Synthesis of a rod–coil block copolymer incorporating PCBM†
Erika
Bicciocchi
ab,
Ming
Chen
ab,
Ezio
Rizzardo
ab and
Kenneth P.
Ghiggino
*a
aSchool of Chemistry, The University of Melbourne, Victoria 3010, Australia. E-mail: ghiggino@unimelb.edu.au
bCSIRO Materials Science and Engineering, Clayton, Victoria 3168, Australia
First published on 21st August 2012
Abstract
A widely applicable synthetic procedure is demonstrated for an electro-active rod–coil block copolymer that incorporates an electron donating conjugated block with pendant electron accepting fullerene units. The copolymer exhibits enhanced thermal stability compared to a blend of the components.
The performance of an organic photovoltaic (OPV) cell depends on the properties of the active layer, which usually consists of a blend of electron donor and acceptor materials.1,2 The donor (D) is often a π-conjugated polymer3–5 that also acts as the light absorbing medium, while the acceptor (A) is commonly a soluble fullerene derivative [6,6]-phenyl-C61-butyric acid methyl ester (PCBM).6–8 Since both donor and acceptor materials tend to phase separate on a macroscopic scale, the control of the morphology of this active layer is of utmost importance.9,10 Recently, studies have focused on developing rod–coil block copolymers that can self-assemble in well defined nano-domains in order to stabilize polymer blends.11,12 However their potential use in modifying OPV material morphology has, apart from a few examples, been mainly neglected.13–15 These examples are mainly based on specific synthesis protocols such as Siegrist polycondensation and Grignard methathesis polymerization that are not applicable to the current state of the art high performance polymers typically made by Suzuki and Stille polycondensation.
We describe here a synthetic approach for D–A block copolymers, where the donor and acceptor blocks are made from a conjugated polymer and a coil macromolecule with an attached C60-pendant respectively (scheme shown in Fig. SI 1†). Suzuki or Stille coupling reactions can be used for the synthesis of the conjugated rigid block component. For the coil block, reversible addition–fragmentation chain transfer (RAFT) polymerization was employed because it can be used for a large variety of monomers and avoids the use of a transition metal catalyst.16 After RAFT polymerization, most polymer chains will have the thiocarbonylthio and R′ groups derived from the RAFT agent as terminal groups (Fig. 1). If a reactive functionality, such as phenyl bromide, is introduced into the R′ group, the resulting polymer can be used as a mono-functional capping agent for the cross-coupling polymerizations. By using unsaturated monomers with pendant functional groups, fullerene based electron acceptors can be introduced.
 | ||
| Fig. 1 The overall process of RAFT polymerisation. Pn is the polymer chain. | ||
A combination of Suzuki and RAFT techniques was chosen for the synthesis of the D–A block copolymers. As a proof of concept, poly(9,9-dihexylfluorene-alt-bithiophene) (F6T2) was chosen as a model system for the conjugated polymer block. The monomers are commercially available and the polymer has been well characterized.17 The synthetic method adopted for the rod–coil block copolymer is outlined in Scheme 1.
 | ||
| Scheme 1 Synthetic procedure for the rod–coil block copolymer. | ||
The coil polymer was obtained by copolymerization of styrene and vinylbenzyl chloride under the control of a trithiocarbonate RAFT-agent 3. As previously reported,18,19 the trithiocarbonate functionality can be readily cleaved by the presence of mild nucleophiles such as primary or secondary amines. Thermolysis and chemical removal techniques have been reported.19,20 We applied the latter method which uses N-ethylpiperidine hypophosphite and a radical initiator as described elsewhere.19 The RAFT end group removal did not affect the polymer chain length as shown by the identical gel permeation chromatography (GPC) elution curves before and after the procedure (Fig. SI 2A†). Comparison of the UV absorption spectrum of polymers 4 and 5 confirmed the complete removal of the trithiocarbonate end group (Fig. SI 2B†). After removing the RAFT end group the benzyl chloride moieties were converted to the corresponding phthalimide groups (6) that were subsequently hydrolyzed following the Ing–Manske procedure, leading to the polymer 7 that contains free amine groups.21,22
In order to use the coil polymer as the end-capping agent in a Suzuki polycondensation and to avoid interaction with the catalyst, the free amine groups were protected with di-tert-butyldicarbonate (DIBOC) resulting in 8. The DIBOC protection allows ready cleavage after polycondensation to restore the previous amine functionality.17,23 The possible origins of a small shoulder in the chromatogram for polymer 8 (see Fig. 2A) are discussed in the ESI.†
 | ||
| Fig. 2 (A) GPC elution profiles of polymer 8, F6T2 and 9; (B) UV absorption spectra of polymer 6, F6T2 and 9 recorded at 27, 25, 24 min elution respectively (normalized at the conjugated polymer absorption spectrum maximum). | ||
The conjugated rod block was synthesized as reported elsewhere using a Suzuki coupling reaction from 5,5′-dibromo-2,2′ bithiophene and 9,9-dihexylfluorene2,7-diboronic acid bis(1,3 propanediol)ester.24 The presence of a small excess of diboronic ester enabled the presence of terminal boronic ester end-groups,25 which was confirmed by 1H NMR of a model reaction (Fig. SI 3†) and were subsequently end capped with the coil polymer 8 to obtain the rod coil block copolymer 9 (Scheme 1).
Purification of 9 was carried out by repeated washing using acetone in order to remove unreacted coil polymer. The presence of the linkage between the rod and coil sections was verified through GPC-PDA measurements (Fig. 2) in combination with 1H-NMR (Fig. SI 4†).
The chromatogram in Fig. 2A shows a shift of the GPC traces to a lower retention time as a result of an increase in the molecular weight of the rod–coil block copolymer compared to its coil and conjugated blocks. The observation that the elution profile of the rod–coil block copolymer 9 is mono-modal in shape and does not exhibit a shoulder around 27 min suggests that any unreacted coil polymer has been efficiently removed. The linking of the rod and coil blocks can be confirmed by the UV absorption spectrum where, in addition to the absorption of the coil block at 230 nm, the emergence of a new peak at 450 nm corresponding to the conjugated polymer block can be observed (Fig. 2B). In addition to the UV absorption spectra, the 1H-NMR (Fig. SI 4†) shows peaks corresponding to both polymer fragments (e.g. 0.84–0.45 ppm CH2–CH3 of the alkyl chain of F6T2 and 4.30–4.05 ppm the CH2–NHCOO of the coil component).
After Suzuki-coupling, the deprotection of the amino group was carried out using trifluoroacetic acid. The polymer obtained was reacted with a small excess of the acid chloride of PCBM (PCBM-Cl), synthesized as reported in the literature,8 leading to the product 11.
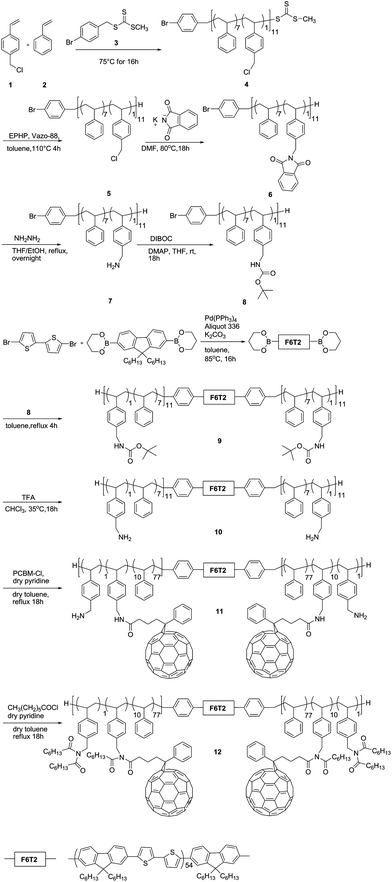
Due to its poor solubility the purification of the resulting polymer proved to be difficult, likely due to the presence of some unreacted amino groups (around 10% of functional groups as detected by 1H-NMR). An additional reaction was conducted using heptanoyl chloride to ensure the complete conversion of the amino groups. Indeed, the solubility dramatically improved and 1H-NMR showed the complete disappearance of the peak at 3.74 ppm attributed to CH2–NH2 and the appearance of a new peak at 4.8 ppm attributed to CH2–NCO. GPC-PDA analysis further confirmed the linkage between the rod and the coil blocks and the successful incorporation of PCBM. The chromatogram shows a shift of the GPC traces to a lower retention time as a result of an increase in the molecular weight of the rod–coil block copolymer compared to the copolymer without functionalisation (Fig. 3A). Additionally, overlying and normalizing the UV absorption spectra of 9 with 12, reveals additional absorption at wavelengths below 370 nm attributable to the presence of PCBM in 12 (Fig. 3B). The amount of PCBM incorporated was determined by 1H NMR to be 90% of the available amino groups.
 | ||
| Fig. 3 (A) chromatograms of polymer 9 and 12; (B) UV absorption spectra of room temperature THF solutions of rod–coil block copolymer 9 (1.13 × 10−3 mg mL−1), 12 (1.11 × 10−2 mg mL−1) and PCBM (4.3 × 10−3 mg mL−1). | ||
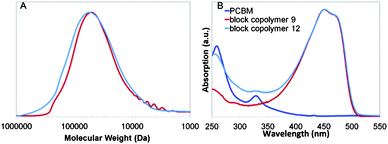
A further confirmation of the successful synthesis of block-copolymer 12 was inferred from fluorescence spectroscopy in solution (Fig. SI 5†) as well as in thin film. In films the presence of the C60 acceptor quenches the fluorescence of the conjugated polymer donor F6T2 attributable to an electron transfer process (Fig. 4). Additionally, fluorescence studies of thin films of rod–coil block copolymer 12 as well as blends of F6T2 with PCBM, were carried out to give insights into the thermodynamic stability. Fig. 4 shows a comparison between the fluorescence spectra of the different films (spin coated 100 nm thickness). At room temperature the fluorescence of F6T2 in the block copolymer and the blend is completely quenched by the fullerene component. However, upon annealing the blend at 150 °C for 2 × 10 min the emission increases, while the fluorescence spectrum of block copolymer 12 is almost unchanged.
 | ||
| Fig. 4 Photoluminescence spectra of thin films cast from chlorobenzene solutions corrected for UV absorption at the excitation wavelength (420 nm) for F6T2, a blend of F6T2 with PCBM, and rod–coil block copolymer 12 at room temperature (RT) and after annealing at 150 °C for 10 min (×2). | ||
The increase in fluorescence intensity exhibited by the blend suggests phase separation of the blend components occurs when the film undergoes thermal treatment. On the contrary, annealed films of block-copolymer 12 exhibit similar (slightly lower) emission, demonstrating their excellent thermodynamic stability. The surface morphology was also characterised by using atomic force microscopy (AFM). A comparison was made between 12 and the blend of F6T2-PCBM. The images (Fig. SI 6†) were obtained at room temperature and after thermal annealing at 150 °C for 2 × 10 min for both samples. The blend at room temperature exhibited higher surface roughness that decreased upon annealing (see Fig. SI 6A and SI 6B†). On the other hand, block copolymer 12 exhibits smoother surface roughness at room temperature and the surface image remains almost unchanged after annealing at 150 °C (see Fig. SI 6C and SI 6D†). These results provide further evidence of the thermodynamic stability of the rod–coil block copolymer compared with the blend.
Conclusions
In summary we have successfully demonstrated a synthetic method for obtaining a donor–acceptor rod–coil block copolymer incorporating a conjugated polymer and PCBM. The polymer exhibits improved thermodynamic stability compared to the component blend suggesting these materials may be useful in modifying the morphology and performance of OPV materials, an aspect currently under investigation.KPG acknowledges the Australian Research Council for support of this research under Discovery Project DP0986166.
References
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef.
- M. C. Scharber, D. Wuhlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. L. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- P. A. Lane and Z. H. Kafafi, Organic Photovoltaics: Mechanisms, Materials, and Devices, CRC Press, Taylor & Francis Group, 2005 Search PubMed.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474 CAS.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS.
- S. A. Backer, K. Sivula, D. F. Kavulak and J. M. J. Frechet, Chem. Mater., 2007, 19, 2927 CrossRef CAS.
- L. M. Popescu, P. van't Hof, A. B. Sieval, H. T. Jonkman and J. C. Hummelen, Appl. Phys. Lett., 2006, 89, 213507 CrossRef.
- J. C. Hummelen, B. W. Knight, F. Lepeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532 CrossRef CAS.
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740 CrossRef CAS.
- C. Groves, O. G. Reid and D. S. Ginger, Acc. Chem. Res., 2010, 43, 612 CrossRef CAS.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217 CrossRef CAS.
- U. Stalmach, B. de Boer, C. Videlot, P. F. van Hutten and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 5464 CrossRef CAS.
- M. H. van der Veen, B. de Boer, U. Stalmach, K. I. van de wetering and G. Hadziioannou, Macromolecules, 2004, 37, 3673 CrossRef CAS.
- C. Yang, J. K. Lee, A. J. Heeger and F. Wudl, J. Mater. Chem., 2009, 19, 5416 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- D. M. Shendage, R. Frohlich and G. Haufe, Org. Lett., 2004, 6, 3675 CrossRef CAS.
- S. Harrisson, Macromolecules, 2009, 42, 897 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446 CrossRef CAS.
- M. Chen, G. Moad and E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6704 CrossRef CAS.
- H. R. Ing and R. H. F. Manske, J. Chem. Soc., 1926, 129, 2348 RSC.
- S. M. Gravano, M. Borden, T. von Werne, E. M. Doerffler, G. Salazar, A. Chen, E. Kisak, J. A. Zasadzinski, T. E. Patten and M. L. Longo, Langmuir, 2002, 18, 1938 CrossRef CAS.
- E. A. Englund, H. N. Gopi and D. H. Appella, Org. Lett., 2004, 6, 213 CrossRef CAS.
- J. Sakamoto, M. Rehahn, G. Wegner and A. D. Schluter, Macromol. Rapid Commun., 2009, 30, 653 CrossRef CAS.
- N. R. Evans, L. S. Devi, C. S. K. Mak, S. E. Watkins, S. I. Pascu, A. Kohler, R. H. Friend, C. K. Williams and A. B. Holmes, J. Am. Chem. Soc., 2006, 128, 6647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental details, GPC, absorption and luminescence spectra of the compounds, AFM. See DOI: 10.1039/c2py20507f |
| This journal is © The Royal Society of Chemistry 2013 |