 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular recognition between functionalized gold nanoparticles and healable, supramolecular polymer blends – a route to property enhancement†‡
Rajendran
Vaiyapuri
a,
Barnaby W.
Greenland
b,
Howard M.
Colquhoun
a,
Joanne M.
Elliott
a and
Wayne
Hayes
*a
aDepartment of Chemistry, University of Reading, Whiteknights, RG6 6AD, Reading, UK. E-mail: w.c.hayes@reading.ac.uk; Fax: +44 (0)118 3786331
bSchool of Pharmacy, University of Reading, Whiteknights, RG6 6AD, Reading, UK
First published on 4th March 2013
Abstract
A new, healable, supramolecular nanocomposite material has been developed and evaluated. The material comprises a blend of three components: a pyrene-functionalized polyamide, a polydiimide and pyrene-functionalized gold nanoparticles (P-AuNPs). The polymeric components interact by forming well-defined π–π stacked complexes between π-electron rich pyrenyl residues and π-electron deficient polydiimide residues. Solution studies in the mixed solvent chloroform–hexafluoroisopropanol (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) show that mixing the three components (each of which is soluble in isolation), results in the precipitation of a supramolecular, polymer nanocomposite network. The precipitate thus formed can be re-dissolved on heating, with the thermoreversible dissolution/precipitation procedure repeatable over at least 5 cycles. Robust, self-supporting composite films containing up to 15 wt% P-AuNPs could be cast from 2,2,2-trichloroethanol. Addition of as little as 1.25 wt% P-AuNPs resulted in significantly enhanced mechanical properties compared to the supramolecular blend without nanoparticles. The nanocomposites showed a linear increase in both tensile moduli and ultimate tensile strength with increasing P-AuNP content. All compositions up to 10 wt% P-AuNPs exhibited essentially quantitative healing efficiencies. Control experiments on an analogous nanocomposite material containing dodecylamine-functionalized AuNPs (5 wt%) exhibited a tensile modulus approximately half that of the corresponding nanocomposite that incorporated 5 wt% pyrene functionalized-AuNPs, clearly demonstrating the importance of the designed interactions between the gold filler and the supramolecular polymer matrix.
:1, v/v) show that mixing the three components (each of which is soluble in isolation), results in the precipitation of a supramolecular, polymer nanocomposite network. The precipitate thus formed can be re-dissolved on heating, with the thermoreversible dissolution/precipitation procedure repeatable over at least 5 cycles. Robust, self-supporting composite films containing up to 15 wt% P-AuNPs could be cast from 2,2,2-trichloroethanol. Addition of as little as 1.25 wt% P-AuNPs resulted in significantly enhanced mechanical properties compared to the supramolecular blend without nanoparticles. The nanocomposites showed a linear increase in both tensile moduli and ultimate tensile strength with increasing P-AuNP content. All compositions up to 10 wt% P-AuNPs exhibited essentially quantitative healing efficiencies. Control experiments on an analogous nanocomposite material containing dodecylamine-functionalized AuNPs (5 wt%) exhibited a tensile modulus approximately half that of the corresponding nanocomposite that incorporated 5 wt% pyrene functionalized-AuNPs, clearly demonstrating the importance of the designed interactions between the gold filler and the supramolecular polymer matrix.
1 Introduction
Healable polymeric materials have received increasing interest in recent years with the ultimate aim of improving the resilience of safety-critical components.1–12 Two distinct classes of materials have been developed – autonomously healing systems that repair in response to the fracture damage itself13–16 and healable materials that require an external intervention to promote healing.17–20 Various stimuli have been demonstrated to provide the energy required to promote healing including pressure,21,22 light23–27 and heat.7,17,28–35An expanding field of healable materials research has targeted the use of reversible supramolecular interactions36–39 such as hydrogen bonding,13 metal–ligand interactions23 and π–π stacking to produce healable materials.32 We have, for example, recently developed a supramolecular healable polymer blend based on electronically complementary interactions between a low molecular weight polyamide, end-capped with π-electron rich pyrene groups, and a linear polymer with multiple π-electron deficient diimide residues.31 Small-molecule spectroscopic studies have shown that pairs of diimide residues are held in a well-defined chain-fold40–46 around the pyrenyl residue during complexation (Fig. 1).47
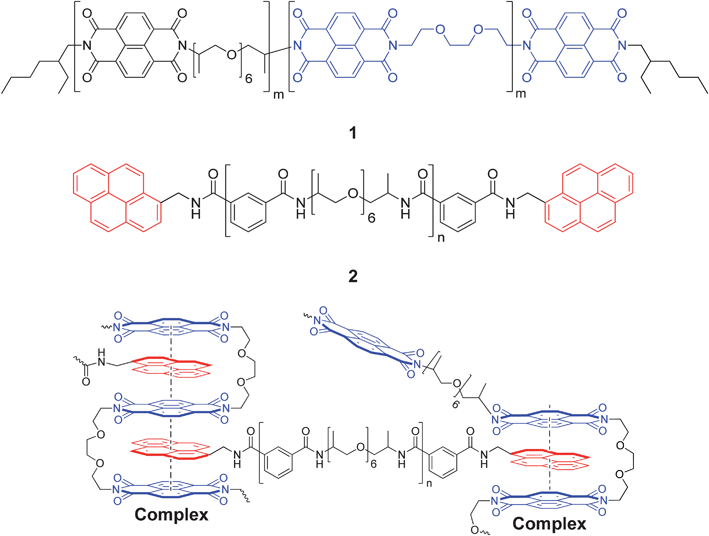 | ||
| Fig. 1 Structures of the chain-folding polydiimide 1 and the pyrenyl end-capped polymer 2. A schematic of the chain-folding π–π stacking interaction47 between the bisdiimide units in 1 and pyrenyl end-groups of 2 as part of a small section of the proposed29 supramolecular polymer network. | ||
One of the major deficiencies of many supramolecular materials is that, in order to achieve healing at accessible temperatures and within usable timeframes, the materials are frequently elastomeric in nature with glass transition temperatures (Tg) well below ambient.7 These properties preclude the use of most supramolecular materials as structural components in high-value engineering applications such as in the aerospace industry.48,49
Inspired by the success of polymer-composite materials in replacing metals across a range of high value products, we have investigated the possibility of producing nanoparticle-reinforced, supramolecular, healable composites.21,22 In collaboration with MacKay, Rowan and co-workers, we have previously reported supramolecular nanocomposites containing cellulose nanocrystals showing tensile moduli up to 20 times greater than the supramolecular matrix polymer, whilst still retaining good healing characteristics.50 In an effort to generate healable materials with the functionality exhibited by metal nanoparticles,51–53 we have also developed a supramolecular nanocomposite containing pyrene-functionalized gold nanoparticles (P-AuNPs) and a complementary chain-folding polydiimide. However, disappointingly this non-covalently cross-linked network proved to be brittle in nature and did not show thermal-healing characteristics.54 In a related study, Tee et al. have recently reported a healable material comprising nickel microparticles embedded within an hydrogen bonded polymer matrix. In this study, the metal filler facilitated electrical conduction, suggesting the possibility of a new route to providing the energy needed for healing damage to the bulk material.55 In addition, Guan et al. have reported recently the synthesis and healing ability of multiphase polymer systems which are conceptually related to this study.56–58
In the present work we demonstrate that addition of the P-AuNP filler to a supramolecular polymer blend of the electronically complementary polymers 1 and 2 (Scheme 1) results in a doubling of the strength of these materials, whilst retaining attractive thermally healable characteristics. In comparison to our previous study on supramolecular gold nanocomposites,54 the new material incorporates the pyrene end-capped polyamide 2 which, in the light of its low Tg value (−7 °C)29 was predicted to afford malleable self-supporting films with potential healable characteristics. We proposed that healing would be achieved through thermally activated disassociation of the π–π stacking interactions between polymers 1, 2 and pyrenyl ligands of the gold nano-particles – the supramolecular interactions can then reform upon cooling, regenerating the strength of the material.
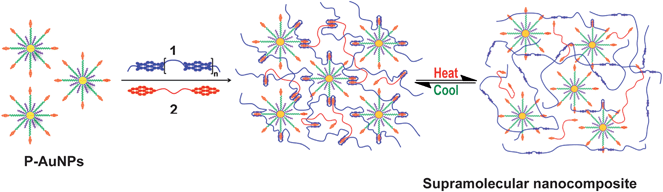 | ||
| Scheme 1 Schematic representation of the formation/dissociation of a supramolecular nanocomposite from chain-folding polymer 1, pyrene end-capped polymer 2 and the incorporation of a pyrene functionalized AuNP (P-AuNP) filler. The supramolecular nanocomposite network can be disrupted upon heating (ca. 60 °C) and interestingly this can be completely re-formed upon cooling to ambient temperature. | ||
2 Experimental
The chain-folding polydiimide (1), pyrenyl end-capped polymer (2) and pyrenethiol/octanethiol functionalized AuNPs (P-AuNPs) were synthesized as reported previously.29,542.1 Instrumental analyses
Ultraviolet-visible spectra were acquired using a Varian Cary 300 spectrometer. For heating–cooling cycle studies, the polymer solution was heated from 20 to 60 °C at ∼25 °C min−1, and then allowed to cool from 60 to 20 °C over 24 hours. Differential scanning calorimetry (DSC) was conducted using a TA-Q2000 instrument. Samples were heated to 170 °C to remove any residual solvents, cooled, and then re-scanned from −20 to 325 °C. Energy-dispersive X-ray spectroscopy (EDX) analysis was carried out on a Cambridge SEM360 instrument operating at 20 kV and 10−5 torr, with a working distance of 24 mm. The EDX images were analysed by ImageJ software to count the number of gold rich areas. Variable temperature healing studies using light microscopy were carried with a LEICA DM2500M instrument at a heating rate of 5 °C min−1.Tensile testing was carried out on TA.XT.Plus texture analyser (from Stable Micro Systems) using a tensile grips probe. Polymer nanocomposite films were cut into strips with dimensions 30.0 × 3.5 mm. Four repeat measurements per sample were carried out, at an extension rate of 0.1 mm s−1. Tensile moduli were calculated as the stress–strain gradient between 0.5 and 1.0% strain (linear region).
2.2 Polymer nanocomposite film preparation
Composite films were cast from solutions in 2,2,2-trichloroethanol. The films were dried by a stepwise process: first at ambient atmosphere for 24 hours, then in a vacuum oven at 50 °C for 24 hours, and finally maintained at 80 °C under vacuum for a further 24 hours.2.3 Healing studies
Polymer composite strips were bisected crosswise using a razor blade and then the edges were overlapped by ∼5 mm and gently pressed together. The samples were then placed onto a PTFE plate and transferred into an oven, pre-heated to 50 °C, for 10 minutes. The samples were then removed from the oven and cooled to room temperature before being subjected to tensile testing.3 Results and discussion
3.1 Complexation studies
As shown in Scheme 1, the target material was designed to incorporate three components: the chain-folding polymer 1, the pyrenyl end-capped polyamide (2) and the P-AuNPs 3. Prior to the healing studies it was necessary to verify that mixing the presence of the nanoparticles did not adversely affect the inter-polymer π–π stacking interactions that were previously studied in detail.47 This was achieved by investigating the behaviour of the three materials in solution as single components and as blends.Fig. 2 shows five vials, three of which contain solutions of either (A) polydiimide 1, (B) pyrenyl end-capped polymer 2, or (C) P-AuNPs 3. It can be seen that they all exhibit distinct colours, especially the P-AuNPs that appear dark red-brown as a consequence of their characteristic surface plasmon resonance absorption. As seen previously,29 mixing polymers 1 and 2 results in the formation of supramolecular polymer network that precipitates from solution (vial D). The red colour of the network is as a consequence of a new absorption band at ∼520 nm which is characteristic of an aromatic, π–π stacked, charge-transfer complex.47,59 Crucially, mixing the solutions of polymers 1, 2 and P-AuNPs also results in a reddish brown precipitate and essentially colourless supernatant (vial E). This demonstrates that the P-AuNPs are being incorporated into the supramolecular cross-linked network and thus removed from solution.
 | ||
| Fig. 2 (A) Polymer 1 (1 mg mL−1); (B) polymer 2 (3 mg mL−1); (C) P-AuNPs (0.125 mg mL−1); (D) polymer 1 (1 mg mL−1) + polymer 2 (3 mg mL−1) after 24 hours; (E) polymer 1 (1 mg mL−1) + polymer 2 (3 mg mL−1) + P-AuNPs (0.125 mg mL−1) after 24 hours. All of these solutions were prepared in CHCl3–HFIP (6:1, v/v). | ||
To investigate the thermo-reversible nature of the supramolecular interactions within the novel nanocomposite, the three-component vial (E) was heated to ∼60 °C, whereupon the heterogeneous system rapidly became a homogeneous solution. Over the following 24 h, the supramolecular nanocomposite re-formed and re-precipitated, leaving again an almost colourless supernatant. The process could be followed by monitoring the intensity of the plasmon resonance absorption band of the P-AuNPs at 513 nm, which was observed to reduce in intensity by approximately 90% from its maximum value on cooling from 60 °C to ambient temperature, indicating that a high proportion of the P-AuNPs had been incorporated in the precipitated nanocomposite.
The rate of precipitation of the polymer blend (1+2) and the nanocomposite (1+2+3) could be followed by measuring the change in the absorbance profile of the solution as a function of time. These data are plotted in Fig. 3 [see also ESI, Fig. S1,‡ for raw data] which shows that the absorbance intensity of the solution at 513 nm drops to around 20% of its initial value after 1600 minutes (ca. 27 h). It is striking that precipitation of the polymer blend containing the P-AuNPs is much more rapid than that observed for the simple polymer blend (1+2). For example, after 60 min, the absorbance of the sample containing P-AuNPs had fallen by over 60%, compared to a reduction of only 20% for the sample containing only polymers 1 and 2. This observation may be accounted for on the basis that the P-AuNPs provide multiple additional crosslinking points, resulting in more rapid formation of an insoluble material.
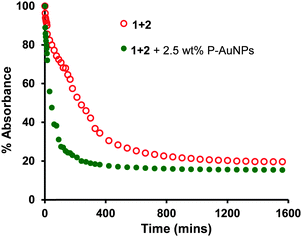 | ||
| Fig. 3 UV/visible spectroscopic analysis of supramolecular nanocomposite precipitation as a function of time. Absorbance change over 27 hours at 521 nm for polymer composite (polymer 1 (1 mg mL−1) + polymer 2 (3 mg mL−1)) precipitation (red); absorbance change over 27 hours at 513 nm for polymer nanocomposite (polymer 1 (1 mg mL−1) + polymer 2 (3 mg mL−1) with 2.5 wt% P-AuNPs) precipitation (green). All the solutions were prepared in CHCl3–HFIP (6:1, v/v) and measurements carried at 25 °C. | ||
In order to produce a thermally healable supramolecular polymer system it is important that the non-covalent interactions are reversible over an appropriate temperature range. Heating the sample containing the precipitated polymer nanocomposite to 60 °C resulted in an almost instantaneous generation of the reddish brown solution. This precipitation/dissolution procedure could be maintained over at least 5 heating and cooling cycles (Fig. 4 and ESI Fig.S2‡ for raw data) demonstrating that the system can be switched repeatedly between the bound and ‘free’ states without loss of the supramolecular interactions.
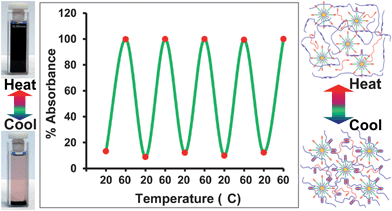 | ||
| Fig. 4 UV-visible analysis of supramolecular nanocomposite precipitation and dissolution during thermal cycling experiment. Polymer 1 (1 mg mL−1) + polymer 2 (3 mg mL−1) mixed with 2.5 wt% P-AuNPs. Absorbance change at 513 nm was measured over 5 heat–cool cycles (at 20 °C UV absorbance = 0.3777 ± 0.0302 (95% confidence interval quoted, σ = 0.0544, n = 5) and at 60 °C, 3.3374 ± 0.004 (95% confidence interval quoted, σ = 0.0072, n = 5)). The photographs (left) show the precipitation and dissolution at 20 and 60 °C, respectively. The schematic (right) indicates how the supramolecular polymer network forms and dissociates at 20 and 60 °C, respectively. | ||
3.2 Polymer nanocomposite films – casting and properties
We have observed previously54 that films cast from a chain-folding polymer with a similar structure to 1 and P-AuNPs are extremely brittle, and could not be subjected to stress–strain testing to study healable characteristics. However, films cast from (1+2) and varying quantities of P-AuNPs (up to 15 wt%) proved flexible, free standing and tough, demonstrating the importance of each of the components in the polymer blend (Fig. 5A for 10 wt% and ESI Fig. S3‡ for 0 wt% nanocomposite film).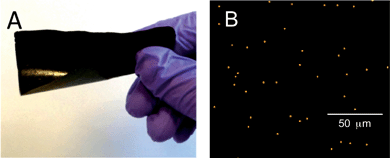 | ||
| Fig. 5 (A) Supramolecular polymer nanocomposite film formed from (1+2) containing 10 wt% P-AuNPs; (B) elemental mapping of a polymer nanocomposite film containing 1.25 wt% P-AuNPs, using EDX analysis. The film was solution cast as described in the Experimental section. Gold dots show the nanoparticles dispersed throughout the polymer film. | ||
The 100 μm thick films were studied by energy-dispersive X-ray spectroscopy (EDX) analysis (see ESI Fig. S4a–c‡ for all the nanocomposite films) which allows the gold rich areas of the surface of the film to be visualised. For example, the composite film containing 1.25 wt% P-AuNPs showed that the film contained a relatively homogeneous dispersion of gold (Fig. 5B, 1400 counts per mm2), and that the casting procedure did not result in significant clustering of P-AuNPs that50 would lead to area of the films possessing higher and lower densities of P-AuNPs and therefore inhomogeneous film properties. For the samples containing the 10 and 15 wt% P-AuNPs the surface gold count observed during EDX analysis dropped from 2200 to 1000 counts per mm2 suggesting that significant clustering of P-AuNPs was occuring at these concentrations.
Studying the thermal properties of the nanocomposite materials also provided insight into the dispersion of P-AuNPs. A DSC thermogram for the nanocomposite containing 1.25 wt% P-AuNPs is shown in Fig. 6, together with thermograms for the individual polymers 1, 2 and for the P-AuNPs themselves. It can be seen that the chain-folding polyimide 1 exhibits a broad melting endotherm at ca. 275 °C, and that the pyrene end-capped polymer 2 has a Tg close to ambient temperature. Analysis of the P-AuNPs gave a thermogram with a sharp exothermic peak at 235 °C, which is generally attributed to a sintering process which occurs as a consequence of energy minimization through coalescence of the AuNPs.54,60,61 Above the sintering temperature, a broad exotherm was evident, consistent with desorption of the octanethiol ligands from the AuNPs surface. In contrast to the individual components, the nanocomposite blend exhibits no really significant thermal transition over the temperature range studied (data for the composite containing 1.25 wt% P-AuNPs in Fig. 6 – for other compositions see ESI Fig. S5‡). In particular, there is no evidence for sintering of the P-AuNPs in these studies, indicating that the gold nanoparticle are relatively site-isolated, and therefore not able to undergo coalescence, in agreement with the EDX map shown in Fig. 5B.
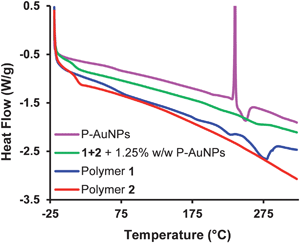 | ||
| Fig. 6 DSC analysis (second heating cycle) of polymers 1 (blue) and 2 (magenta), and the polymer nanocomposite prepared from 1.25 wt% P-AuNPs (green) and P-AuNPs (red). | ||
The dramatic change in thermal properties of the composite polymers when compared to the starting components clearly demonstrates that the supramolecular interactions observed in solution are maintained in the solid films.
Nanocomposite films were subjected to tensile testing to investigate the effect of added P-AuNPs on the stiffness and healing characteristics of the material. Fig. 7 shows stress–strain data for pristine samples of polymer nanocomposites containing up to 15 wt% P-AuNPs. Films were cast from 2,2,2-trichloroethanol and were cut into strips approximately 30 × 3.5 mm and 100 μm thick. It can be seen that both the tensile modulus (the initial slope) and the yield-stress (the maximum on the stress–strain curve) increase continuously with the proportion of P-AuNPs incorporated into the films. The films that contain between 5 and 15 wt% P-AuNPs all failed at a strain of approximately 18%. Furthermore, a control sample that included 5 wt% dodecylamine-stabilized AuNPs (i.e. AuNPs that did not contain the pyrenyl ligand – see ESI S6‡ for experimental/preparation and ESI Fig. S7‡ for comparative DSC thermograms between composites featuring the two types of AuNPs),54 was clearly the weakest film, demonstrating the need for the presence of pyrenyl residues on the AuNPs, to bind to the matrix and so impart enhanced mechanical properties to these nanocomposite materials.
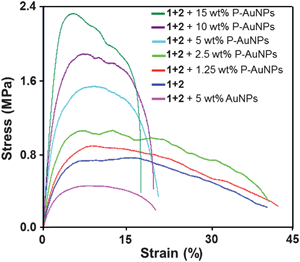 | ||
| Fig. 7 Stress–strain curves for composite films containing gold nanoparticle fillers. | ||
3.3 Composite healing studies
In an initial study of the healing potential of these nanocomposite films, a sample containing 1.25 wt% P-AuNPs was pierced with a razor blade and the damaged film analysed by hot-stage microscopy (Fig. 8). At the beginning of the experiment, a hole is apparent as a white area caused by backlighting of the red film. As the film was heated from ambient to 75 °C the hole diminished progressively in size, eventually vanishing altogether.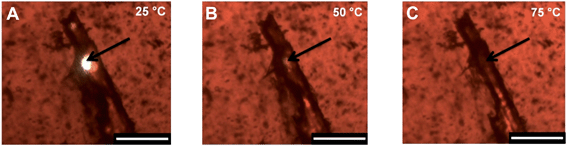 | ||
| Fig. 8 Optical microscopic images of the damaged area of a polymer nanocomposite film containing 1.25 wt% of the P-AuNPs: (A) 25 °C; (B) 50 °C; (C) 75 °C. Heating rate 5 °C min−1. Scale bar 300 μm. Sample thickness 120 μm. | ||
More quantitative healing data was acquired using established break/heal test protocols,28,29 rather than by visual analysis. During these tests, stress–strain data was generated for samples that were first cut into two sections, their damaged edges overlapped and then heated at 50 °C for 10 min. The mechanical properties of the healed films were then compared to those of the pristine samples.28,29,32 Tensile (Young's) moduli for each sample, plotted as a function of increasing P-AuNPs concentration for both pristine and healed samples, are shown in Fig. 9. All of the samples showed very good healing characteristics, except for the material containing the highest loading of P-AuNPs (15 wt%). This result may be attributed to clustering of the P-AuPNs, as observed during EDX analysis, inhibiting movement of the polymers during healing.
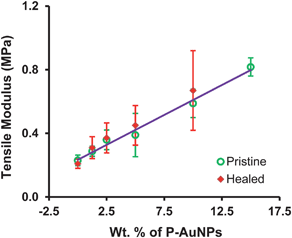 | ||
| Fig. 9 Comparison of tensile moduli of pristine and healed polymer nanocomposites. The line of best fit (r2 = 0.99) includes only data for the pristine samples (error bars shown 1 standard deviation from a mean of 4 repeat measurements). | ||
The strength of the composite materials increased linearly with the P-AuNP filler loading (r2 = 0.99). This result contrasts with our findings for healable nanocomposites containing high aspect-ratio cellulose nanocrystal (CNC) fillers.50 In these materials the strength of the sample increased with the log of the filler content, in accordance with percolation-theory models. In such materials the strength and stiffness of the composite is derived from stress-transfer across the macroscopic test sample by a continuous mesh of cellulose nanocrystals.
Analysis of the tensile moduli for the healed samples showed an excellent correlation with the data for the pristine samples. Indeed, the mean healing efficiency (calculated as the ratio of the tensile moduli of the pristine and healed samples) determined to be ca. 108%. An increase in modulus after healing has been observed previously50 and may be attributed to small changes in the dimensions of the samples or to the thermal treatment resulting in a new, mechanically stronger arrangement of the supramolecular network.
To further demonstrate healing in these samples, the ultimate tensile strength (UTS) of the pristine and healed samples were compared. These data are plotted in Fig. 10 and show a linear increase in UTS as a function of increasing P-AuNPs incorporation (r2 = 0.99), i.e. exactly the same trend as is observed for the tensile moduli. In all cases where healing could be measured, the UTS of the healed sample was within the error of the measurements for the UTS of the pristine sample with the same loading level of P-AuNPs.
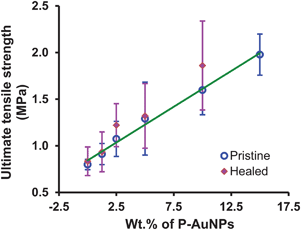 | ||
| Fig. 10 Plot of the ultimate tensile strength of pristine and healed polymer nanocomposites as a function of P-AuNPs loading. The line of best fit includes only data for the pristine samples (r2 = 0.99). Error bars shown one standard deviation from the mean of 4 repeat measurements. | ||
As a final demonstration of the importance of the new design element in this system, i.e. the use of pyrene-functionalized AuNPs that enhance the cross-linking density within the polymer blend, a control experiment was carried out whereby a composite contained 5 wt% dodecylamine-functionalized AuNPs was subjected to the same break/heal tests as described above. It can be seen from Fig. 11 that, although healing was observed, the material containing AuNPs that lacked pyrenyl ligands had a tensile modulus approximately half that measured for the P-AuNPs–composite system. For comparison: this control sample exhibited essentially the same tensile modulus supramolecular polymer blend without P-AuNPs (0 wt% nanocomposite film) (Fig. 11). These results demonstrate that the π–π-stacking interactions between polymers 1, 2 and P-AuNPs contribute significantly to the mechanical properties of these composite materials.
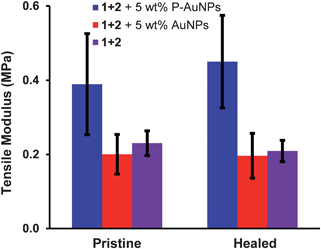 | ||
| Fig. 11 Control experiment: comparison of tensile modulus of pristine and healed polymer nanocomposites prepared from 5 wt% P-AuNPs (blue), 5 wt% dodecylamine-stabilized AuNPs (red) and 0 wt% nanocomposite (purple) (error bars are one standard deviation from the mean of 4 repeat measurements). | ||
4 Conclusions
A new healable supramolecular nanocomposite polymer system has been designed and synthesised. The material contains a pyrenyl-endcapped polyamide which interacts through π–π stacking interactions with a chain-folding polydiimide. Introduction of pyrenyl functionalized AuNPs (P-AuNPs) into this polymer blend results in a healable polymer-composite system. Complexation studies in solution demonstrate that the introduction of P-AuNPs results in more rapid formation of an insoluble supramolecular network when compared to control samples that did not contain the P-AuNPs. Films of the nanocomposite are tough and flexible, and contain a relatively homogeneous dispersion of P-AuNPs as shown by EDX mapping and DSC analysis. Films containing P-AuNPs are stronger and stiffer than those cast from the same polymers but without P-AuNPs, and also than films containing AuNPs that lacked the pyrenyl motif. Healing studies using a classic break/heal test, followed by stress–strain analysis, showed – remarkably – that materials containing up to 10 wt% P-AuNPs can even exhibit healing efficiencies of more than 100%.Acknowledgements
We thank the University of Reading for a studentship in support of RV, and EPRSC for support of BWG (grants EP/G026203/1 and EP/D07434711). The authors are grateful to Dr Vitaliy V. Khutoryanskiy for providing access to mechanical testing facilities in his research laboratory. We also thank Dr Saeed Mohan from the Department of Chemistry, University of Reading for assistance with EDX analysis. Spectroscopic and thermal data were acquired using equipment based in the Chemical Analysis Facility (CAF) of the University of Reading.References
- Self Healing Materials: an Alternative approach to 20 centuries of materials science, ed. S van der Zwaag, Springer, Dordrecht, 2007 Search PubMed.
- Self Healing Materials: Fundamentals, Design Strategies and Applications, ed. S. K. Ghosh, J. W. Wiley and sons Ltd, Weinheim, 2009 Search PubMed.
- Adaptive Structures: Engineering Applications, ed. D. Wagg, I. Bond, P. Weaver and M. Friswell, J. W. Wiley and sons Ltd, Chichester, 2007 Search PubMed.
- Healable Polymer Systems, ed. W. Hayes and B. W. Greenland, RSC, Cambridge, 2013 Search PubMed.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- Y. C. Yuan, T. Yin, M. Z. Rong and M. Q. Zhang, eXPRESS Polym. Lett., 2008, 2, 238–250 Search PubMed.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- T. C. Mauldin and M. R. Kessler, Int. Mater. Rev., 2010, 55, 317–346 Search PubMed.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- M. W. Urban, Nat. Chem., 2012, 4, 80–82 CrossRef CAS.
- A. Ciferri, Polym. Chem., 2013 10.1039/c3py21156h.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, in Annual Review of Materials Research, ed. D. R. Clarke, M. Ruhle and F. Zok, 2010, vol. 40, pp. 179–211 Search PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J. M. Lehn, Chem.–Eur. J., 2009, 15, 1893–1900 CrossRef CAS.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS.
- Z. S. Kean and S. L. Craig, Polymer, 2012, 53, 1035–1048 CrossRef CAS.
- K. Haraguchi, K. Uyama and H. Tanimoto, Macromol. Rapid Commun., 2011, 32, 1253–1258 CrossRef CAS.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 Search PubMed.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. G. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264 RSC.
- S. Burattini, B. W. Greenland, W. Hayes, M. E. Mackay, S. J. Rowan and H. M. Colquhoun, Chem. Mater., 2011, 23, 6–8 CrossRef CAS.
- X. X. Chen, F. Wudl, A. K. Mal, H. B. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- Y. L. Liu and C. Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 Search PubMed.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013 10.1039/c3py00081h.
- Z. Zhu, C. J. Cardin, Y. Gan, C. A. Murray, A. J. P. White, D. J. Williams and H. M. Colquhoun, J. Am. Chem. Soc., 2011, 133, 19442–19447 CrossRef CAS.
- Z. Zhu, C. J. Cardin, Y. Gan and H. M. Colquhoun, Nat. Chem., 2010, 2, 653–660 Search PubMed.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, M. G. B. Drew and Y. Gan, Faraday Discuss., 2009, 143, 205–220 RSC.
- H. M. Colquhoun, Z. Zhu, C. J. Cardin, Y. Gan and M. G. B. Drew, J. Am. Chem. Soc., 2007, 129, 16163–16174 CrossRef CAS.
- H. M. Colquhoun and Z. X. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040–5045 CrossRef CAS.
- H. M. Colquhoun, Z. X. Zhu, C. J. Cardin and Y. Gan, Chem. Commun., 2004, 2650–2652 RSC.
- B. W. Greenland, M. B. Bird, S. Burattini, R. Cramer, R. K. O'Reilly, J. P. Patterson, W. Hayes, C. J. Cardin and H. M. Colquhoun, Chem. Commun., 2013, 49, 454–456 RSC.
- B. W. Greenland, S. Burattini, W. Hayes and H. M. Colquhoun, Tetrahedron, 2008, 64, 8346–8354 CrossRef CAS.
- J. W. C. Pang and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791–1799 CrossRef CAS.
- R. S. Trask and I. P. Bond, Smart Mater. Struct., 2006, 15, 704–710 CrossRef CAS.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS.
- R. Vaiyapuri, B. W. Greenland, S. J. Rowan, H. M. Colquhoun, J. M. Elliott and W. Hayes, Macromolecules, 2012, 45, 5567–5574 Search PubMed.
- B. C. K. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832 Search PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 Search PubMed.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561–10565 Search PubMed.
- Y.-X. Lu and Z. Guan, J. Am. Chem. Soc., 2012, 134, 14226–14231 Search PubMed.
- R. Foster, in Organic Charge Transfer Complexes, Academic Press Inc, London, 1969, pp. 33–93 Search PubMed.
- M. J. Coutts, M. B. Cortie, M. J. Ford and A. M. McDonagh, J. Phys. Chem. B, 2009, 113, 1325–1328 Search PubMed.
- Y. L. Wu, Y. N. Li, P. Liu, S. Gardner and B. S. Ong, Chem. Mater., 2006, 18, 4627–4632 CrossRef CAS.
Footnotes |
| † Submitted to the themed issue on self-healing polymers. |
| ‡ Electronic supplementary information (ESI) available: Details of the UV-visible study of supramolecular polymer nanocomposite precipitation over 27 hours. Experimental data for the UV-visible analysis of nanocomposite cooling and heating cycles. Photographs of the film cast from the blend of 1+2 (1:3, w/w ratio). EDX analyses of all the nanocomposites films. DSC analyses of polymer nanocomposites prepared from 0–15 wt% P-AuNPs. Preparation of dodecylamine stabilized AuNPs. Comparison of DSC thermogram of polymer nanocomposites prepared from 5 wt% P-AuNPs and 5 wt% dodecylamine stabilized AuNPs. See DOI: 10.1039/c3py00086a |
| This journal is © The Royal Society of Chemistry 2013 |