprotection of the UPy group. The SEC traces of P5a at 1 mg mL−1 in THF before and after deprotection are shown in Fig. 4. A clear increase in the retention time from 14.3 (Mn = 24.9 kDa, PDI = 1.12) to 14.6 min (Mn = 20.3 kDa, PDI = 1.17) is observed, corresponding to a 19% decrease in apparent molecular weight. Since 1H-NMR clearly shows the formation of UPy dimers under these conditions, this is consistent with a collapse of the polymer chain as a result of H-bond formation. This reduction in apparent molecular weight is in good agreement with earlier reported, UPy based nanoparticles.14
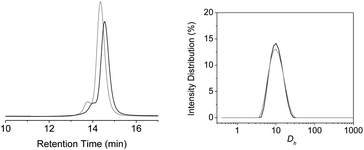 | ||
| Fig. 4 SEC measurements on polymer P5a in THF (left) and intensity distribution versus hydrodynamic diameter (Dh) from DLS measurements on polymer P5a in THF (right), before (grey) and after deprotection (black) of the photocleavable group. | ||
To assess if the UPy dimerization is indeed an intramolecular process, DLS measurements were performed on solutions of P5a before and after deprotection in THF (Fig. 4). From the intensity distributions, the hydrodynamic radius (Rh) was determined. The Rh stayed constant at 5.0 nm, indicating that the UPy dimerization is indeed an intramolecular process; the lack of significant change in Rh can be explained by the small size of the particle, making small differences hard to detect. These combined results allow us to conclude that particles of nanometer-sized dimensions and containing one polymer chain only are formed in a relatively polar solvent like THF. SEC in DMF reveals only small changes for P5a and an almost negligible collapse of 2% is observed (Table S1†).
In chloroform, the behaviour of P5a is strikingly different, as was already indicated by the broader signals of the 4[1H]-pyrimidinone dimer in 1H-NMR (vide supra). SEC in CHCl3 (c = 1 mg mL−1) shows that Mn = 14.3 kDa and PDI = 1.33 before deprotection (Fig. 5). After deprotection, an increase in the apparent molecular weight is observed to Mn = 18.1 kDa, PDI = 1.24. The lower molecular weight observed in CHCl3 compared to THF before deprotection could indicate that P5a has more interactions with the column, which results in an increase in the retention time. In DLS, in contrast, a decrease in Rh (Fig. 5) can be observed from 7.1 to 6.6 nm after deprotection, but the polydisperse peaks observed before and after deprotection indicate a significant degree of interparticle interactions. While an apolar solvent like chloroform enables strong UPy dimerization, interparticle interactions are more pronounced than in the more polar solvent THF. These results indicate that solvent is an important parameter in the formation of well-defined SCPNs starting from P5a.
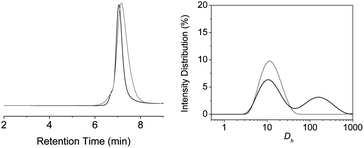 | ||
| Fig. 5 SEC measurements on polymer P5a in CHCl3 (left) and intensity distribution versus hydrodynamic diameter (Dh) from DLS measurements on polymer P5a in CHCl3 (right), before (grey) and after deprotection (black) of the photocleavable group. | ||
The ability of the UPy group to dimerize after deprotection in polybutylacrylate-based P6, polystyrene-based P7a and polynorbornene-based P8 polymers is quite comparable to that of P5a, as evidenced by 1H-NMR. Spectra for deprotected polymers are shown in Fig. S16–S19;† clear formation of the 4[1H]-pyrimidinone dimer in THF and CHCl3 can be observed. This allows us to conclude that backbone rigidity does not have a significant influence on the ability of the polymers to form UPy dimers.
SEC-measurements in THF show a substantial decrease in apparent molecular weight (Fig. 6, full data in Table 3) for P6, P7a and P8 (20% for P6, 19% for P7a and 8% for P8) after deprotection of the photolabile nitrobenzyl group, indicative of a collapse of the polymer chains upon UPy dimerization. In addition, DLS measurements in THF indicate a decrease of the Rh. For example, the Rh of P8 decreases from 7.3 nm to 6.3 nm, while it decreases for P7a from 4.8 nm to 4.4 nm (Fig. S14 and S12†). Although a small increase in Rh is observed for P6, it is accompanied by a decrease in the presence of larger aggregates (Fig. S11†), indicating a better defined system. The difference with the SEC-measurements in this latter observation can be rationalized since the shear-forces involved in SEC-measurements probably disrupt the weak interchain interactions in such a polar solvent. In DMF, we also see small decreases in Rh in DLS and apparent hydrodynamic volume in SEC for P7a (Table S1, Fig. S5 and S12,† a change of 32% in SEC and a decrease in Rh from 4.6 to 4.4 nm in DLS after deprotection). Due to a poor match in refractive index difference for P6, and P8 with DMF, DLS and SEC were not possible.
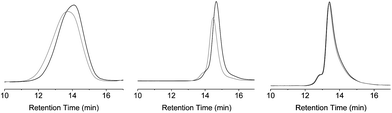 | ||
| Fig. 6 SEC measurements on polymer P6 in THF (left), P7a in THF (middle) and P8 in THF (right), before (grey) and after deprotection (black) of the photocleavable group. | ||
| Solvent | Backbone | THF | CHCl3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | Mn,p | PDIp | Mn,d | PDId | Collapse (%) | Mn,p | PDIp | Mn,d | PDId | Collapse (%) | |
| a Mn,p = Mn photoprotected polymer in Da. Mn,d = Mn deprotected polymer in Da; Mn relative to PS standards. | |||||||||||
| P5a | Methacrylate | 24900 | 1.12 | 20300 | 1.17 | 18.6 | 14300 | 1.33 | 18100 | 1.24 | −27.1 |
| P5b | Methacrylate | 67900 | 1.43 | 60100 | 1.46 | 11.5 | 366000 | 2.58 | 86100 | 2.54 | 76.5 |
| P5c | Methacrylate | 24100 | 1.16 | 21500 | 1.19 | 10.6 | 254300 | 2.54 | 167200 | 2.94 | 34.3 |
| P6 | Acrylate | 48700 | 1.84 | 39100 | 1.65 | 19.8 | 465800 | 2.90 | 107200 | 6.14 | 77.0 |
| P7a | Styrene | 23600 | 1.17 | 19100 | 1.18 | 18.9 | 59400 | 2.72 | 20200 | 1.63 | 66.1 |
| P7b | Styrene | 28700 | 1.17 | 23300 | 1.22 | 18.8 | 19200 | 4.96 | 14400 | 1.59 | 25.1 |
| P8 | Norbornene | 57000 | 1.26 | 52500 | 1.42 | 8.0 | 101800 | 1.81 | 54500 | 1.55 | 46.5 |
In chloroform we also observe a similar behaviour for urethane containing polymers P6, P7a and P8 to that for P5a. In SEC measurements, the polymers show large polydispersities before and after deprotection of the photolabile group and activation of the hydrogen bonds, and also show large decreases in apparent hydrodynamic volume (changes from 47–77%, Table 3), indicating that also P6, P7a and P8 show relevant interparticle interactions in chloroform.
This behaviour is also observed in DLS measurements for P6 (Fig. S11†), where also aggregates are visible before and after deprotection of the photolabile protecting group. Surprisingly, for the stiffer backbones, polystyrene-based P7a and polynorbornene-based P8, a collapse can be observed in DLS after deprotection and relative narrow distributions are obtained (Fig. S12 and S14†). From these experiments we conclude that the exact nature of the polymeric backbone is not the most important parameter in the formation of well-defined SCPNs but that the choice of solvent is a far more important parameter. First, the polymer has to be well-soluble in the chosen solvent, second, the solvent must allow for a substantial amount of H-bonding inside the particle and third, interparticle interactions have to be suppressed. A possible explanation for the ability of THF to suppress the interparticle interactions can be that THF, a H-bond acceptor, is in competition with interparticle interactions, thus limiting the formation of large aggregates.
Influence of Mn on the formation of SCPNs
The influence of molecular weight on the folding behaviour was investigated by comparing polymethacrylate based P5a (DP = 230) with its heavier analogue P5b (DP = 470). Although P5b is twice as large as P5a, it has a quite comparable number of phUPys. The behaviour of both polymers is quite similar, although the larger size of P5b makes interpretation of the data more straightforward. In THF the apparent hydrodynamic volume of P5b decreases with 12% (Table 3 and Fig. S2†) in SEC measurements while also a significant decrease in Rh can be observed from 12.6 nm to 11.8 nm after deprotection (Table 4 and Fig. S9†). A similar behaviour is visible in DMF; an apparent decrease in hydrodynamic volume of 20% in SEC and a significant decrease in Rh from 9.1 nm to 8.5 nm in DLS after deprotection (Table S1 and Fig. S2 and S9†). Also in chloroform the behaviour of P5b is similar to that of P5a; very broad peaks are observed in SEC, while also broad distributions are visible in DLS (Tables 3 and 4 and Fig. S2 and S9†), again indicating that interparticle interactions are present. These results allow us to conclude that increasing the polymer length does not influence the ability of the polymer to form a SCPN.| Solvent | THF | CHCl3 | ||
|---|---|---|---|---|
| Polymer | Rh,p (nm) | Rh,d (nm) | Rh,p (nm) | Rh,d (nm) |
| a nd = not determined. | ||||
| P5a | 5.0 | 5.0 | 7.1 | 6.6 |
| P5b | 12.6 | 11.8 | 15.3 | 18.1 |
| P5c | nd | nd | 62.5 | 52.1 |
| P6 | 9.3 | 9.9 | 8.2 | 16.5 |
| P7a | 4.8 | 4.4 | 5.4 | 5.2 |
| P7b | 6.3 | 5.7 | 6.4 | 5.5 |
| P8 | 7.3 | 6.3 | 9.9 | 9.0 |
Influence of linker unit on the formation of SCPNs
To investigate the influence of the linker unit between the UPy and the polymer backbone on the folding we compared urethane containing polymethacrylate-based P5a and polystyrene-based P7a with their urea containing counterparts P5c and P7b, respectively. Polymers P5c and P7b have comparable Mn, PDI and number of phUPys as their urethane counterparts (Table 2). Polymer P5c shows a similar collapse in THF-SEC to P5a (19% versus 12%, Table 3), and a clear decrease in Rh can be observed from the DLS measurements (Table 4). Also in CHCl3 the behaviour is similar for P5c to that for P5a; polydisperse aggregates can be observed in both SEC and DLS measurements (Tables 3 and 4 and Fig. S3 and S10†).The comparison of P7a with P7b shows similar results. In THF, P7b collapses into a smaller particle after deprotection as is evidenced by SEC (Table 3 and Fig. S6†) and DLS measurements (Table 4 and Fig. S13†), while in SEC measurements in chloroform (Table 3 and Fig. S6†) large aggregates are present, although the DLS measurements show a collapse in DLS with relative narrow distributions (Fig. S13†). These results allow us to conclude that the linker between the polymeric backbone and the UPy does not significantly influence the ability to form SCPNs.
Atomic force microscopy
AFM is a widely used technique to study nanoparticles; to visualize the nanoparticles obtained in this study we selected two polymers differing in polymer flexibility. Polymethacrylate based P5c has a relatively flexible backbone, while polystyrene based P7b has a relatively stiff backbone. Dilute solutions (c = 10−4 mg mL−1, Fig. 7) of deprotected nanoparticles in dioxane were dropcast on a freshly cleaved mica-surface and the resulting nanoparticles were measured. Dioxane was chosen as a solvent that resembles THF in terms of chemical nature and polarity but evaporates slower, thus decreasing potential dewetting phenomena.26 | ||
| Fig. 7 AFM height-micrographs of polymers P5c (left) and P7b (right). | ||
The AFM micrographs show comparable features for both polymers; representative height images are shown in Fig. 7. The micrographs show spherical particles, no large, undefined aggregates are visible. As expected, some polydispersity is present in the size of the particles for both polymers. The nanoparticles are on average 25–30 nm in diameter, which is in good agreement with earlier reported sizes for UPy-based nanoparticles.14
Conclusions
In conclusion, we have shown that a modular post-functionalization approach is an efficient way to produce a large library of polymers capable of forming SCPNs. Using dynamic light scattering (DLS) and size exclusion chromatography (SEC) techniques the change in hydrodynamic radius of these particles upon removal of the photoprotecting group has been shown. 1H-NMR showed the dimerisation of the UPy-moieties. Last, in atomic force microscopy (AFM) studies, the appearance of well-defined particles after formation of the H-bonds is visualised. Interestingly, neither the difference in backbone rigidity or the molecular weight nor the linking moiety between the different polymeric backbones results in notable differences in the formation of the SCPNs. Solvent is however a crucial parameter in the formation of well-defined SCPNs. Well-defined SCPNs are formed in solvents that show some competition for H-bonding (THF), and particles with a 8–20% lower hydrodynamic volume than the protected polymers are observed. These particles form no networks even though a large number of UPy-groups are available. In less competitive solvents (e.g. CHCl3), the tendency to form defined particles is less pronounced and more interparticle interactions are observed in DLS and SEC. This illustrates the versatility and freedom of choice in selecting polymeric backbones when SCPNs for advanced applications like catalysis and sensing are pursued.Acknowledgements
The authors would like to thank R. Bovée, E. B. Berda, T. F. E. Paffen and G. van Straaten (Eindhoven University of Technology) for assistance with synthesis and measurements. The ICMS Animation Studio (Eindhoven University of Technology) is acknowledged for providing the art-work. This work was supported by the European Research Council (ERC Advanced grant no. 246829) and the Netherlands Organisation for Scientific Research (NWO-TOP grant: 10007851).Notes and references
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS.
- A. Bertrand, F. Lortie and J. Bernard, Macromol. Rapid Commun., 2012, 33, 2062 Search PubMed.
- (a) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS; (b) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS.
- (a) P. Y. W. Dankers, J. M. Boomker, A. Huizinga-van der Vlag, E. Wisse, W. P. J. Appel, F. M. M. Smedts, M. C. Harmsen, A. W. Bosman, E. W. Meijer and M. J. A. van Luyn, Biomaterials, 2011, 32, 723 CrossRef CAS; (b) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS.
- (a) J.-L. Wietor, A. Dimopoulos, L. E. Govaert, R. A. T. M. van Benthem, G. de With and R. P. Sijbesma, Macromolecules, 2009, 42, 6640 CrossRef CAS; (b) P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS; (c) M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334 CrossRef CAS; (d) G. M. L. van Gemert, J. W. Peeters, S. H. M. Söntjens, H. M. Janssen and A. W. Bosman, Macromol. Chem. Phys., 2012, 213, 234 CrossRef.
- See for example: (a) L. L. de Lucca Freitas and R. Stadler, Macromolecules, 1987, 20, 2478 CrossRef CAS; (b) C. Hilger, R. Stadler and L. L. de Lucca Freitas, Polymer, 1990, 31, 818 Search PubMed; (c) C. Hilger and R. Stadler, Makromol. Chem., 1991, 192, 805 Search PubMed; (d) C. Hilger, M. Drager and R. Stadler, Macromolecules, 1992, 25, 2498 CrossRef CAS.
- (a) R. Deans, F. Ilhan and V. M. Rotello, Macromolecules, 1999, 32, 4956 CrossRef CAS; (b) F. Ilhan, M. Gray and V. M. Rotello, Macromolecules, 2001, 34, 2597 CrossRef CAS; (c) R. Shenhar, H. Xu, B. L. Frankamp, T. E. Mates, A. Sanyal, O. Uzun and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 16318 CrossRef CAS.
- (a) J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193 RSC; (b) M. Weck, Polym. Int., 2007, 56, 453 CrossRef CAS.
- See for example: (a) A. K. Sharma, M. Caricato, E. Quartarone, S. Edizer, A. G. Schieroni, R. Mendichi and D. Pasini, Polym. Bull., 2012, 69, 911 Search PubMed; (b) L. R. Rieth, R. F. Eaton and G. W. Coates, Angew. Chem., Int. Ed., 2001, 40, 2153 CrossRef CAS.
- (a) M. K. Aiertza, I. Odriozola, G. Cabañero, H.-J. Grande and I. Loinaz, Cell. Mol. Life Sci., 2012, 69, 337 CrossRef CAS; (b) O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958 CrossRef CAS; (c) M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917 CrossRef CAS.
- M. Seo, J. B. Beck, J. M. J. Paulusse, C. J. Hawker and S. Y. Kim, Macromolecules, 2008, 41, 6413 CrossRef CAS.
- (a) T. Terashima, T. Mes, T. F. A. de Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742 CrossRef CAS; (b) T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085 CrossRef CAS; (c) E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013 DOI:10.1002/anie.201207123.
- (a) E. J. Foster, E. B. Berda and E. W. Meijer, J. Am. Chem. Soc., 2009, 131, 6964 CrossRef CAS; (b) E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430 CrossRef CAS; (c) N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501 Search PubMed.
- M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166 RSC.
- E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185 CrossRef CAS.
- O. Altintas, E. Lejeune, P. Gerstel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 640 RSC.
- B. T. Tuten, D. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068 RSC.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242 CAS.
- P. Englebienne, P. A. J. Hilbers, E. W. Meijer, T. F. A. de Greef and A. J. Markvoort, Soft Matter, 2012, 8, 7610 RSC.
- (a) R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS; (b) S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487 CrossRef.
- B. J. B. Folmer, E. Cavini, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 1998, 1847 RSC.
- A. T. ten Cate, P. Y. W. Dankers, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2003, 125, 6860 CrossRef CAS.
- H. M. Keizer, R. P. Sijbesma and E. W. Meijer, Eur. J. Org. Chem., 2004, 12, 2553 Search PubMed.
- P. J. M. Stals, T. N. T. Phan, D. Gigmes, T. F. E. Paffen, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 780 Search PubMed.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 118 CrossRef CAS.
- M.-P. Chien, M. W. Thompson and N. C. Gianneschi, Chem. Commun., 2011, 47, 167 RSC.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS.
- (a) N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270 CrossRef CAS; (b) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS; (c) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133 CrossRef CAS.
- (a) D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189 RSC; (b) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS; (c) J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013 DOI:10.1016/j.progpolymsci.2012.06.002.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS.
- H. W. I. Peerlings and E. W. Meijer, Tetrahedron Lett., 1999, 40, 1021 CrossRef CAS.
- G. Moad and C. Barner-Kowollik, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) R. Nicolaÿ, Y. Kwak and K. Matyjaszewski, Angew. Chem., Int. Ed., 2010, 49, 541 CAS; (b) R. Nicolaÿ and Y. Kwak, Isr. J. Chem., 2012, 52, 288 Search PubMed.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, John Wiley & Sons, New York, 4th edn, 1999, p. 2005 Search PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables. See DOI: 10.1039/c3py00094j |
| This journal is © The Royal Society of Chemistry 2013 |