 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic excitation and structural relaxation of the adenine dinucleotide in gas phase and solution†
Felix
Plasser
*a and
Hans
Lischka
*ab
aInstitute for Theoretical Chemistry, University of Vienna, Währingerstr. 17, 1090 Vienna, Austria
bDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, Texas 79409-1061, USA. E-mail: felix.plasser@univie.ac.at; hans.lischka@ttu.edu
First published on 8th May 2013
Abstract
The excited states and potential surfaces of the adenine dinucleotide are analyzed in gas phase and in solution using a correlated ab initio methodology in a QM/MM framework. In agreement with previous studies, a rather flat S1 surface with a number of minima of different character is found. Specifically, our results suggest that exciplexes with remarkably short intermolecular separation down to ∼2.0 Å are formed. A detailed analysis shows that due to strong orbital interactions their character differs significantly from any states present in the Franck–Condon region. The lowest S1 energy minimum is a ππ* exciplex with only a small amount of charge transfer. It possesses appreciable oscillator strength with a polarization almost perpendicular to the planes of the two adenine molecules.
Introduction
In the last few years, several experiments studying the photophysics of DNA fragments showed a strong difference between the excited state behaviour of nucleobase stacks as opposed to isolated nucleobases or mononucleotides. Whereas all isolated nucleobases decay on a picosecond timescale,1–3 additional transients with a lifetime of 10–100 ps and even nanosecond components were found in single and double stranded DNA oligonucleotides.4–9 Several possible reasons have been put forward to explain this intriguing situation. As one option, processes were considered where the excitation was strictly localized on one DNA base and it was conjectured from quantum chemical computations that the DNA environment could have a significant influence in this case.10 However, in a dynamical study no significant increase in lifetime due to the stacking interaction was found.11 Secondly, hydrogen bonding was considered as a major factor. In this sense a proton-coupled electron transfer between Watson–Crick (WC) paired bases was considered as a possible decay channel.12–14 Furthermore, dynamics simulations emphasized the role of structural effects related to hydrogen bonding.11,15,16 Thirdly, stacking interaction was considered as the main influence on the collective excited state behaviour in DNA. The importance of stacking was highlighted by the observation that single and double stranded DNA oligonucleotides showed very similar transients after photoabsorption, highlighting the relative unimportance of the second strand.4 Experimental signatures for two types of processes due to stacking interactions were found pertaining to (i) electron dynamics leading to excitation energy transfer,8,17,18 and (ii) structural relaxation leading to a trapping of the defects.4,19 The nature of the trapping sites in the second case, which remains a major open question, will be the main focus of this study. These sites were initially assumed to be charge transfer exciplexes.19,20 But also computational evidence for neutral or mixed exciplexes was given.21,22 Aside from their structure, also the fate of such exciplexes is unknown. There was experimental evidence for a decay through charge recombination19 and computational evidence for deactivation mediated by a shortening of the intermolecular distance,23 but also a restoration of localized states has been considered.24 In light of these unknowns, further studies are needed to fully understand this important phenomenon.Having recently analysed the effects of base–base stacking on the absorbing states in DNA,25 it is the aim of this paper to consider structural relaxation processes. It has been observed by experiment that long-lived transients are already present in the ribodinucleotides (diribonucleoside monophosphates) and, in particular, that the adenine dinucleotide (ApA, see Fig. 1) shows identical bleach recovery signals as compared to longer adenine strands.19 Moreover, it was pointed out that after UV excitation in adenine strands the excitation is localized on not more than two bases after 1 ps and that there is not even conclusive experimental evidence for invoking delocalized absorbing states.26 Furthermore, there is recent computational evidence that the absorbing states are rather localized.25,27 Specifically, the RNA (rather than DNA) dinucleotide ApA was chosen in order to be able to match a detailed experimental investigation of this system.19 However, we believe that due to similar stacking geometries the presented results should also be relevant to DNA analogues. In light of all these considerations we conclude that ApA is a highly interesting model system possessing many parallels to larger RNA and DNA oligonucleotides.
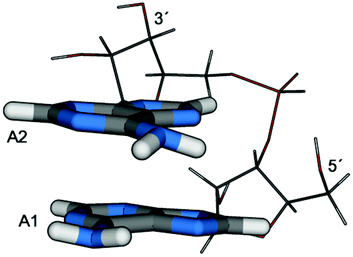 | ||
| Fig. 1 Depiction of the adenine dinucleotide (as optimized at the MP2/SV(P)-SV level in solution). The labelling of the two units (A1, A2) and the position of the 5′- and 3′-ends of the backbone are indicated. | ||
When studying structural relaxation and excited state dynamics in DNA fragments, special challenges are present related to (i) the system size, (ii) a treatment of the environment, in particular for describing charge transfer states, (iii) providing a reliable description even for strongly distorted structures, (iv) giving an accurate interaction potential, especially for exciplexes with short intermolecular separations, and (v) an instructive analysis of the nature of the excited states. A number of computational strategies with different merits and shortcomings in light of these demands are, in principle, applicable and a wide range of types of calculations have been performed. These amount to wavefunction based ab initio calculations,21 time-dependent density functional theory (TDDFT) using a polarizable continuum model,22,28,29 semi-empirical methods,23,30 and model Hamiltonians.31 For more information about calculations performed on DNA fragments see also ref. 25 and 32. This study is based on ab initio theory in an attempt to eliminate uncertainties related to unusual molecular structures, which may not be accounted for properly in approaches relying on parameterization such as, aside from purely semi-empirical methods, also the empirical dispersion correction for DFT33 or the M06 method.34 The algebraic diagrammatic construction to second order (ADC(2))35 using the resolution of the identity approximation (RI)36 is used as an efficient and reliable method for the description of excited state energies, properties, and gradients (see also ref. 25 and 37 for experiences on DNA fragments with this method and the related second order coupled cluster (CC2) method). These calculations are supplemented by multi-reference configuration interaction (MR-CI) computations, which provide reference values for strongly distorted geometries and structures with a small S0/S1 gap, cases in which the reliability of a single reference method is limited. Solvation effects are treated using explicit solvation in an electrostatic quantum mechanics–molecular mechanics (QM/MM) coupling scheme. To allow for averaging over several different solvent configurations the concept of an averaged solvent electrostatic potential (ASEP)38 was used. Mutual polarization between the core region and the solvent is treated in an iterative manner. A newly developed scheme for analysing excitonic and charge transfer (CT) interactions was used to allow for a meaningful description of the excited states even in challenging cases of partially delocalized orbitals and several interacting configurations.39
Computational details
Following ref. 19 the adenine ribodinucleotide (diriboadenosine monophosphate, ApA) was considered. This system was solvated in 3000 water molecules and the negative charge of the phosphate group was counterbalanced with one Na+ ion. RI-ADC(2)35 calculations were performed in TURBOMOLE36,40,41 using the SV(P),42 TZVP, TZVPP, and QZVPP basis sets.43 MR-CISD calculations were carried out with COLUMBUS44–46 using the 6-31G* basis set47 where CI expansions of about 200 million configurations were considered by applying the parallel version48,49 running on 64 CPUs. All MR-CISD results are reported with the size consistency correction suggested by Pople (+P).50Electrostatic embedding, QM/MM calculations51 were performed following ref. 25 and 52. For the MM interactions the Amber-99 forcefield53 as implemented in TINKER54 was used. The different potentials were combined using the Newton-X55–57 hybrid gradient facility.52 Initial structures were generated using PACKMOL.58 The QM region consisted either of the whole ApA system or just the two adenines. The remaining system (i.e. the water molecules, the Na+ ion and in the second case also the backbone) was treated as point charges using Amber-99 values. The connection of the adenine molecules to the sugar/phosphate backbone in the second case was achieved through the link hydrogen atom technique,51,59 where the link hydrogen was placed on the broken C–N bond but moved closer to the N atom with the distance reduced by a factor of 0.68922 obtained by considering individual optimizations of the isolated base and the nucleotide. The charge of the carbon atom forming the link atom on the MM side was set to zero, and to maintain the original neutral charge of the overall system, the excess charge (i.e., the sum of the deleted charge of the MM-link atom and the partial charge corresponding to the atoms in the respective QM region) were equally distributed onto the three atoms bonded to the MM-link atom (cf. ref. 60).
Specifically the following levels were considered with or without additional solvation in water:
• “SV(P)-SV” denoting that the whole dinucleotide was in the QM region with the two adenine molecules described with the SV(P) basis set and the backbone with SV,
• “SV(P)-MM”, “TZVPP-MM”, “QZVPP-MM”, etc. denoting that the adenine molecules were considered at the respective basis set while the backbone was considered only as MM point charges,
• “SV(P)-x” denoting that the backbone was not considered at all, used for test purposes.
Gas phase geometry optimizations were carried out at the SV(P)-MM and TZVP-MM levels allowing for a more efficient treatment. Optimizations in aqueous solution were performed in general with the whole dinucleotide in the QM region (SV(P)-SV) to simplify the computation of interaction terms.
The solvated QM/MM single point and geometry optimization calculations were performed using an averaged solvent electrostatic potential (ASEP, cf.ref. 38, 61 and 62). The idea of this approach is to combine the (properly scaled) point charges of several MD frames to obtain an averaged potential describing the solvent influence. Mutual polarization of the solvent field and the central molecule can be obtained by using a coupled method where charges obtained in the QM calculation are fed back to the MD.61 Geometry optimizations in such a potential actually amount to approximate free energy optimizations with respect to the solvent coordinates.62 An important consideration for a practical application of this approach is that the number of point charges has to be reduced for the QM/MM calculation, in particular when gradients are considered. For this purpose we use a coarse graining of the point charges (in a somewhat different approach as presented in ref. 38). Our method considers a dynamical threshold, which for point charges i and j takes on the value
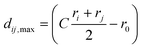
To represent the intermolecular separation between the two adenine molecules, the mutual distance between the C6 atoms, i.e. the C atoms bonded to the respective amino groups (Fig. 2), labelled dC6C6, was in most cases considered. Additional geometric parameters are presented in the ESI.†
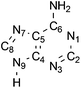 | ||
| Fig. 2 The numbering scheme used in the adenine molecule. | ||
In some test calculations the counterpoise (CP) correction65 for basis set superposition error (BSSE) was used, considering gas phase calculations at the SV(P)-MM, TZVP-MM, and QZVPP-MM levels. However, due to the problems of this approach for correlated methods (as described below) the main results reported are all without CP correction. The CP correction term was computed as
| ΔECP = −E(A1[A2]) + E(A1) − E(A2[A1]) + E(A2) | (2) |
where E(A1) is the energy of a QM calculation of adenine A1 alone and E(A1[A2]) the energy of a QM calculation of A1 where additionally the basis functions of A2 were present, and vice versa. The ΔECP term was added to the hybrid QM/MM energy for BSSE correction. CP corrected gradients for geometry optimizations were simply obtained by combining the gradients of the individual terms with the normal QM/MM gradient expression using the Newton-X hybrid gradient facility.
For the analysis of the excited states a scheme developed in ref. 39 (partially based on work by Luzanov and Zhikol66 as well as Tretiak and Mukamel67) was applied. The scheme proceeds by analyzing the transition density matrix in the atomic orbital AO basis. Using different summations over the blocks of this matrix, several different quantities can be computed:
• POS, the position of the excitation;
• PR, the excitonic participation ratio, representing delocalization;
• CT, the total amount of charge separation (including charge resonance and directed transfer contributions); and
• CTnet, the net charge shifted.
In addition it is also of interest to consider the natural transition orbitals (NTO),68–70 which can be obtained from a singular value decomposition of the transition density matrix. Aside from pictorial representations, also the NTO participation ratio PRNTO was considered, counting the number of non-vanishing singular values.
Results and discussion
Franck–Condon point and vertical excitations
In this section the ground state potential around the Franck–Condon (FC) point, the vertical excitations, and effects of solvation will be discussed. In addition, several methodological points related to the reliability of our method will be considered.In a first step the gas phase ground state potential of the system and the adequacy of the present methods to describe it were considered. Particular focus was laid on BSSE as it has been pointed out that this may have important effects for the binding energies of excimers.21,71 For this purpose structures were first optimized at the MP2 level considering the adenine molecules at the TZVP level and the backbone as an MM potential (i.e. the MP2/TZVP-MM level in the notation described above). Optimizing at this level without CP correction, a tightly stacked minimum “Stack A” with dC6C6 = 3.00 Å (as a representative stacking distance) was found. By contrast, when the CP correction was applied the stacking distance was increased significantly, yielding “Stack B” with dC6C6 = 3.42 Å. Furthermore, with CP correction, an additional “V-shaped” minimum stabilized by about 0.05 eV with respect to “Stack B” was found exhibiting dC6C6 = 4.31 Å. These structures are depicted in the ESI (S1–S3†). To understand these discrepancies and to get more insight into the ground state potential, an interpolation in internal coordinates between these three structures was performed and potential curves at the MP2 level using the SV(P), TZVP, and QZVPP basis sets with and without CP correction were computed and plotted against the respective energy of the V-shaped system (Fig. 3). By considering the same set of structures for these different cases, a direct comparison of the different methods is possible. However, it should be noted that these curves have been obtained by linear interpolation between the three structures (see above) and are not geometrically relaxed curves. Using the QZVPP basis set a very flat potential curve was obtained with and without CP correction showing differences between the minima and maxima on the curve of only ∼0.05 eV. The situation is significantly different with the TZVP basis set. Without CP correction “Stack A” exists as a minimum at 0.13 eV below the energy of the V-shaped structure. By contrast, when applying the CP correction at this level “Stack A” is destabilized lying 0.14 eV above the V-shaped structure. Both values differ strongly from the QZVPP reference. The results are even somewhat further off when applying the CP correction at the SV(P) basis set level, yielding a destabilization of “Stack A” by 0.22 eV. Interestingly the uncorrected SV(P) curve shows a very similar behaviour to the QZVPP curves exhibiting a similar flat potential, which is only somewhat biased toward the tightly stacked “Stack A” structure.
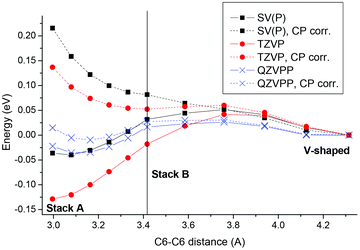 | ||
| Fig. 3 Interpolated ground state potential energy curves for ApA plotted against the intermolecular C6–C6 distance for three basis sets considered with (dotted lines) and without (solid lines) CP correction. | ||
The strong basis set dependence of CP corrected MP2 interaction energies is in agreement with previous experience.72 However, an interesting observation in the present context is that the CP correction may even worsen the result. This phenomenon in the context of the different curves will now be discussed briefly. Firstly, the results suggest that as expected BSSE at the TZVP level is rather strong, explaining the drop in energy at lower stacking distances. Secondly, the good performance of the SV(P) basis set may be related to the fact that this basis set is simply too small to have significant superposition at the stacking distances considered here or possibly to other kinds of error compensation. Thirdly, a possible reason of the overcompensation of the CP correction has been given in ref. 73. Briefly, the virtual space in the “ghost” calculation (containing 265 orbitals in the SV(P) case) is larger than in the dimer calculation (230 orbitals) due to the smaller number of electrons present. This phenomenon, in addition to different energetics of these orbitals following from missing electron repulsion, leads to additional terms in the MP2 energy expression providing an overestimation of the CP correction term at the correlated level.
In summary we have shown that at the MP2/QZVPP-MM level the system possesses a rather flat potential energy surface with respect to a variation in the degree of stacking, suggesting that a wide distribution of such structures should exist at room temperature in agreement with experimental interpretations.19,26 At the MP2/SV(P)-MM level without CP correction the flatness of the QZVPP-MM surface is reproduced remarkably well. Therefore, this basis set and no CP correction will be used for geometry optimizations in the further course of this study. In addition, results will be verified at the QZVPP-MM level. As described for example in ref. 74 the MP2 method tends to overestimate somewhat the interaction strength in nucleobase stacks and therefore yields somewhat too tightly stacked structures. For example, binding energies are too large by ∼0.1 eV and the stacking distances are too small by ∼0.1 Å as compared to CCSD(T) results (which are in turn about 0.1 Å shorter than experimental values).74,75
We have recently discussed the UV absorption of oligonucleotides in detail.25 Therefore, in this contribution the absorbing states will be considered only briefly. For this purpose the ground state structure of ApA was optimized in solution at the MP2/SV(P)-SV level using an ASEP solvent field included through electrostatic embedding as explained in more detail in the Computational details section and the vertical excitations were computed (Table 1). Aside from excitation energies and oscillator strengths, also descriptors characterizing the electronic excitation as defined in the Computational details section were determined. The first four states were of ππ* character, all showing at least partial delocalization (PR > 1.5). The brightest state at this level was S4 followed closely in oscillator strength by S3. Then two nπ* states localized on either molecule follow. The S7 and S8 states are combinations of opposing CT states, yielding charge resonance states, i.e. states with a high amount of charge separation (CT ≈ 0.75) but a low amount of net transfer (|CTnet| < 0.30), also characterized by a concurrent high delocalization degree (PR ≈ 1.9). It may be noted here that the current computational protocol neglects to include dynamic solvent polarizability of the surrounding water, which may somewhat stabilize the CT states and increase net charge transfer as compared to the results presented in Table 1. But due to the small dynamic polarizability of water this effect should be rather small. After the CT states, two more localized nπ* states follow. An observation to be made at this point is that many of the states are not pure Frenkel excitons but contain small CT contributions, showing that already at the Franck–Condon point, orbital interactions mixing these types of states (see also ref. 76) come into play. This is in agreement with our recent study on alternating DNA strands.25 Another interesting fact is that the delocalized states exhibit a PRNTO significantly larger than one, which is consistent with the statement that several configurations are needed to describe excitonic delocalization (see also ref. 39, 77 and 78).
| State | ΔE | f | POS | PR | CT | CTnet | PRNTO | Type |
|---|---|---|---|---|---|---|---|---|
| S1 | 5.044 | 0.033 | 1.26 | 1.61 | 0.13 | 0.02 | 2.12 | ππ* |
| S2 | 5.051 | 0.085 | 1.77 | 1.56 | 0.06 | 0.00 | 1.58 | ππ* |
| S3 | 5.133 | 0.160 | 1.51 | 2.00 | 0.09 | −0.03 | 2.32 | ππ* |
| S4 | 5.259 | 0.189 | 1.46 | 1.99 | 0.10 | −0.04 | 2.41 | ππ* |
| S5 | 5.396 | 0.004 | 1.03 | 1.05 | 0.04 | 0.01 | 1.03 | nπ* |
| S6 | 5.424 | 0.014 | 1.97 | 1.06 | 0.04 | −0.03 | 1.03 | nπ* |
| S7 | 5.742 | 0.072 | 1.48 | 1.85 | 0.75 | −0.28 | 1.73 | CT (ππ*) |
| S8 | 5.914 | 0.031 | 1.53 | 1.92 | 0.76 | 0.21 | 1.88 | CT (ππ*) |
| S9 | 5.931 | 0.002 | 1.05 | 1.10 | 0.07 | 0.04 | 1.06 | nπ* |
| S10 | 6.018 | 0.002 | 1.94 | 1.12 | 0.07 | −0.03 | 1.12 | nπ* |
To get a more detailed insight into the effect of the environment and of the computational model chosen, the adenine dinucleotide was considered at three different levels: (i) as two isolated molecules (SV(P)-x), (ii) two molecules with the backbone included at the MM level (SV(P)-MM), and (iii) the full system at the QM level (SV(P)-SV). Furthermore, models (ii) and (iii) were considered with and without H2O solvation. In Fig. 4 the individual effects of these models on the first six states are summarized. In all of these cases the geometry optimized in solution (as used in Table 1) was considered. The largest effect visible is the shift of the nπ* states due to H-bonding. Such a shift is a well known phenomenon in these types of systems.25,79–81 Further trends are that an inclusion of the backbone through point charges (moving from SV(P)-x to SV(P)-MM) somewhat destabilized the nπ* states as well. A full QM treatment of the backbone (moving to SV(P)-SV) had only a minor effect, somewhat lowering all the excitation energies. But in summary the glycosidic bond has a rather low influence (cf. ref. 81). In solution the difference between SV(P)-SV and SV(P)-MM is somewhat enhanced: the nπ* state on A2 is shifted down by a significant amount when the backbone is only considered at the MM level. However, a more detailed analysis of the character of this state revealed that the large apparent shift is probably enhanced by mixing with the ππ* states. In summary we can state that the standard link-atom model used here is reliable but in critical cases the results may have to be checked carefully. An alternative link atom model for these types of systems including also two atoms of the adjacent sugar into the QM region has been suggested.10 Such an approach would, however, significantly increase the size of the QM region and add complications due to the need of two link atoms per nucleobase. Fortunately, in the further course of the present study we were able to perform full QM calculations on the whole dinucleotide; the link-atom models are only used for increased basis sets and MR-CISD calculations.
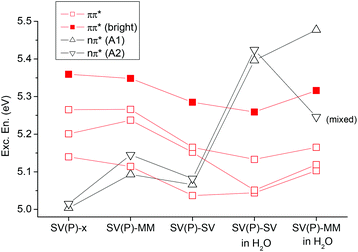 | ||
| Fig. 4 Vertical excitation energies of the first six excited states of ApA using different models: two isolated adenine molecules (SV(P)-x), the system with the backbone treated at the MM level (SV(P)-MM), the full system at the QM level (SV(P)-SV), and the latter two levels with an additional consideration of water solvation. | ||
Finally, basis set effects on the vertical excitations will be discussed (Fig. 5). The general trend, a rather uniform decrease of excitation energies when the basis set is increased, agrees with previous experience.25,82 This decrease amounts to ∼0.1–0.3 eV when going from SV(P) to QZVPP and is present for all states considered. However, the lowest ππ* state is affected to a somewhat larger extent than the others. Furthermore, for the larger basis sets the brightest state is stabilized and becomes S2.
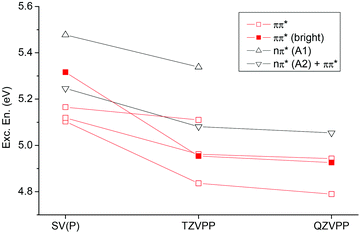 | ||
| Fig. 5 Vertical excitation energies of the first six excited states of ApA in aqueous solution considering different basis sets (always considering the backbone at the MM level). | ||
Excited state geometry relaxation
The second part of this paper will be concerned with processes happening in the excited state subsequent to photoabsorption. In this context it is of particular interest to investigate how interactions between the two nucleobases may give rise to new excited state minima and possible decay channels. In general, a large manifold of structures and a number of modes need to be considered. Here, we will focus on well-defined stacked structures considering that they are connected to the long-lived experimental transients26 whereas unstacked structures will trivially decay according to monomer-like pathways. As far as coordinates are concerned, we will consider the intermolecular distance between the two C6 atoms on the bases (cf. Fig. 2) dC6C6 since this coordinate showed large alterations in the unconstrained optimizations (shown below). The study will focus on excited state minima, which are well accessible with the present computational methods, whereas intersections between electronic states will only be considered indirectly.First, the ApA system was analysed in gas phase. For this purpose a potential curve was computed where the dC6C6 coordinate was fixed at different values between 2.0 and 2.9 Å and the geometry of the remaining system was optimized for the S1 state. Considering these geometries, the adiabatic energies of the S0–S2 states relative to the ground state minimum geometry were computed (Fig. 6). An important feature is the flatness of the S1 curve over the whole range considered, down to a distance of dC6C6 = 2.0 Å where the ground state surface is of course already strongly repulsive. To understand this phenomenon a more detailed inspection of the involved excited states is needed. At larger intermolecular separations the S1 state is described by a local nπ* transition on A1 (PR ≈ 1, CT ≈ CTnet ≈ 0, PRNTO ≈ 1). This state possesses a very low oscillator strength (f ≈ 0.005). There is no significant change down to dC6C6 = 2.4 Å, only that the PR and CT values increase slightly as the π* orbital acquires partial delocalized character. The vertical excitation energy always stays above 3.0 eV. Structurally the system exhibits an out-of-plane motion of the amino group bonded to the C6 atom on A1 whereas almost no changes occur in A2 (see Fig. 7(a)).
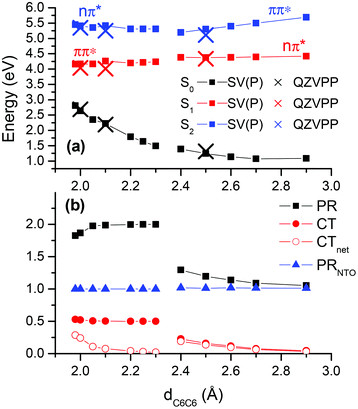 | ||
| Fig. 6 Relaxed potential energy curve computed for the S1 state of ApA at the ADC(2)/SV(P)-MM level in gas phase: (a) energies for the first three electronic states at this level (squares) and at the ADC(2)/QZVPP-MM level (crosses) for selected points; (b) statistical descriptors for the S1 state. | ||
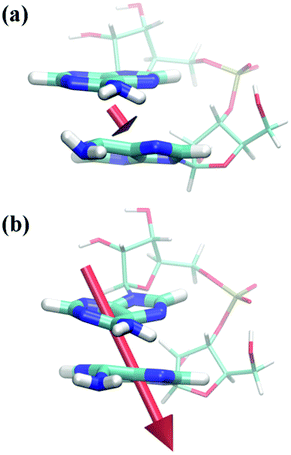 | ||
| Fig. 7 Depiction of ApA structures optimized at the ADC(2)/SV(P)-MM level in gas phase on the S1 surface for fixed dC6C6 distances of (a) 2.6 Å and (b) 2.2 Å. Transition dipole moments corresponding to (a) f = 0.005 and (b) f = 0.035 are plotted as well. | ||
When the dC6C6 distance is decreased to 2.30 Å and below, the geometry optimization converges to a different type of structure. The amino group on A1 is still out-of-plane, similar to the dC6C6 ≥ 2.40 Å case, but now also the planarity of A2 is lifted (see Fig. 7(b)) yielding a structure similar to the π(AA)* minimum in solution as described below. The structures exhibit a strong interbase “tilt” and “roll”. This can for example be understood from the fact that when dC6C6 was fixed at 2.20 Å, the intermolecular distances between the neighbouring atoms were much larger with dC5C5 = 2.50 Å and dN1N1 = 2.89 Å. The states in this area can be characterized as delocalized ππ* states (PR ≈ 2). A more detailed consideration of their excited state structure reveals that their nature is different from any of the states at the FC point as presented in Table 1. This may be immediately seen by considering the number of independent transitions needed to describe the excited state (i.e. the PRNTO value). At the FC point there were always several transitions involved for delocalized states (PRNTO ≈ 2), in agreement with the model of two independent transitions needed to form a delocalized Frenkel exciton.39,77,78 By contrast the S1 state computed for structures on this potential curve exhibited always PRNTO ≈ 1.00 despite the delocalization present for dC6C6 < 2.30 Å. In fact, this difference could already be seen in the canonical orbital representation, which showed that the excited state was always largely dominated by a simple transition between the highest occupied molecular orbital (HOMO) and the lowest unoccupied MO (LUMO).‡ A different way to analyse these states can be achieved by considering the charge transfer character. The values CT ≈ 0.5 and CTnet ≈ 0, which are present for 2.05 Å ≤ dC6C6 ≤ 2.25 Å, show that this is a state with 50% charge separation but no net transfer of charge. Another way to interpret this is by the presence of a homogeneous excited state over the whole system with an equal probability of the electron and hole being on the same or different fragments, a condition which is in turn consistent with the simple picture of only one transition. This situation is very similar to the naphthalene dimer.39 In that case we could show that the excimer state could formally be constructed as a mixture between the Frenkel excitonic and charge resonance states present at larger intermolecular separations. However, at the excimer minimum the state was of neither type and was determined by direct interactions between the orbitals on the two molecules, rather than just electrostatic interactions. Due to the ππ* nature and the lack of charge transfer of this excimer state in ApA it possesses appreciable oscillator strength (e.g. f = 0.035 for dC6C6 = 2.2 Å). An interesting phenomenon arising from the excited state structure is an out-of-plane polarization of the transition moment (Fig. 7(b)). It should be noted here that ππ* states in planar systems show strict in-plane polarization for symmetry reasons. Furthermore, if these states are coupled by purely electrostatic interactions to form Frenkel excitons, the polarization vectors still have to remain parallel to the molecular planes of the aromatic molecules. In this sense the out-of-plane transition moments are an observable manifestation of the unusual excited state structure described here. An extended analysis of this potential curve is presented in the ESI (Fig. S8†). In particular, it should be noted that for the ππ* state orbital interactions come into play already at dC6C6 = 2.90 Å.
In a one-dimensional picture one would expect to notice a lowering of the S1/S2 gap to an avoided crossing as these two states exchange their character around dC6C6 = 2.35 Å. Such a feature is only rudimentarily visible in Fig. 6 where the S1/S2 gap always stays above 0.8 eV. This fact is probably related to the multidimensional nature of the potential surface and the fact that the optimization procedure moves the structures away from the avoided crossing when relaxing the S1 state of the respective character. In fact, when searching for a signature of the S1/S2 avoided crossing a different type of constrained local S1 minimum was found in the area between 2.25 Å ≤ dC6C6 ≤ 2.35 Å. Those structures were determined by strong ring puckering of C6 on A1 and the amino group was shifted out-of-plane pointing in a direction almost perpendicular to the ring, a structure resembling the C6-puckered intersection in the monomer,83 only that the virtual orbital was partially delocalized here (PR = 1.33). The S0/S1 gap was lowered to ∼0.5 eV (while the S1/S2 gap moved up to ∼1.5 eV). The MR-CISD reference value was somewhat higher at 1.1 eV, which is, however, still in agreement with the picture of a close neighbourhood of an S0/S1 intersection structure. This finding suggests that monomer-like decay pathways to the ground state may play a role even in tightly stacked structures.
Fig. 6 shows that the S1 potential is very flat and in fact no minimum related to the exciplex could be located by unconstrained optimizations. By contrast, these optimizations lead to a decrease of dC6C6 below 2 Å and a subsequent strong destabilization of the ground state with very small S0/S1 gaps and a concomitant failure of the ADC(2) method. These results suggest that a new deactivation channel arises in the adenine dimer, which is accessed through decreasing the intermolecular distance between the respective C6 atoms to below 2.0 Å, similar to results obtained by semi-empirical calculations,23 only that in those cases the mutual C2 distance was shortened to below 2.0 Å. In ref. 23 a non-reactive deactivation between the two adenine molecules was observed. By contrast, an analogous intersection between two thymine molecules may be responsible for photodimerization in this case.84 The importance of this type of intersection and the relation to potential photoproduct formation in different base stacks requires further investigations.
To verify our results against basis set effects, selected calculations at the ADC(2)/QZVPP-MM level were performed (Fig. 6, crosses). These showed very good agreement with the SV(P) results, highlighting in particular the relative unimportance of BSSE in this case. MR-CISD calculations performed at the structures with dC6C6 = 2.0 Å and dC6C6 = 2.1 Å gave excitation energies, which were very similar to the ADC(2)/SV(P)-MM results (about 0.2 eV higher) highlighting that also multi-reference effects do not play an important role.
The computation of potential curves in solution is not feasible with the present computational protocol due to the iterative nature of the procedure (see the Computational details section for more information). Therefore, the focus was on locating different S1 minima in solution. Three minima possessing entirely different character were found. They are depicted in Fig. 8 and the major properties are collected in Table 2. The first minimum, which will be denoted AA*, is described by a local nπ* excitation on A2 (POS = 1.97, PR = 1.06, orbitals shown in Fig. S9†). This nucleobase (Fig. 8, top) shows puckering around the C2 atom exhibiting a very similar structure to a corresponding S1 minimum in the isolated adenine molecule (see e.g.ref. 83). The value of dC6C6 used as a measure of the intermolecular separation is 3.05 Å, which is similar to the ground state distance at this level, also reflecting that there is only negligible interaction.
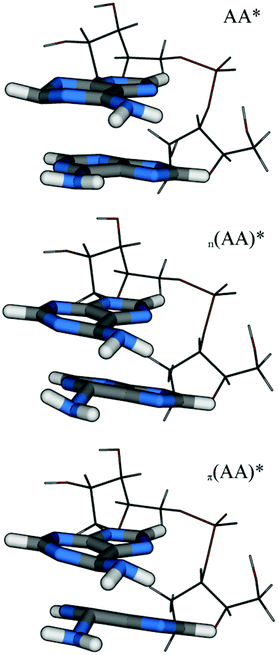 | ||
| Fig. 8 Molecular structures of the different minima of ApA located by optimizations at the ADC(2)/SV(P)-SV level in aqueous solution. | ||
| Struct. | d C6C6 | S0 En. | ΔE | f | POS | PR | CT | CTnet | PRNTO | Type |
|---|---|---|---|---|---|---|---|---|---|---|
| AA* | 3.05 | 1.2 | 3.221 | 0.016 | 1.97 | 1.06 | 0.06 | −0.06 | 1.01 | nπ* |
| n (AA)* | 2.21 | 2.2 | 2.438 | 0.010 | 1.25 | 1.53 | 0.45 | 0.41 | 1.00 | nπ* |
| π (AA)* | 2.00 | 2.3 | 1.843 | 0.061 | 1.40 | 1.77 | 0.53 | 0.32 | 1.00 | ππ* |
The second S1 minimum n(AA)* is related to the structures present in the gas phase potential curve for dC6C6 ≥ 2.4 Å (Fig. 6, right part). However, the molecules are now moved closer together in the geometry optimizations, in particular around the C6 atoms, leading to dC6C6 = 2.21 Å and the structure could indeed be called an exciplex. The vertical excitation energy of 2.438 eV shows the strong energetic stabilization in the excited state. As opposed to the gas phase, the excited state is strongly polarized here, showing CTnet = 0.41. This value follows from an excitation from the n orbital on A1 to a π* orbital which is delocalized between both adenine molecules, resulting also in partial delocalization (PR = 1.53, see also Fig. S10†).
The third minimum denoted π(AA)* is related to the gas phase potential curve at distances dC6C6 ≤ 2.30 Å (Fig. 6, left part). It is structurally similar to n(AA)*, only that the distortions of the rings are enhanced. A strong exciplex interaction leads to a decrease of the separation to dC6C6 = 2.00 Å and to a vertical excitation energy of 1.843 eV. The excitation is described by a single transition between a π and π* orbital both delocalized over both adenine molecules (Fig. S11†). Being of ππ* character, this structure shows an appreciable oscillator strength of 0.061 and it can be expected that its fluorescence should be visible. The transition moment has again a strong out-of-plane component similar to the gas phase calculation (cf. Fig. 7(b)). As described above, the presence of only one transition (PRNTO = 1.00) in a strongly delocalized state (PR = 1.77) is in contrast to the picture of a Frenkel exciton and only orbital interactions can lead to this situation, highlighting the marked difference between the character of the state at the exciplex structure and any state at the FC point. This situation is also represented by CT = 0.53. Due to solvation the excited state is partially polarized (CTnet = 0.32). The comparable gas phase structure at dC6C6 = 2.00 Å exhibits ΔE = 1.534, CTnet = 0.24 and an S0 energy of 2.633 eV (as presented in Fig. 6). It thus appears that the solvent has an important effect on the structural relaxation leading to exciplex structures, which are more strongly polarized and not so strongly distorted as opposed to the gas phase.
The relative adiabatic ground state energies are presented in Table 2 as well (these were converged up to a few tenths of an eV in the iterative procedure described in the Computational details section). When adding these to the vertical energies given also in the table, it follows that the AA* and n(AA)* minima are located at about 4.5 eV with respect to the ground state minimum or in other words they are reached by a relaxation of about 0.7 eV from the bright excited states at the FC point. The π(AA)* structure is situated lower at around 4.1 eV.
In a next step we aim to verify the results in light of reference calculations (Table 3). The backbone was treated at the MM level in these calculations and it was first established that this only had a minor effect, verifying that the ADC(2)/SV(P)-MM results always reproduced the full QM (SV(P)-SV) ones well, deviating by at most 0.1 eV in excitation energies. Increasing the basis set to the QZVPP-MM level somewhat lowered the excitation energies (by about 0.1–0.2 eV) but produced no systematic changes. MR-CISD excitation energies were always somewhat higher, which is however a known phenomenon for this method related in part to size consistency errors present in a system of this size and to orbital relaxation (see e.g.ref. 85 and 86). It is however worth noting that at least the energetic trend is reproduced here. For comparison, also calculations at the PBE087 level were performed, showing quite good agreement with the ADC(2) results.
| AA* | n (AA)* | π (AA)* | |||
|---|---|---|---|---|---|
| Vert. S0–S1 | ADC(2) | SV(P)-SV | 3.221 | 2.438 | 1.843 |
| ADC(2) | SV(P)-MM | 3.259 | 2.476 | 1.898 | |
| ADC(2) | QZVPP-MM | 3.157 | 2.351 | 1.715 | |
| MR-CISD | 6-31G*-MM | 3.651 | 3.008 | 2.295 | |
| PBE0 | TZVP-MM | 3.307 | 2.533 | 1.908 | |
| Vert. S0–S2 | ADC(2) | SV(P)-SV | 4.267 | 3.258 | 2.821 |
| ADC(2) | SV(P)-MM | 4.370 | 3.316 | 2.859 | |
| ADC(2) | QZVPP-MM | 4.188 | 3.081 | 2.735 | |
| MR-CISD | 6-31G*-MM | — | 3.523 | — | |
| PBE0 | TZVP-MM | 4.080 | 3.105 | 3.018 | |
In agreement with ref. 21 and 22 we find a number of excited state minima of different degrees of delocalization and charge separation located down to adiabatic energies of about 4 eV relative to the ground state minimum. This presence of a number of different minima and plateaus is consistent with our observation that the potential surface in the ground (Fig. 3) as well as excited (Fig. 6) states is very flat. In ref. 21 and 22 these minima exhibit vertical S0/S1 energy gaps above 2.5 eV and stacking distances above 2.8 Å, whereas the π(AA)* minimum in this study exhibits a gap of only 1.8 eV, which derives from a stronger decrease of the intermolecular separation and an out-of-plane movement of the amino groups. Precisely answering the question of how closely the two adenine molecules approach each other is quite challenging as accurate intermolecular potentials are difficult to compute. In ref. 21 the CASSCF method was used for geometry optimizations (while CASPT2 was only used for single point calculations). Due to a lack of dynamical electron correlation the CASSCF method is not expected to describe dispersion well and should therefore underestimate interaction energies and overestimate binding distances. Furthermore, it is also quite difficult to describe those interactions correctly with density functional theory. Whereas the M06-2X functional used in ref. 22 has been designed to give good interaction energies in the case of standard complexes, it is not clear whether good results can also be obtained for interatomic distances far below 3 Å. By contrast, we use the correlated ab initio methodology, which is, at least in general, able to provide good interaction energies. The MP2 method has been described as producing reasonably accurate binding energies, only that dispersion energies may be somewhat overestimated.72 Additionally, the state loses its single reference character giving a value of 0.063 for the D1 diagnostic,88 which is exceeding the recommended value (0.04). In summary, it can be said that we could show strong evidence for exciplex structures determined by short C6–C6 distances and strong structural distortions even if the interaction strength may be somewhat overestimated. However, it should also be recognized that due to the flatness of the potential energy curves shown in Fig. 6, especially the location of the minima could change by a few tenths of an Å.
The existence of a strong exciplex interaction in ApA is certainly in agreement with the reported slow excited state decay of this system.19 However, in contrast to the original experimental interpretation we find that in this system interactions due to orbital overlap play a dominant role while charge transfer is small. A similar mechanism could also explain the increased stability reported for ApG as opposed to purine–pyrimidine stacks, a phenomenon which could not be accounted for when simply considering ionization potentials and electron affinities of the individual bases.19
An emission energy below 2 eV, which is found for the π(AA)* exciplex in this study, is smaller than the values of fluorescence maxima reported from experimental studies of poly(dA) and d(pApA), which are around 3 eV.6,89–91 It appears therefore that the binding distances may be somewhat too short here. Due to the steep increase in the ground state potential with decreasing intermolecular distance (cf. Fig. 6) even small geometric changes may result in a large difference from an energetic viewpoint. In spite of this difference in quantitative terms our study sheds new light on excimer formation and highlights the importance of orbital interactions, which have rarely been considered in previous investigations. In particular, an out-of-plane polarization of the fluorescence (cf. Fig. 7), which has been reported from experiment,90 is difficult to explain when only pure Frenkel exciton and CT states are considered.
Conclusions
A detailed ab initio QM/MM study of the excited state potential energy surfaces of ApA in solution was performed using the ADC(2) and MR-CISD methods. First, the Franck–Condon point was analyzed. The flat ground state surface represents the structural flexibility well known for dinucleotides. For the vertical excitations Frenkel excitonic states as well as charge transfer states were found with only a small mixing between these two types (less than 10%). As a next step, structural relaxation in gas phase and in solution was considered. In gas phase a potential curve against the intermolecular dC6C6 distance was computed showing a very flat potential down to dC6C6 = 2.0 Å. Three different minima on the S1 surface of different excitation characters (nπ* and ππ*) as well as varying degrees of delocalization (up to about 80%) and charge transfer (up to about 0.4 e) were located. This emergence of several S1 minima of different character and of flat potential surfaces in between them agrees well with previous studies.21,22 A new outcome of this study is the presence of exciplex minima with significantly decreased intermolecular separations, in particular between the C6 atoms, reaching in our calculations distances of dC6C6 ≤ 2.20 Å. The electronic structure at these minima was analyzed in detail using methods recently developed for this purpose.39 The main result of this analysis is that the minima are neither of Frenkel excitonic nor of pure charge transfer type but that orbital interactions lead to coherent homogeneous excited states, which differ significantly from any states present in the Franck–Condon region. These states were in turn dynamically polarized by interactions with the solvent, which led to a net charge transfer of about 0.3–0.4 e for these states. The most stable minimum was of ππ* character, which in connection with the low degree of charge separation led to the presence of appreciable transition strength for this structure. Interestingly the transition showed a high degree of out-of-plane polarization.Acknowledgements
This work was supported by the Austrian Science Fund within the framework of the Special Research Program F41, Vienna Computational Materials Laboratory (ViCoM). We also acknowledge technical support from and computer time at the Vienna Scientific Cluster (Projects 70019 and 70151). Support was also provided by the Robert A. Welch Foundation under Grant No. D-0005.Notes and references
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Ultrafast excited-state dynamics in nucleic acids, Chem. Rev., 2004, 104, 1977–2019 CrossRef.
- C. Canuel, M. Mons, F. Piuzzi, B. Tardivel, I. Dimicoli and M. Elhanine, Excited states dynamics of DNA and RNA bases: characterization of a stepwise deactivation pathway in the gas phase, J. Chem. Phys., 2005, 122, 074316 CrossRef.
- M. Barbatti, A. J. A. Aquino, J. J. Szymczak, D. Nachtigallová, P. Hobza and H. Lischka, Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21453–21458 CrossRef CAS.
- C. Crespo-Hernandez, B. Cohen and B. Kohler, Base stacking controls excited-state dynamics in A-T DNA, Nature, 2005, 436, 1141–1144 CrossRef CAS.
- C. T. Middleton, K. de La Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernandez and B. Kohler, DNA excited-state dynamics: from single bases to the double helix, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS.
- W. M. Kwok, C. S. Ma and D. L. Phillips, Femtosecond time- and wavelength-resolved fluorescence and absorption spectroscopic study of the excited states of adenosine and an adenine oligomer, J. Am. Chem. Soc., 2006, 128, 11894–11905 CrossRef CAS.
- I. Buchvarov, Q. Wang, M. Raytchev, A. Trifonov and T. Fiebig, Electronic energy delocalization and dissipation in single- and double-stranded DNA, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4794–4797 CrossRef CAS.
- D. Markovitsi, T. Gustavsson and F. Talbot, Excited states and energy transfer among DNA bases in double helices, Photochem. Photobiol. Sci., 2007, 6, 717–724 CAS.
- I. Vaya, F. Miannay, T. Gustavsson and D. Markovitsi, High-energy long-lived excited states in DNA double strands, ChemPhysChem, 2010, 11, 987–989 CrossRef CAS.
- I. Conti, P. Altoe, M. Stenta, M. Garavelli and G. Orlandi, Adenine deactivation in DNA resolved at the CASPT2//CASSCF/AMBER level, Phys. Chem. Chem. Phys., 2010, 12, 5016–5023 RSC.
- D. Nachtigallova, T. Zeleny, M. Ruckenbauer, T. Muller, M. Barbatti, P. Hobza and H. Lischka, Does stacking restrain the photodynamics of individual nucleobases?, J. Am. Chem. Soc., 2010, 132, 8261–8263 CrossRef CAS.
- A. L. Sobolewski, W. Domcke and C. Hättig, Tautomeric selectivity of the excited-state lifetime of guanine/cytosine base pairs: the role of electron-driven proton-transfer processes, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17903–17906 CrossRef CAS.
- A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. de Vries, Photochemical selectivity in guanine-cytosine base-pair structures, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20–23 CrossRef CAS.
- K. de La Harpe, C. E. Crespo-Hernandez and B. Kohler, Deuterium isotope effect on excited-state dynamics in an alternating GC oligonucleotide, J. Am. Chem. Soc., 2009, 131, 17557–17559 CrossRef CAS.
- Y. Lu, Z. G. Lan and W. Thiel, Hydrogen bonding regulates the monomeric nonradiative decay of adenine in DNA strands, Angew. Chem., Int. Ed., 2011, 50, 6864–6867 CrossRef CAS.
- T. Zeleny, M. Ruckenbauer, A. J. A. Aquino, T. Muller, F. Lankas, T. Drsata, W. L. Hase, D. Nachtigallova and H. Lischka, Strikingly different effects of hydrogen bonding on the photodynamics of individual nucleobases in DNA: comparison of guanine and cytosine, J. Am. Chem. Soc., 2012, 134, 13662–13669 CrossRef CAS.
- A. Trifonov, M. Raytchev, B. I. M. Rist, J. Barbaric, H. A. Wagenknecht and T. Fiebig, Ultrafast energy transfer and structural dynamics in DNA, J. Phys. Chem. B, 2005, 109, 19490–19495 CrossRef CAS.
- I. Vaya, T. Gustavsson, T. Douki, Y. Berlin and D. Markovitsi, Electronic: excitation energy transfer between nucleobases of natural DNA, J. Am. Chem. Soc., 2012, 134, 11366–11368 CrossRef CAS.
- T. Takaya, C. Su, K. de La Harpe, C. Crespo-Hernandez and B. Kohler, UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10285–10290 CrossRef CAS.
- F. Santoro, V. Barone and R. Improta, Influence of base stacking on excited-state behavior of polyadenine in water, based on time-dependent density functional calculations, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9931–9936 CrossRef CAS.
- G. Olaso-Gonzalez, M. Merchan and L. Serrano-Andres, The role of adenine excimers in the photophysics of oligonucleotides, J. Am. Chem. Soc., 2009, 131, 4368–4377 CrossRef CAS.
- R. Improta and V. Barone, Interplay between “neutral” and “charge-transfer” excimers rules the excited state decay in adenine-rich polynucleotides, Angew. Chem., Int. Ed., 2011, 50, 12016–12019 CrossRef CAS.
- W. Y. Zhang, S. A. Yuan, Z. J. Wang, Z. M. Qi, J. S. Zhao, Y. S. Dou and G. V. Lo, A semiclassical dynamics simulation for a long-lived excimer state of pi-stacked adenines, Chem. Phys. Lett., 2011, 506, 303–308 CrossRef CAS.
- I. Vaya, T. Gustavsson, F. A. Miannay, T. Douki and D. Markovitsi, Fluorescence of natural DNA: from the femtosecond to the nanosecond time scales, J. Am. Chem. Soc., 2010, 132, 11834–11835 CrossRef CAS.
- F. Plasser, A. J. A. Aquino, W. L. Hase and H. Lischka, UV absorption spectrum of alternating DNA duplexes. Analysis of excitonic and charge transfer interactions, J. Phys. Chem. A, 2012, 116, 11151–11160 CrossRef CAS.
- C. Su, C. T. Middleton and B. Kohler, Base-stacking disorder and excited-state dynamics in single-stranded adenine homo-oligonucleotides, J. Phys. Chem. B, 2012, 116, 10266–10274 CrossRef CAS.
- S. Patwardhan, S. Tonzani, F. D. Lewis, L. D. A. Siebbeles, G. C. Schatz and F. C. Grozema, Effect of structural dynamics and base pair sequence on the nature of excited states in DNA hairpins, J. Phys. Chem. B, 2012, 116, 11447–11458 CrossRef CAS.
- R. Improta, F. Santoro, V. Barone and A. Lami, Vibronic model for the quantum dynamical study of the competition between bright and charge-transfer excited states in single-strand polynucleotides: the adenine dimer case, J. Phys. Chem. A, 2009, 113, 15346–15354 CrossRef CAS.
- F. Santoro, V. Barone and R. Improta, Excited states decay of the A-T DNA: A PCM/TD-DFT study in aqueous solution of the (9-methyl-adenine)(2)center dot(1-methyl-thymine)(2) stacked tetramer, J. Am. Chem. Soc., 2009, 131, 15232–15245 CrossRef CAS.
- A. N. Alexandrova, J. C. Tully and G. Granucci, Photochemistry of DNA fragments via semiclassical nonadiabatic dynamics, J. Phys. Chem. B, 2010, 114, 12116–12128 CrossRef CAS.
- E. R. Bittner, Frenkel exciton model of ultrafast excited state dynamics in AT DNA double helices, J. Photochem. Photobiol., A, 2007, 190, 328–334 CrossRef CAS.
- F. Plasser, M. Barbatti, A. J. A. Aquino and H. Lischka, Electronically excited states and photodynamics: a continuing challenge, Theor. Chim. Acta, 2012, 131, 1073 CrossRef.
- S. Grimme, Accurate description of van der Waals complexes by density functional theory including empirical corrections, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS.
- Y. Zhao and D. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and 12 other functionals, Theor. Chem. Acc., 2008, 119, 525–525 CrossRef CAS.
- A. B. Trofimov and J. Schirmer, An efficient polarization propagator approach to valence electron-excitation spectra, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- C. Hättig and F. Weigend, CC2 excitation energy calculations on large molecules using the resolution of the identity approximation, J. Chem. Phys., 2000, 113, 5154–5161 CrossRef.
- A. J. A. Aquino, D. Nachtigallova, P. Hobza, D. G. Truhlar, C. Hattig and H. Lischka, The charge-transfer states in a stacked nucleobase dimer complex: a benchmark study, J. Comput. Chem., 2011, 32, 1217–1227 CrossRef CAS.
- M. L. Sanchez, M. A. Aguilar and F. J. O. delValle, Study of solvent effects by means of averaged solvent electrostatic potentials obtained from molecular dynamics data, J. Comput. Chem., 1997, 18, 313–322 CrossRef CAS.
- F. Plasser and H. Lischka, Analysis of excitonic and charge transfer interactions from quantum chemical calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS.
- C. Hattig, Structure optimizations for excited states with correlated second-order methods: CC2 and ADC(2), Adv. Quantum Chem., 2005, 50, 37–60 CrossRef.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Electronic-structure calculations on workstation computers – the program system turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. Schafer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian-basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- H. Lischka, R. Shepard, F. B. Brown and I. Shavitt, New implementation of the graphical unitary-group approach for multi-reference direct configuration-interaction calculations, Int. J. Quantum Chem., 1981, S15, 91–100 Search PubMed.
- H. Lischka, R. Shepard, I. Shavitt, R. M. Pitzer, M. Dallos, T. Mueller, P. G. Szalay, F. B. Brown, R. Ahlrichs, H. J. Boehm, A. Chang, D. C. Comeau, R. Gdanitz, H. Dachsel, C. Ehrhardt, M. Ernzerhof, P. Hoechtl, S. Irle, G. Kedziora, T. Kovar, V. Parasuk, M. J. M. Pepper, P. Scharf, H. Schiffer, M. Schindler, M. Schueler, M. Seth, E. A. Stahlberg, J.-G. Zhao, S. Yabushita, Z. Zhang, M. Barbatti, S. Matsika, M. Schuurmann, D. R. Yarkony, S. R. Brozell, E. V. Beck, J.-P. Blaudeau, M. Ruckenbauer, B. Sellner, F. Plasser and J. J. Szymczak, COLUMBUS, an ab initio electronic structure program, release 7.0, 2011, http://www.univie.ac.at/columbus..
- H. Lischka, R. Shepard, R. M. Pitzer, I. Shavitt, M. Dallos, T. Muller, P. G. Szalay, M. Seth, G. S. Kedziora, S. Yabushita and Z. Y. Zhang, High-level multireference methods in the quantum-chemistry program system COLUMBUS: analytic MR-CISD and MR-AQCC gradients and MR-AQCC-LRT for excited states, GUGA spin-orbit CI and parallel CI density, Phys. Chem. Chem. Phys., 2001, 3, 664–673 RSC.
- P. C. Hariharan and J. A. Pople, Influence of polarization functions on molecular-orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- H. Dachsel, H. Lischka, R. Shepard, J. Nieplocha and R. J. Harrison, A massively parallel multireference configuration interaction program: the parallel COLUMBUS program, J. Comput. Chem., 1997, 18, 430–448 CrossRef CAS.
- T. Muller, Large-scale parallel uncontracted multireference-averaged quadratic coupled cluster: the ground state of the chromium dimer revisited, J. Phys. Chem. A, 2009, 113, 12729–12740 CrossRef.
- J. A. Pople, R. Seeger and R. Krishnan, Variational configuration interaction methods and comparison with perturbation theory, Int. J. Quantum Chem., 1977, 12, 149–163 CrossRef.
- D. Bakowies and W. Thiel, Hybrid models for combined quantum mechanical and molecular mechanical approaches, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- M. Ruckenbauer, M. Barbatti, T. Muller and H. Lischka, Nonadiabatic excited-state dynamics with hybrid ab initio quantum-mechanical/molecular-mechanical methods: solvation of the pentadieniminium cation in apolar media, J. Phys. Chem. A, 2010, 114, 6757–6765 CrossRef CAS.
- J. M. Wang, P. Cieplak and P. A. Kollman, How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- J. W. Ponder and F. M. Richards, An efficient newton-like method for molecular mechanics energy minimization of large molecules, J. Comput. Chem., 1987, 8, 1016–1024 CrossRef CAS.
- M. Barbatti, G. Granucci, M. Persico, M. Ruckenbauer, M. Vazdar, M. Eckert-Maksic and H. Lischka, The on-the-fly surface-hopping program system Newton-X: application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems, J. Photochem. Photobiol., A, 2007, 190, 228–240 CrossRef CAS.
- M. Barbatti, G. Granucci, M. Ruckenbauer, F. Plasser, J. Pittner, M. Persico and H. Lischka, NEWTON-X: a package for Newtonian dynamics close to the crossing seam, 2011, http://www.newtonx.org..
- M. Barbatti, M. Ruckenbauer, F. Plasser, J. Pittner, G. Granucci, M. Persico and H. Lischka, NEWTON-X: a surface-hopping program for nonadiabatic molecular dynamic, WIREs Comput. Mol. Sci., 2013 DOI:10.1002/wcms.1158.
- J. M. Martinez and L. Martinez, Packing optimization for automated generation of complex system's initial configurations for molecular dynamics and docking, J. Comput. Chem., 2003, 24, 819–825 CrossRef CAS.
- M. J. Field, P. A. Bash and M. Karplus, A combined quantum-mechanical and molecular mechanical potential for molecular-dynamics simulations, J. Comput. Chem., 1990, 11, 700–733 CrossRef CAS.
- R. M. Nicoll, S. A. Hindle, G. MacKenzie, I. H. Hillier and N. A. Burton, Quantum mechanical/molecular mechanical methods and the study of kinetic isotope effects: modelling the covalent junction region and application to the enzyme xylose isomerase, Theor. Chem. Acc., 2001, 106, 105–112 CrossRef CAS.
- M. L. Sanchez, M. E. Martin, M. A. Aguilar and F. J. O. Del Valle, Solvent effects by means of averaged solvent electrostatic potentials: coupled method, J. Comput. Chem., 2000, 21, 705–715 CrossRef CAS.
- I. F. Galvan, M. L. Sanchez, M. E. Martin, F. J. O. del Valle and M. A. Aguilar, Geometry optimization of molecules in solution: joint use of the mean field approximation and the free-energy gradient method, J. Chem. Phys., 2003, 118, 255–263 CrossRef.
- F. Plasser, Quantum Mechanical Simulations of Defect Dynamics in DNA and Model Systems, Doctoral thesis, Vienna (http://othes.univie.ac.at/), 2012 Search PubMed.
- U. C. Singh and P. A. Kollman, An approach to computing electrostatic charges for molecules, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- S. F. Boys and F. Bernardi, Calculation of small molecular interactions by differences of separate total energies – some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- A. V. Luzanov and O. A. Zhikol, Electron invariants and excited state structural analysis for electronic transitions within CIS, RPA, and TDDFT models, Int. J. Quantum Chem., 2010, 110, 902–924 CAS.
- S. Tretiak and S. Mukamel, Density matrix analysis and simulation of electronic excitations in conjugated and aggregated molecules, Chem. Rev., 2002, 102, 3171–3212 CrossRef CAS.
- A. V. Luzanov, A. A. Sukhorukov and V. E. Umanskii, Application of transition density matrix for analysis of excited states, Theor. Exp. Chem., 1976, 10, 354–361 CrossRef.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- I. Mayer, Using singular value decomposition for a compact presentation and improved interpretation of the CIS wave functions, Chem. Phys. Lett., 2007, 437, 284–286 CrossRef CAS.
- G. Olaso-Gonzalez, D. Roca-Sanjuan, L. Serrano-Andres and M. Merchan, Toward the understanding of DNA fluorescence: the singlet excimer of cytosine, J. Chem. Phys., 2006, 125, 231102 CrossRef.
- K. E. Riley, J. A. Platts, J. Rezac, P. Hobza and J. G. Hill, Assessment of the performance of MP2 and MP2 variants for the treatment of noncovalent interactions, J. Phys. Chem. A, 2012, 116, 4159–4169 CrossRef CAS.
- D. B. Cook, J. A. Sordo and T. L. Sordo, Some comments on the counterpoise correction for the basis-set superposition error at the correlated level, Int. J. Quantum Chem., 1993, 48, 375–384 CrossRef CAS.
- J. Sponer, P. Jurecka, I. Marchan, F. J. Luque, M. Orozco and P. Hobza, Nature of base stacking: reference quantum-chemical stacking energies in ten unique B-DNA base-pair steps, Chem.–Eur. J., 2006, 12, 2854–2865 CrossRef CAS.
- C. A. Morgado, P. Jurecka, D. Svozil, P. Hobza and J. Sponer, Reference MP2/CBS and CCSD(T) quantum-chemical calculations on stacked adenine dimers. Comparison with DFT-D, MP2.5, SCS(MI)-MP2, M06-2X, CBS(SCS-D) and force field descriptions, Phys. Chem. Chem. Phys., 2010, 12, 3522–3534 RSC.
- G. D. Scholes and K. P. Ghiggino, Electronic interactions and interchromophore excitation transfer, J. Phys. Chem., 1994, 98, 4580–4590 CrossRef CAS.
- E. C. Lim and A. L. L. East, Naphthalene dimer: electronic states, excimers, and triplet decay, J. Chem. Phys., 2000, 113, 8981–8994 CrossRef.
- I. Mayer, Identifying a pair of interacting chromophores by using SVD transformed CIS wave functions, Chem. Phys. Lett., 2007, 443, 420–425 CrossRef CAS.
- F. Santoro, V. Barone, T. Gustavsson and R. Improta, Solvent effect on the singlet excited-state lifetimes of nucleic acid bases: a computational study of 5-fluorouracil and uracil in acetonitrile and water, J. Am. Chem. Soc., 2006, 128, 16312–16322 CrossRef CAS.
- J. J. Szymczak, T. Muller and H. Lischka, The effect of hydration on the photo-deactivation pathways of 4-aminopyrimidine, Chem. Phys., 2010, 375, 110–117 CrossRef CAS.
- P. G. Szalay, T. Watson, A. Perera, V. Lotrich, G. Fogarasi and R. J. Bartlett, Benchmark studies on the building blocks of DNA. 2. Effect of biological environment on the electronic excitation spectrum of nucleobases, J. Phys. Chem. A, 2012, 116, 8851–8860 CrossRef CAS.
- A. Panda, F. Plasser, A. J. A. Aquino, I. Burghardt and H. Lischka, Electronically excited states in poly(p-phenylene vinylene): vertical excitations and torsional potentials from high-level ab initio calculations, J. Phys. Chem. A, 2013, 117, 2181–2189 CrossRef CAS.
- M. Barbatti, Z. G. Lan, R. Crespo-Otero, J. J. Szymczak, H. Lischka and W. Thiel, Critical appraisal of excited state nonadiabatic dynamics simulations of 9H-adenine, J. Chem. Phys., 2012, 137, 22A503 CrossRef.
- M. Boggio-Pasqua, G. Groenhof, L. V. Schafer, H. Grubmuller and M. A. Robb, Ultrafast deactivation channel for thymine dimerization, J. Am. Chem. Soc., 2007, 129, 10996–10997 CrossRef CAS.
- M. Barbatti, A. J. A. Aquino, J. J. Szymczak, D. Nachtigallova and H. Lischka, Photodynamical simulations of cytosine: characterization of the ultrafast bi-exponential UV deactivation, Phys. Chem. Chem. Phys., 2011, 13, 6145–6155 RSC.
- C. Angeli, On the nature of the pi → pi* ionic excited states: the V state of ethene as a prototype, J. Comput. Chem., 2009, 30, 1319–1333 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. L. Janssen and I. M. B. Nielsen, New diagnostics for coupled-cluster and Moller-Plesset perturbation theory, Chem. Phys. Lett., 1998, 290, 423–430 CrossRef CAS.
- R. Plessow, A. Brockhinke, W. Eimer and K. Kohse-Hoinghaus, Intrinsic time- and wavelength-resolved fluorescence of oligonucleotides: a systematic investigation using a novel picosecond laser approach, J. Phys. Chem. B, 2000, 104, 3695–3704 CrossRef CAS.
- A. Banyasz, T. Gustavsson, D. Onidas, P. Changenet-Barret, D. Markovitsi and R. Improta, Multi-pathway excited state relaxation of adenine oligomers in aqueous solution: a joint theoretical and experimental study, Chem.–Eur. J., 2013, 19, 3762–3774 CrossRef CAS.
- M. C. Stuhldreier, C. Schuler, J. Kleber and F. Temps, Femtosecond fluorescence measurements of the adenine dinucleotide: direct observation of the excimer state, in Ultrafast Phenomena XVII, ed. M. Chergui, D. M. Jonas, E. Riedle, R. W. Schoenlein and A. J. Taylor, Oxford University Press, Oxford, 2011, pp. 553–555 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, selected intermolecular distances, total energies and depictions of the molecular structures used in this work (S1–S7), extended analysis of the gas phase potential curve (S8), and NTOs at the S1 minima (S9–S11). See DOI: 10.1039/c3pp50032b |
| ‡ The presence of only one transition between two canonical orbitals is a sufficient but not a necessary criterion for PRNTO = 1. If more than one transition is present they may still disappear due to the NTO transformation. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |