Pattern of sensitivity of progressive cutaneous squamous cell carcinoma cells to UVB and oxidative stress-induced cell death†‡
Kathleen
Barrette
a,
Nele
Zutterman
a,
Sofie
Van Kelst
a,
Charlotte
Proby
b and
Marjan
Garmyn
*a
aLaboratory of Dermatology, Catholic University of Leuven, Leuven, Belgium. E-mail: Marjan.Garmyn@med.kuleuven.be
bThe Wellcome Trust Centre for Molecular Medicine, Ninewells Hospital & Medical School, University of Dundee, Dundee, UK
First published on 23rd May 2012
Abstract
Previous investigations have demonstrated that isogenic cutaneous squamous cell carcinoma cell lines (cSCC), isolated from highly dysplastic skin (PM1), primary invasive SCC (MET1) and its lymph node metastasis (MET4), show an increasing resistance to cisplatin-induced apoptosis in the increasingly malignant MET1 and MET4 cells. To investigate whether cell death sensitivity in progressive stages of skin carcinogenesis is dependent on the kind of stress we examined the sensitivity of PM1, MET1 and MET4 cells to apoptosis in response to a single UVB-dose (mixture of genotoxic and oxidative stress), or to hydrogen peroxide and hypericin photodynamic treatment (both pure oxidative stresses). MET1 cells, followed by the MET4 cells, were more sensitive to UVB, resulting in more cell death and more apoptosis in comparison with the PM1 cells. A similar pattern of sensitivity was observed when we exposed the SCC cells to hydrogen peroxide or hypericin photodynamic treatment, which both generate mainly oxidative stress. The MET1 cells were the most sensitive to all stresses examined. The pattern of cell death sensitivity in a model of progressive cutaneous squamous cell carcinoma is dependent on the kind of stress. While more advanced skin cancer cells like MET1 and MET4 cells lose their sensitivity to the chemotherapeutic agent cisplatin, they remain sensitive to hydrogen peroxide or physical treatments, which induce major oxidative stress. This differential sensitivity could have implications for the treatment of advanced cSCC.
Introduction
Chronic exposure to UVB (280–315 nm) in sunlight is the most important risk factor for cutaneous squamous cell carcinomas (cSCCs). The underlying mechanisms of the carcinogenic effect of UVB include damage to DNA of the skin cells, via both direct and indirect mechanisms.1,2 Direct damage to DNA occurs as a consequence of direct absorption of UVB photons by the DNA, resulting in the formation of pyrimidine dimers, mainly cyclobutyl pyrimidine dimer (CPD) and pyrimidine (6–4) pyrimidinone photoproducts, which may lead to mutations if not repaired correctly via nucleotide excision repair.3 Besides the direct structural damage to DNA, UVB photons are also absorbed by other chromophores in the cell, resulting in the formation of radicals and reactive oxygen species (ROS).4 These ROS can damage proteins and DNA (modification of purines and pyrimidines, and DNA/protein cross-links) in an indirect way. To protect itself against permanent DNA damage/mutation, the cell has developed a fail-safe mechanism, whereby damage beyond repair triggers apoptotic cell death through the apoptotic process, which is evidenced by the formation of sunburn cells (SBCs).5–7 The formation of sunburn cells, arising from severe DNA damage or damage to other organelles which results in the generation of ROS, is critically regulated by several signaling cascades. Skin cells that failed to repair the UV-induced damage may die as sunburn cells, thereby escaping the risk of becoming malignant. Therefore it is important that apoptosis is induced soon after UVB irradiation.8Using a unique model of cell lines representing different tumor progression stages, previous studies in our laboratory have shown that tumor progression parallels increasing resistance to cisplatin, a therapeutic agent that causes DNA damage with inter- and intrastrand DNA crosslinks, used in the clinic for advanced cSCC.9 In this study we investigated the effect of tumor progression on the cell death response to UVB, since apoptosis is an important protection against UVB-induced mutations, involved in the initiation and progression of carcinogenesis. We tested this effect in the same unique model of isogenetic cell lines representing different tumor progression stages, namely dysplastic skin (PM1), a primary tumor (MET1) and its metastasis (MET4), and show that, in contrast to the sensitivity pattern to cisplatin, the primary cancer cells (MET1) cells are more sensitive to UVB than the PM1 cells.10
To investigate if the cell death response at different tumor progression stages is influenced by the type of stress used, we also tested the response of the cells against the pure oxidative stressors hydrogen peroxide and hypericin photodynamic treatment.
Materials and methods
Cells and cell culture
PM1, MET1 and MET4 SCC cell lines were respectively derived from forehead skin which showed moderate to severe dysplasia, from a primary squamous cell carcinoma from dorsal hand and from its metastasis within axillary lymph nodes, all from a single immunosuppressed individual.10 These cell lines were grown in Dulbecco's modified Eagle's medium plus HAMS F12 medium containing 10% fetal calf serum, hydrocortisone, mouse EGF and antibiotics in a 37 °C incubator at 5% CO2.Reagents
Hypericin was prepared, purified, and dissolved in DMSO as described in Chen et al.11 All experiments were performed using a concentration of 90 nM hypericin. Hydrogen peroxide was obtained from Sigma-Aldrich (St. Louis, MO) and all experiments were performed using a final concentration of 1 mM H2O2.UVB irradiation and hydrogen peroxide and hypericin photodynamic therapy treatment
Prior to the UVB-exposure, cell culture medium was removed and cells were rinsed twice with phosphate buffered saline (PBS). PM1, MET1 and MET4 cell lines were exposed to a single UVB dose (120 mJ cm−2, Philips TL 20W12 tubes, peak output 310 nm). After irradiation, cells were incubated again in cell culture medium for the indicated time periods.For H2O2 treatments, cells were rinsed twice with PBS and respectively incubated for the indicated time period with 1 mM H2O2 (dissolved in the cell culture medium).
For the photodynamic therapy, SCC cells were pre-incubated with hypericin for 24 hours, in subdued light conditions (<1 μW cm−2) and subsequently irradiated (2 J cm−2) in PBS by placing the samples on a plastic diffuser sheet 5 cm above a set of seven L18W30 fluorescent lamps (Osram; maximal emission between 530 and 620 nm, coinciding with the absorption peak of hypericin at 595 nm). At the surface of the diffuser, the uniform fluence rate was 4.5 mW cm−2, as measured with an IL 1400 radiometer (International Light, Newburyport, MA).
Viability assays
Metabolic activity was assessed using the tetrazolium salt MTT (Sigma, St. Louis, MO). 16 or 24 hours after treatment, cells were incubated with MTT (5 mg mL−1) in cell culture medium for 1 hour. Cleavage of MTT by dehydrogenase enzymes of metabolically active cells yields a blue formazan product. The formazan product was dissolved in DMSO and the absorbance was measured at 550 nm by spectrophotometry. In addition, cell viability was assessed using the trypan blue exclusion assay. Cells were harvested using trypsin-EDTA and resuspended in PBS. Viability was measured by The Countess™ automated cell counter (Invitrogen, Belgium).Western blot analysis
Cells were scraped in medium on ice, spun down and lysed using lysis buffer (25 mM hepes [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], 0.3 mM NaCl, 1.5 mM MgCl2, 20 mM β-glycerolphosphate, 2 mM EDTA and 2 mM EGTA (pH 7.5)) containing 1% Triton, 10% glycerol, 1 mM Na3VO4, 0.5 mM dithiothreitol, 10 μg mL−1 Leupeptin, 10 μg mL−1 aprotinin and 10 μg mL−1 antipain and phosphatase inhibitor cocktail (phosSTOP, Roche, Germany). After incubation of the lysate on ice for 20 minutes and spinning down at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 minutes, protein concentration was determined using the BCA Protein Assay Reagent (Pierce Chemical Company, Rockford, IL). Equal amounts of protein from each sample were separated by electrophoresis through SDS-PAGE gels (Invitrogen, Belgium) and transferred to a nitrocellulose membrane (Whatman Protran, UK). Equal loading of proteins was verified using Ponceau-S. Membranes were blocked for 1 hour at room temperature in Tris-buffered saline containing 0.1% Tween-20 and 5% non-fat dry milk. The membrane was incubated overnight at 4 °C with the primary antibody diluted in 5% non-fat dry milk or 5% BSA in 1 × TBS plus 0.1% Tween20. We purchased the PARP antibody from BD Biosciences (Canada). Antibodies against caspase-3 were obtained from Cell Signaling Technology (Beverly, MA). The primary antibody against actin (JLA20) was purchased from Developmental Studies Hybridoma Bank at the University of Iowa. The membranes were washed and incubated for 1 hour at room temperature with the peroxidase-conjugated secondary antibody (Cell Signaling Technology, Beverly, MA). Protein bands were visualized using enhanced chemiluminescence as described by the supplier (Amersham Pharmacia Biotech, Piscataway, NJ).
000 rpm for 20 minutes, protein concentration was determined using the BCA Protein Assay Reagent (Pierce Chemical Company, Rockford, IL). Equal amounts of protein from each sample were separated by electrophoresis through SDS-PAGE gels (Invitrogen, Belgium) and transferred to a nitrocellulose membrane (Whatman Protran, UK). Equal loading of proteins was verified using Ponceau-S. Membranes were blocked for 1 hour at room temperature in Tris-buffered saline containing 0.1% Tween-20 and 5% non-fat dry milk. The membrane was incubated overnight at 4 °C with the primary antibody diluted in 5% non-fat dry milk or 5% BSA in 1 × TBS plus 0.1% Tween20. We purchased the PARP antibody from BD Biosciences (Canada). Antibodies against caspase-3 were obtained from Cell Signaling Technology (Beverly, MA). The primary antibody against actin (JLA20) was purchased from Developmental Studies Hybridoma Bank at the University of Iowa. The membranes were washed and incubated for 1 hour at room temperature with the peroxidase-conjugated secondary antibody (Cell Signaling Technology, Beverly, MA). Protein bands were visualized using enhanced chemiluminescence as described by the supplier (Amersham Pharmacia Biotech, Piscataway, NJ).
Statistical analysis
The data were expressed as ±SD. Statistical analysis was performed using Student's t-test (two-tailed). The criterion for statistical significance was taken as p < 0.05.Results
Cell death response after UVB irradiation differs between progressive/consecutive cSCC cell lines
We studied the effect of UVB, which induces both DNA damage (genotoxic stress) as well as oxidative stress, on progressive stages of keratinocyte transformation, derived from dysplastic forehead skin (PM1), primary cSCC of the back of the left hand (MET1) and its lymph node metastasis (MET4) of the same immunosuppressed patient. This model represents a unique tumor progression of the skin with PM1 as an early, MET1 as a late and MET4 as a very late stage of cutaneous SCC.10 The aim of this study was to test whether skin tumor progression demonstrates increasing resistance to UVB.24 hours after UVB irradiation (120 mJ cm−2) the cancerous MET1 and MET4 cells show morphologically the most cell death compared to the precancerous PM1 cells (Fig. 1a). UVB decreased cell viability (Fig. 1b) and metabolic activity (Fig. 1c) in all cell lines, with the decrease most pronounced in the primary cancer cells (MET1) and progressively less in, respectively, MET4 and PM1 cells. Since apoptosis is known to be the main cell death modality following UVB stress in keratinocytes, we investigated whether the detected decrease in viability and metabolic activity in the cSCCs after UVB irradiation was due to the induction of apoptosis.6 To this end we measured PARP cleavage and caspase-3 activation, as biochemical markers of apoptosis.
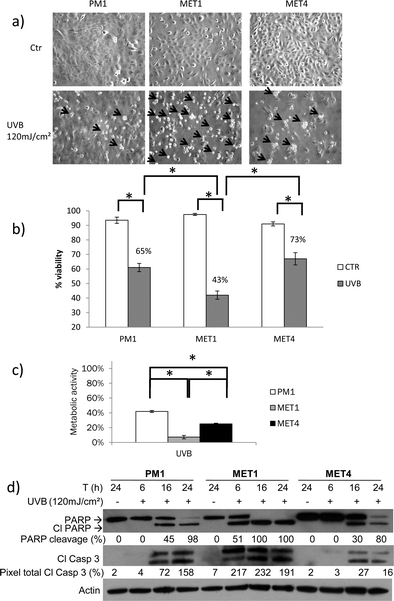 | ||
| Fig. 1 Cell death response after UVB irradiation differs between progressive/consecutive cSCC cell lines. (a) Morphologic representation of cell death 24 hours after 120 mJ cm−2 UVB. Arrows indicate dead cells. Arrows were drawn proportional to the amount of floating dead cells present. The criteria used to evaluate the morphologic representation of cell death are the amount of floating cells and the amount of cells which display membrane blebbing.23 (b) 24 hours after exposure to a single UVB dose (120 mJ cm−2), cell viability was assessed by trypan blue exclusion assay. A representative graph of at least 3 independent experiments is shown. Columns show mean of an experiment performed in duplicate, bars denote standard deviation; * p < 0.05. Numbers given are percentages of control. (c) PM1, MET1 and MET4 were exposed to a single UVB dose (120 mJ cm−2). Metabolic activity was analyzed by MTT assay and was expressed relatively to the untreated conditions (percent of control). A representative graph of at least 3 independent experiments is shown. Columns show mean of an experiment performed in triplicate, bars denote standard deviation; * p < 0.05. Metabolic activity of PM1, MET1 and MET4 after UVB irradiation was significantly less compared to control (not shown in picture). (d) SCC cells were UVB irradiated (120 mJ cm−2) and cell lysates were collected after 24 hours and were analyzed by immunoblotting with antibodies against PARP, cleaved caspase-3. Percentage of PARP cleavage (pixel total of cleaved product/(pixel total of total PARP + pixel total of cleaved product)) and total pixel of cleaved caspase-3 (pixel total of cleaved caspase 3/pixel total of actin) was calculated using UnScanIt. Actin was used a loading control. A representative blot of at least 3 independent experiments is shown. | ||
An earlier activation of the apoptotic machinery was detected after 120 mJ cm−2 UVB-irradiation in the MET1 cells (51% PARP cleavage after 6 hours) in comparison with the precancerous PM1 and the MET4 cells (PARP cleavage after 16 hours), indicating that the MET1 cells are the most sensitive to UVB stress, which is in concordance with the data obtained by MTT assay and trypan blue exclusion assay. Complete PARP cleavage was accomplished after 16 hours in the MET1 cells, whereas MET4 cells did not achieve complete PARP cleavage after 24 hours. These data are in agreement with the data obtained on the metabolic activity of the surviving cells and cell death level (MTT and trypan blue assay) suggesting that the main cell death modality is apoptosis.
We can conclude that the primary cancer cells (MET1) and, to a lesser extent, the MET4 cells, are more sensitive to UVB induced cell death than the precancerous PM1 cells, with an earlier activation of the apoptotic machinery. This is in contrast with previous findings, demonstrating that PM1 are most sensitive to CDDP (ESI‡) and that sensitivity to CDDP-induced cell death diminishes as the tumor progresses.9 To investigate whether the difference in cell death response in PM1, MET1 and MET4 is dependent on the stressor we investigated the response of the cells to two additional stressors which cause only oxidative stress and thus indirect DNA damage: H2O2 and hypericin photodynamic therapy.
Primary SCCs (MET1) are more sensitive to H2O2 than precancerous PM1 and metastatic MET4 cells
The different cancer cell lines were exposed to the non-radical ROS hydrogen peroxide for 24 hours and cell death could be seen morphologically predominantly in the MET1 cells followed by the MET4 cells (Fig. 2a). The viability, using a trypan blue assay (Fig. 2b), and the metabolic activity, using a MTT assay (Fig. 2c), of the surviving cells was assessed. The cytotoxic effect of H2O2 was most pronounced in the MET1 cells, followed by the MET4 cells and significantly less in the PM1 cells (Fig. 2b and 2c). To investigate whether the observed cell death is mainly by apoptosis, PARP cleavage and caspase-3, activation was assessed as a function of time using a Western blot analysis (Fig. 2c). PM1 cells underwent almost no apoptosis after 24 hours (1% PARP cleavage) while MET1 cells activated the apoptotic machinery after only 6 hours exposure with a 27% PARP-cleavage, with a lesser and delayed apoptosis response seen in MET4 cells. These data are in concordance with the data obtained after UVB irradiation where we also have shown the later activation of the apoptotic machinery in the precancerous PM1 cells. The primary cSCC (MET1) displayed more PARP and caspase-3 cleavage compared to the metastatic cSCC (MET4) and the precancerous PM1 cells at each timepoint. For PM1 and MET1 cells, the Western blot analysis confirmed the data observed with the MTT assay, suggesting that the main cause of cell death is apoptosis. However, MET4 cells display less apoptosis detected by the Western blot compared with the level of metabolic activity of the surviving cells (MTT assay) and cell death trypan blue exclusion assay, suggesting that MET4 cells probably also die by another cell death modality.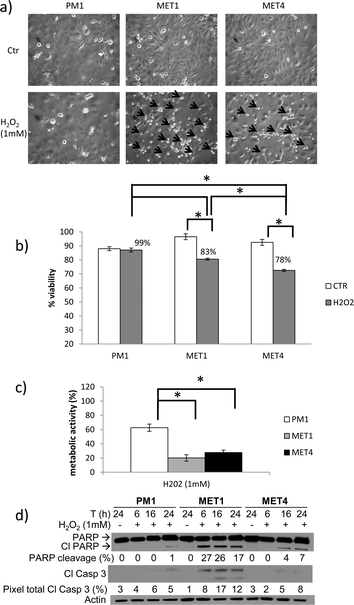 | ||
| Fig. 2 Primary SCCs (MET1), followed by metastatic MET4 cells, are more sensitive to H2O2 than precancerous PM1. PM1, MET1 and MET4 cells were treated with 1 mM of hydrogen peroxide (H2O2). (a) Morphologic representation of cell death 24 hours after treatment with H2O2. Arrows indicate dead cells. Arrows were drawn proportional to the amount of floating dead cells present. The criteria used to evaluate the morphologic representation of cell death are the amount of floating cells and the amount of cells which display membrane blebbing.23 (b) Cell viability was assessed by trypan blue exclusion assay after 24 hours of 1 mM H2O2 exposure. A representative graph of at least 3 independent experiments is shown. Columns show mean of an experiment performed in duplicate, bars denote standard deviation; * p < 0.05. Numbers given are percentages of control. (c) Metabolic activity after 24 hours, as measured by MTT assay. A representative graph of at least 3 independent experiments is shown. The metabolic activity was expressed relative to the untreated controls. Columns show mean of an experiment performed in triplicate, bars denote standard deviation; * p < 0.05. Metabolic activity of PM1, MET1 and MET4 cells treated with 1 mM H2O2 was significantly less compared to control (not shown in picture). (d) Lysates were collected at the indicated timepoints and analyzed for PARP and caspase-3 cleavage by Western blot analysis. Actin was used to confirm equal loading of proteins. Percentage of PARP cleavage (pixel total of cleaved product/(pixel total of total PARP + pixel total of cleaved product)) and total pixels of cleaved caspase-3 (pixel total of cleaved caspase 3/pixel total of actin) was calculated using UnScanIt. A representative blot of at least 3 independent experiments is shown. | ||
Primary SCCs (MET1 cells) are more sensitive to photodynamic therapy (PDT) than the precancerous PM1 and metastatic MET4 cells
PDT involves the light activation of a photosensitizing agent, which in the presence of molecular oxygen leads to the formation of reactive oxygen species (ROS) causing cellular damage. We assessed the sensitivity of the different cell lines to hypericin-mediated photodynamic therapy 24 hours after light exposure. Morphologically, we could detect the same sensitivity pattern of the cells as with the H2O2 treatment, whereby the MET1 cells show the most cell death followed by the MET4 cells (Fig. 3a). A trypan blue exclusion assay (Fig. 3b) and an MTT assay (Fig. 3c) also indicated that the MET1 and MET4 cells are significant more sensitive to the oxidative stress generated by hypericin-mediated photosensitization compared to the PM1 cells. Because hypericin-mediated photosensitization results in cell killing through either apoptosis or necrosis, depending on the intensity of the generated photo-oxidative stress (in a concentration- and light dose-dependent fashion), we also monitored the induction of apoptosis.12 MET1 cells display the most apoptosis induction, whereby the PARP cleavage is already 35% after 6 hours. The PM1 and MET4 cells display almost the same percentage of PARP cleavage after 6 hours, however the MET4 cells seem to switch to another form of cell death after 16 hours, demonstrated by the lack of PARP and caspase-3 cleavage.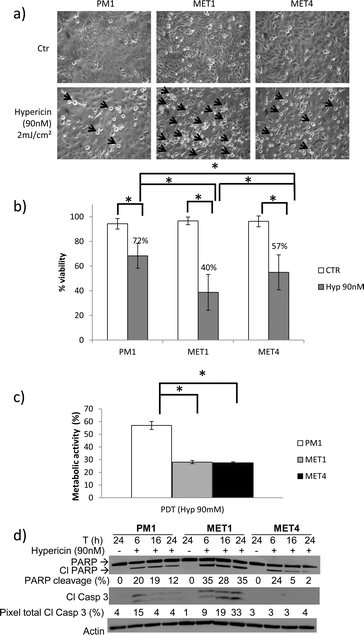 | ||
| Fig. 3 Primary SCCs (MET1), followed by MET4 cells are more sensitive to PDT than precancerous PM1 cells. PM1, MET1 and MET4 cells were exposed to a hypericin-mediated photodynamic therapy by treating the cells 24 hours before illuminating (2 J cm−2) with hypericin (90 nM). (a) Morphologic representation of cell death 24 hours after photodynamic therapy (PDT). Arrows indicate dead cells. Arrows were drawn proportional to the amount of floating dead cells present. The criteria used to evaluate the morphologic representation of cell death are the amount of floating cells and the amount of cells which display membrane blebbing.23 (b) Cell viability was assessed by trypan blue exclusion assay 24 hours after PDT. A representative graph of at least 3 independent experiment is shown. Columns show mean of an experiment performed in duplicate, bars denote standard deviation; * p < 0.05. Numbers given are percentages of control. (c) Metabolic activity was measured after 24 hours by MTT assay. A representative graph of at least 3 experiments is shown. The metabolic activity was expressed relatively to the untreated controls. Columns show mean of an experiment performed in triplicate, bars denote standard deviation; * p < 0.05. Metabolic activity of PM1, MET1 and MET4 after PDT was significantly less compared to control (not shown in picture). (d) Lysates were collected at the indicated time points and analyzed for PARP and caspase-3 cleavage by Western blot analysis. Actin was used to confirm equal loading of proteins. Percentage of PARP cleavage (pixel total of cleaved product/(pixel total of total PARP + pixel total of cleaved product)) and total pixels of cleaved caspase-3 (pixel total of cleaved caspase 3/pixel total of actin) was calculated using UnScanIt. A representative blot of at least 3 independent experiments is shown. | ||
In this study, we have shown that MET1 cells are the most sensitive to UVB, H2O2 and hypericin PDT induced cell death, followed by the MET4 cells. This is in contrast with the observed sensitivity of the progressive stages of cSCC whereby the dysplastic PM1 cells are the most sensitive and the advanced SCC were most resistant to cisplatin.9
Discussion
Cell death is an important mechanism for prevention of UVB-damage related initiation of the carcinogenic process. We investigated the effect of tumor progression on the cell death response to UVB. Surprisingly, and in contrast with the previous study, which studied the cell death response to cisplatin in this model of skin tumor progression,9 the primary cancer cells (MET1) were more sensitive to UVB-induced apoptosis in comparison with the early malignant PM1 cells, or the later MET4 metastatic cells, indicating that the pattern of sensitivity in progressive skin tumorigenesis is stress dependent.Different factors could be responsible for the loss in cell death sensitivity to cisplatin in cells representative of progressive skin tumorigenesis, including reduced uptake, increased inactivation and increased efflux of cisplatin, which could not be the case with the physical stressor UVB and hydrogen peroxide.13 Cisplatin is a genotoxic agent inducing direct DNA damage by adduct formation. Increased adduct repair and molecular mechanisms, such as activation of the AKT pathway, that inhibit propagation of DNA damage signals that the apoptotic machinery could further be responsible for loss in cell death response.14,15 Of interest in a former study we demonstrated in this model that skin tumor progression parallels enhanced AKT activation and increased resistance to cisplatin-induced apoptosis, while AKT inhibition sensitizes the apoptosis-resistant MET1 and, to a lesser extent, MET4 cells to cisplatin-mediated cell death.9
UVB not only causes direct DNA damage but UVB photons are also absorbed by other chromophores in the cell, resulting in the formation of radicals and reactive oxygen species. We hypothesize that the observed difference in the sensitivity pattern of the cSCC cells detected in cisplatin-treated versus UVB-irradiated cells may be at least partly due to the distinct response of the cells to oxidative versus genotoxic stress, because UVB generates oxidative and genotoxic stress whereas cisplatin predominantly generates genotoxic stress. This hypothesis is supported by our observation that the same pattern of sensitivity (maximal sensitivity in the MET1 cells) is observed when cells are treated with two oxidative stressors, hydrogen peroxide (H2O2) and hypericin photodynamic therapy.
Cells may die by apoptosis or via another form of cell death. After the treatment with H2O2 or hypericin PDT, MET4 cells must have died, at least in part, by another cell death modality since the MET4 cells display excess cell death (morphologically, MTT and TB assay) compared to the observed PARP and caspase-3 cleavage. The type of cell death initiated by UVB irradiation in the MET4 cells is probably apoptosis, since the observed PARP and caspase-3 cleavage concur with the observed cell death (morphologically, MTT and TB-assay). One explanation for the different cell death patterns in MET4 cells in the context of different stressors could be the formation of different forms of ROS (radical versus non-radical) since both oxidative stressors (H2O2 and hypericin PDT) form mostly non-radical ROS, while UVB irradiation leads to the formation of radical and non-radical forms of ROS which are mainly O2 and H2O2.8 The observed alternative cell death could be mediated by a caspase-independent pathway, like necrosis.
Notwithstanding the different forms of ROS formation by UVB irradiation versus H2O2 treatment and hypericin-PDT,16 the sensitivity pattern towards cell death remains the same with the MET1 cells displaying the most cell death, followed by the MET4 cells.
A second explanation for the alternative cell death evident in MET4 cells could be the difference between mainly oxidative stress (H2O2 and hypericin PDT) compared to the UVB stress, which causes oxidative but also genotoxic stress.
Our hypothesis that the observed difference in sensitivity of cSCC cells treated with cisplatin versus UVB is due to the distinct response of the cells to oxidative versus genotoxic stress is further supported by our observation that the same pattern of sensitivity (maximal sensitivity in the MET1 cells) is observed when cells are treated with two oxidative stressors, hydrogen peroxide (H2O2) and hypericin photodynamic therapy, which can however also induce indirect DNA damage. However the underlying mechanisms need to be further investigated.
We could exclude the hypothesis that sensitivity to the different stressors may be a result of differential proliferative rate between the cells since the doubling time is almost the same in het PM1 (34 hours), MET1 (30 hours) and MET4 (29 hours) cells. We also showed that the sensitivity of the PM1, MET1 and MET4 cells is different according to the type of stressor which may indicate that the sensitivity is dependent on the kind of stressor regardless the doubling time of the cells.
Sensitization of cells to ROS-mediated cell death via AKT, as an explanation for the difference in sensitivity pattern between CDDP and UVB, as previously shown in other cells,17 is probably not the mechanism involved in our experimental setting. Although AKT activation is increased in MET1 and MET4,9 we could demonstrate that AKT inhibition sensitizes our cells to UVB-induced cell death (data not shown).
The strength of this study is the use of a unique isogenic tumor progression model in which we can investigate the response of the cells to stress, excluding variations due to interindividual factors. A limitation is the rareness of this model.
Cancer research is focused on identifying new strategies for treating cancer and especially aggressive metastatic cancers. Metastatic cSCCs have a very poor prognosis due to a high recurrence rate after surgery and radiotherapy and resistance to chemotherapy. Transplantation patients who undergo chronic immunosuppression have a major risk to develop these advanced cSCCs with a high cancer burden and a significant risk of death.18 Irrespective of the underlying mechanisms involved, we were able to confirm our hypothesis that the pattern of cell death sensitivity in a model of progressive skin tumor progression is similar in response to agents which cause major oxidative stress (UVB, H2O2 and PDT). This could argue for the use of oxidative stress in chemotherapy, and is further supported by previous findings showing that oxidative stress also exerts antitumorigenic actions as it has been linked to senescence and apoptosis, two major mechanisms that counteract tumor development.19 More advanced cancer cells have a higher metabolic activity rate (Warburg effect) and this could easily translate into higher rates of formation of ROS such as H2O2, features that could be important in the development of new anticancer strategies.20,21 Cancer cells often compensate with overactivation of antioxidant systems, such as superoxide dismutases (SOD), but an overload of oxidative stress could induce more rapid cell death in cancer cells compared to normal cells. Cancer cells with increased oxidative stress are likely to be more vulnerable to damage by further ROS insults induced by exogenous agents compared to normal cells.21 There is experimental evidence that cancer cells are more susceptible to H2O2-induced cell death than normal cells when the concentration of H2O2 becomes toxic.20
However, a low concentration of oxidative stress may have several protumorigenic effects like increasing DNA mutation rate, inducing indirect DNA damage, genome instability and cell proliferation as a low H2O2 concentration.20 Also under persistent intrinsic oxidative stress, many cancer cells become well-adapted to such stress and develop an enhanced endogenous antioxidant capacity, which makes the malignant cells resistant to exogenous stress. Increased ROS stress below a toxic dose correlates in cancer cells with the aggressiveness of tumours and poor patient outcome.21 ROS, such as low levels of H2O2, also appear crucial in regulating several normal cellular functions, whereby regulated changes in ROS concentration has been found critical for functions like respiration, control of enzymatic activities, transcriptional regulation, modulation of signal transduction pathways, cell cycle progression and inflammatory response, resulting in survival of the cancer cells.20
So we believe that the toxicity of ROS is concentration dependent, with low levels of ROS stimulating carcinogenesis, whereas high levels of ROS are cytotoxic to the cancer cell.
Because advanced cancer cells such as the primary (MET1) and metastatic (MET4) SCC cells are more sensitive to oxidative stressors compared to the dysplastic cells, oxidative stress could have therapeutic implications in the treatment of advanced SCC, especially in cells which have lost their sensitivity to genotoxic strategies like cisplatin. Direct administration of H2O2 to cancer patients is not an appropriate therapy. There is increasing evidence that raising the cellular levels of H2O2 using H2O2-generating systems or by inhibition of ROS scavenging may be an efficient way of killing cancer cells. Mangafodipir, which increases H2O2 levels specifically in cancer cells, is undergoing clinical trial in a phase II studies in combination with chemotherapy in liver cancer. Chemotherapy like cisplatin is able to inhibit thioredoxin, which results in disrupted ROS scavenging. So combining cisplatin and thioredoxin inhibitors (such as flavanols) in synergistic combinations may prove to be the most useful approach.22
Conclusion
In addition to classical therapeutic agents, which induce predominantly direct DNA damage, stressors or compounds which induce mainly oxidative stress could be promising in advanced skin cancer.Acknowledgements
This work has been supported by the K.U. Leuven (grant: OT/04/42/BOF) and the F.W.O. (grant: G-08-00425).References
- S. Claerhout, L. A. Van, P. Agostinis and M. Garmyn, Pathways involved in sunburn cell formation: deregulation in skin cancer, Photochem. Photobiol. Sci., 2006, 5, 199–207 RSC.
- F. R. de Gruijl, Photocarcinogenesis: UVA vs. UVB radiation, Skin Pharmacol. Appl. Skin Physiol., 2002, 15, 316–320 CrossRef CAS.
- L. Marrot and J. R. Meunier, Skin DNA photodamage and its biological consequences, J. Am. Acad. Dermatol., 2008, 58, S139–S148 CrossRef.
- G. H. Jin, Y. Liu, S. Z. Jin, X. D. Liu and S. Z. Liu, UVB induced oxidative stress in human keratinocytes and protective effect of antioxidant agents, Radiat. Environ. Biophys., 2007, 46, 61–68 CrossRef CAS.
- B. J. Nickoloff and M. Denning, Life and death signaling in epidermis: following a planned cell death pathway involving a trail that does not lead to skin cancer, J. Invest. Dermatol., 2001, 117, 1–2 CrossRef CAS.
- A. Van Laethem, S. Claerhout, M. Garmyn and P. Agostinis, The sunburn cell: regulation of death and survival of the keratinocyte, Int. J. Biochem. Cell Biol., 2005, 37, 1547–1553 CrossRef CAS.
- B. J. Nickoloff, J. Z. Qin, V. Chaturvedi, P. Bacon, J. Panella and M. F. Denning, Life and death signaling pathways contributing to skin cancer, J. Invest. Dermatol. Symp. Proc., 2002, 7, 27–35 Search PubMed.
- A. Van Laethem, M. Garmyn and P. Agostinis, Starting and propagating apoptotic signals in UVB irradiated keratinocytes, Photochem. Photobiol. Sci., 2009, 8, 299–308 RSC.
- S. Claerhout, L. Verschooten, K. S. Van, V. R. De, C. Proby, P. Agostinis and M. Garmyn, Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma, Int. J. Cancer, 2010, 127, 2790–803 CrossRef CAS.
- C. M. Proby, K. J. Purdie, C. J. Sexton, P. Purkis, H. A. Navsaria, J. N. Stables and I. M. Leigh, Spontaneous keratinocyte cell lines representing early and advanced stages of malignant transformation of the epidermis, Exp. Dermatol., 2000, 9, 104–117 CrossRef CAS.
- B. Chen, T. Roskams, Y. Xu, P. Agostinis and P. A. de Witte, Photodynamic therapy with hypericin induces vascular damage and apoptosis in the RIF-1 mouse tumor model, Int. J. Cancer, 2002, 98, 284–290 CrossRef CAS.
- P. Agostinis, A. Vantieghem, W. Merlevede and P. A. de Witte, Hypericin in cancer treatment: more light on the way, Int. J. Biochem. Cell Biol., 2002, 34, 221–241 CrossRef CAS.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Targeting multidrug resistance in cancer, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef CAS.
- T. G. Oliver, K. L. Mercer, L. C. Sayles, J. R. Burke, D. Mendus, K. S. Lovejoy, M. H. Cheng, A. Subramanian, D. Mu, S. Powers, D. Crowley, R. T. Bronson, C. A. Whittaker, A. Bhutkar, S. J. Lippard, T. Golub, J. Thomale, T. Jacks and E. A. Sweet-Cordero, Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer, Genes Dev., 2010, 24, 837–852 CrossRef CAS.
- D. J. Peng, J. Wang, J. Y. Zhou and G. S. Wu, Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells, Biochem. Biophys. Res. Commun., 2010, 394, 600–605 CrossRef CAS.
- M. Dewaele, T. Verfaillie, W. Martinet and P. Agostinis, Death and survival signals in photodynamic therapy, Methods Mol. Biol., 2010, 635, 7–33 CrossRef CAS.
- V. Nogueira, Y. Park, C. C. Chen, P. Z. Xu, M. L. Chen, I. Tonic, T. Unterman and N. Hay, Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis, Cancer Cell, 2008, 14, 458–470 CrossRef CAS.
- S. Euvrard, J. Kanitakis and A. Claudy, Skin cancers after organ transplantation, N. Engl. J. Med., 2003, 348, 1681–1691 CrossRef.
- R. Visconti and D. Grieco, New insights on oxidative stress in cancer, Curr. Opin. Drug Discov. Devel., 2009, 12, 240–245 Search PubMed.
- M. Lopez-Lazaro, Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy, Cancer Lett., 2007, 252, 1–8 CrossRef CAS.
- D. Trachootham, J. Alexandre and P. Huang, Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS.
- J. P. Fruehauf and F. L. Meyskens Jr., Reactive oxygen species: a breath of life or death?, Clin. Cancer Res., 2007, 13, 789–794 CrossRef CAS.
- U. Ziegler and P. Groscurth, Morphological features of cell death, News Physiol. Sci., 2004, 19, 124–128 Search PubMed.
Footnotes |
| † This article is published as part of a themed issue on current topics in photodermatology. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c2pp25064k |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |