 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of protectin D1: a potent anti-inflammatory and proresolving lipid mediator†
M.
Aursnes
a,
J. E.
Tungen
a,
A.
Vik
a,
J.
Dalli
b and
T. V.
Hansen
*a
aSchool of Pharmacy, Department of Pharmaceutical Chemistry, University of Oslo, PO Box 1068 Blindern, N-0316 Oslo, Norway. E-mail: t.v.hansen@farmasi.uio.no
bCenter for Experimental Therapeutics and Reperfusion Injury, Department of Anesthesiology, Perioperative and Pain Medicine, Harvard Institutes of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts 02115, USA
First published on 1st November 2013
Abstract
A convergent stereoselective synthesis of the potent anti-inflammatory, proresolving and neuroprotective lipid mediator protectin D1 (2) has been achieved in 15% yield over eight steps. The key features were a stereocontrolled Evans-aldol reaction with Nagao's chiral auxiliary and a highly selective Lindlar reduction of internal alkyne 23, allowing the sensitive conjugated E,E,Z-triene to be introduced late in the preparation of 2. The UV and LC/MS–MS data of synthetic protectin D1 (2) matched those obtained from endogenously produced material.
Introduction
Polyunsaturated fatty acids (PUFAs), such as docosahexaenoic acid (1, DHA), play a major role in the physiology of living organisms.1 Recent efforts by the Serhan research group have established that DHA (1) is a substrate for the biosynthesis of several potent anti-inflammatory proresolving mediators, such as protectin D1 (2),2 maresin 1,3 resolvin D1 and resolvin D3.2a,4 All of these compounds have enabled new research areas related to many disease states associated with inflammation.5 It was reported that protectin D1 (2) is biosynthesized from DHA (1) via a lipoxygenase-mediated pathway that converts 1 by 15-lipoxygenase (15-LO) to the 17S-hydroperoxide intermediate (3), which is rapidly converted into the 16,17-epoxide (4), followed by enzymatic hydrolysis to the anti-inflammatory and proresolving oxygenated lipid 2 (Fig. 1).6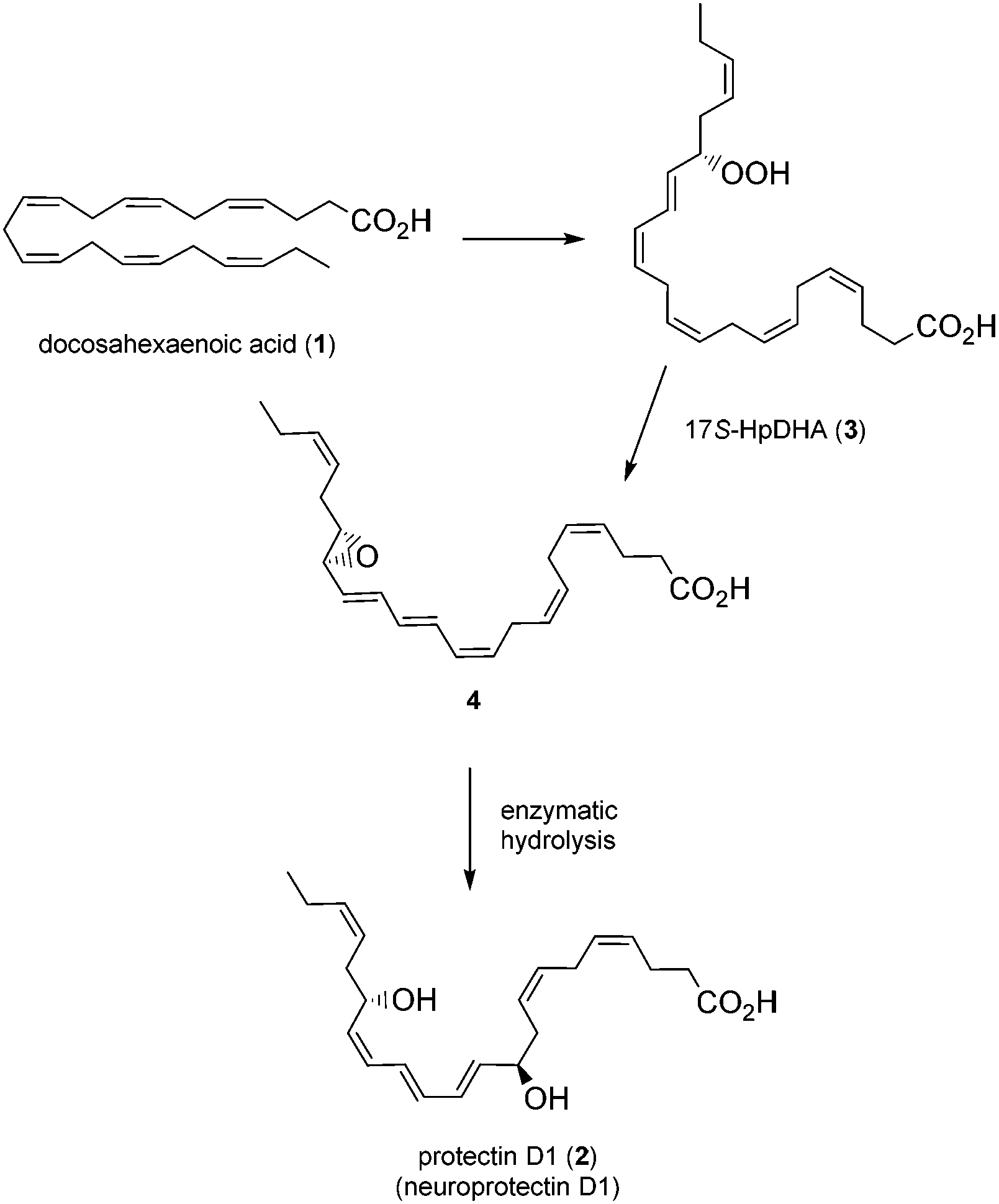 | ||
| Fig. 1 Biosynthesis of protectin D1 (2). | ||
This compound has been reported to exhibit strong in vivo protective activity in several inflammatory6 as well as many other disease models.7–10 For example, the oxygenated polyunsaturated fatty acid 2 protects the retina and the brain from oxidative stress with very potent agonist activities.7 It is noteworthy that 2 was observed to be several orders of magnitude more potent in vivo than its precursor DHA.2c Moreover, additional biological effects have recently been reported for this C22-oxygenated metabolite.11 Hence, protectin D1 (2) is very interesting as a lead compound for the development of potential new anti-inflammatory drugs.12 The prefix neuro is added when this oxygenated PUFA is formed by neural tissues.2a As of today, two syntheses of protectin D1 (2) have appeared.6,13 In connection with our interest in the synthesis of biologically active PUFA-derived natural products,14 as well as the many interesting biological activities of protectin D1 (2), we decided to prepare the DHA derived product 2. A common structural feature for several of the lipid mediators isolated by the Serhan group2–4 is the chemically unstable E,E,Z-triene connected to either one or two secondary allylic alcohols. In the retrosynthetic analysis of 2, Fig. 2, the aldehyde 6 is a key intermediate.
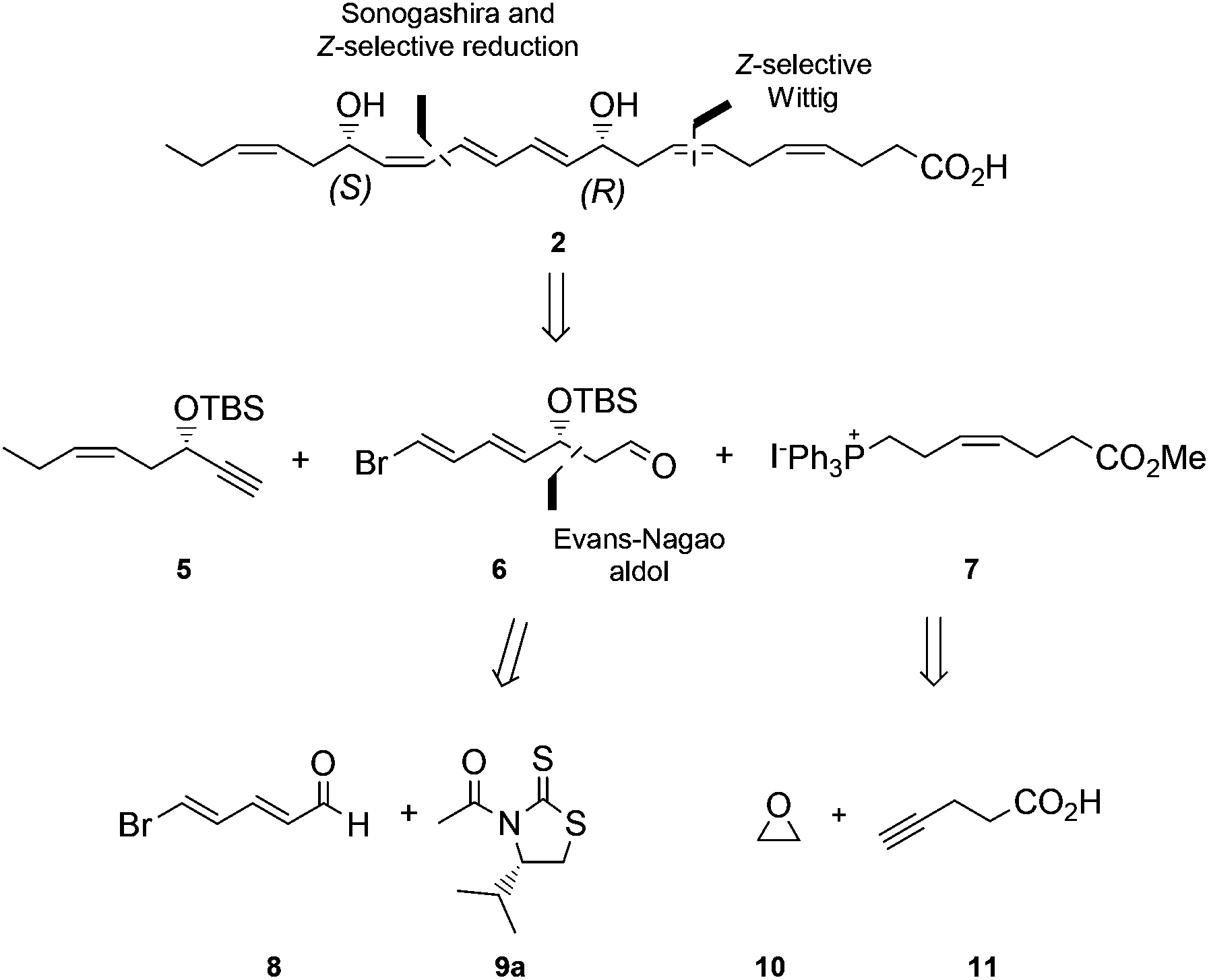 | ||
| Fig. 2 Retrosynthetic analysis of protectin D1 (2). | ||
Results and discussion
Our synthesis of 2 commenced with the preparation of 5, essentially as previously reported,15 from 1-butyne (12) and THP-protected (R)-glycidol 13 (Scheme 1).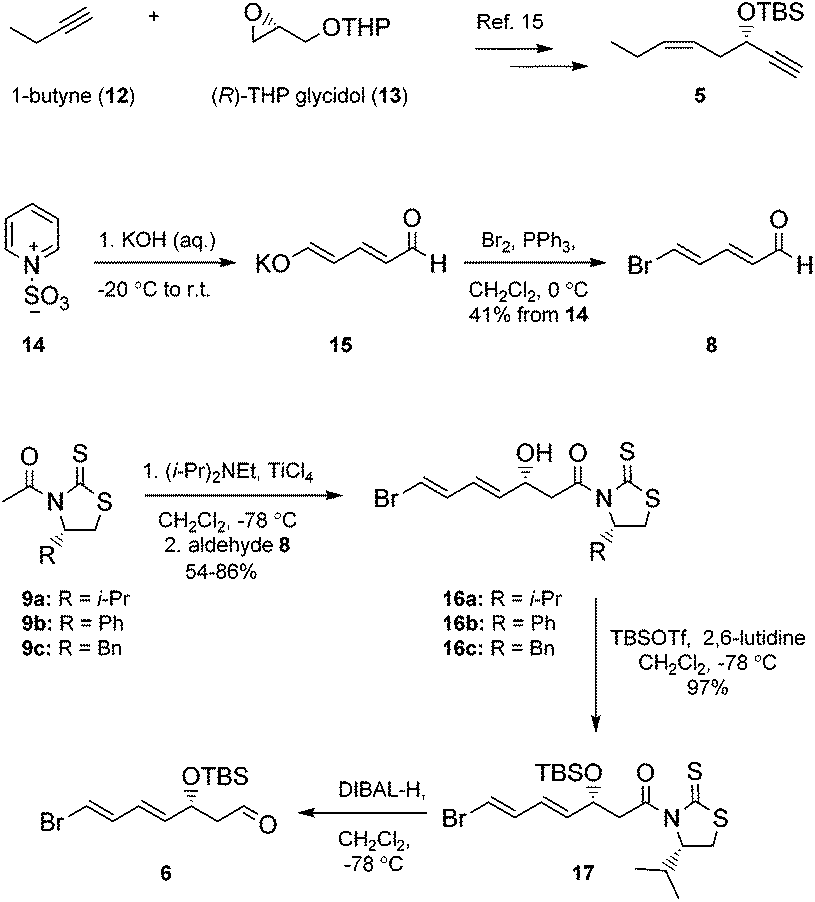 | ||
| Scheme 1 Synthesis of alkyne 5 and aldehyde 6. | ||
Aldehyde 8 was prepared by a slightly modified and improved literature protocol.16 Commercially available pyridinium-1-sulfonate (14) was treated with aqueous potassium hydroxide at −20 °C to yield glutaconaldehyde potassium salt 15 that was transformed further with the Br2/PPh3 complex to (2E,4E)-5-bromopenta-2,4-dienal (8) in 41% yield over the two steps. This sensitive aldehyde was then reacted with thiazolidinone 9a, developed by Nagao and co-workers,17 in an Evans-aldol18 type reaction using conditions developed by Olivo and co-workers (TiCl4, Et(i-Pr)2N, CH2Cl2, −78 °C).19 This smoothly produced the intermediate 16a in a 15.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio as determined by HPLC and 1H NMR analyses. We also investigated reactions using thiazolidinones 9b and 9c, with the phenyl and the benzyl group, respectively, which afforded 16b and 16c with lower diastereoselectivity (4.5:1 and 9.8:1). Purification by chromatography yielded diastereomeric pure 16a in 86% isolated yield. Protection of the alcohol functionality in 16a to compound 17 was achieved using standard conditions.20 Then DIBAL-H-reduction of 17 in CH2Cl2 at −78 °C afforded the sensitive aldehyde 6 (Scheme 1).
:1 diastereomeric ratio as determined by HPLC and 1H NMR analyses. We also investigated reactions using thiazolidinones 9b and 9c, with the phenyl and the benzyl group, respectively, which afforded 16b and 16c with lower diastereoselectivity (4.5:1 and 9.8:1). Purification by chromatography yielded diastereomeric pure 16a in 86% isolated yield. Protection of the alcohol functionality in 16a to compound 17 was achieved using standard conditions.20 Then DIBAL-H-reduction of 17 in CH2Cl2 at −78 °C afforded the sensitive aldehyde 6 (Scheme 1).
Next, the Wittig-salt 7 was synthesized. The dianion of 4-pentynoic acid (11) in HMPA,21 prepared by treatment with excess n-BuLi, was reacted with ethylene oxide (10). This afforded 7-hydroxy-hept-4-ynoic acid which was directly esterified to 18 (MeOH, catalytic H2SO4), see Scheme 2. Reduction of the internal alkyne in 18 using the Lindlar reaction gave (Z)-methyl 7-hydroxyhept-4-enoate (19) with high stereochemical purity as determined by 1H NMR analyses. Then an Appel reaction22 provided the iodide 20 which was treated with PPh3 in acetonitrile to provide the Wittig-salt 7 in a total yield of 42% from 11. Conditions for the Z-stereoselective Wittig reaction between the key aldehyde 6 and the salt 7 were then investigated. Different bases, i.e. LiHMDS, KHMDS, NaHMDS, temperatures as well as altering the concentrations of 6 and 7, with or without different amounts of HMPA in THF, all resulted in lower Z-selectivity. The best result was obtained when aldehyde 6 and the ylide of 7, the latter obtained after treatment with NaHMDS in THF, were reacted at −78 °C. This afforded the bromo-E,E,Z,Z-tetraene ester 21 (Scheme 2).
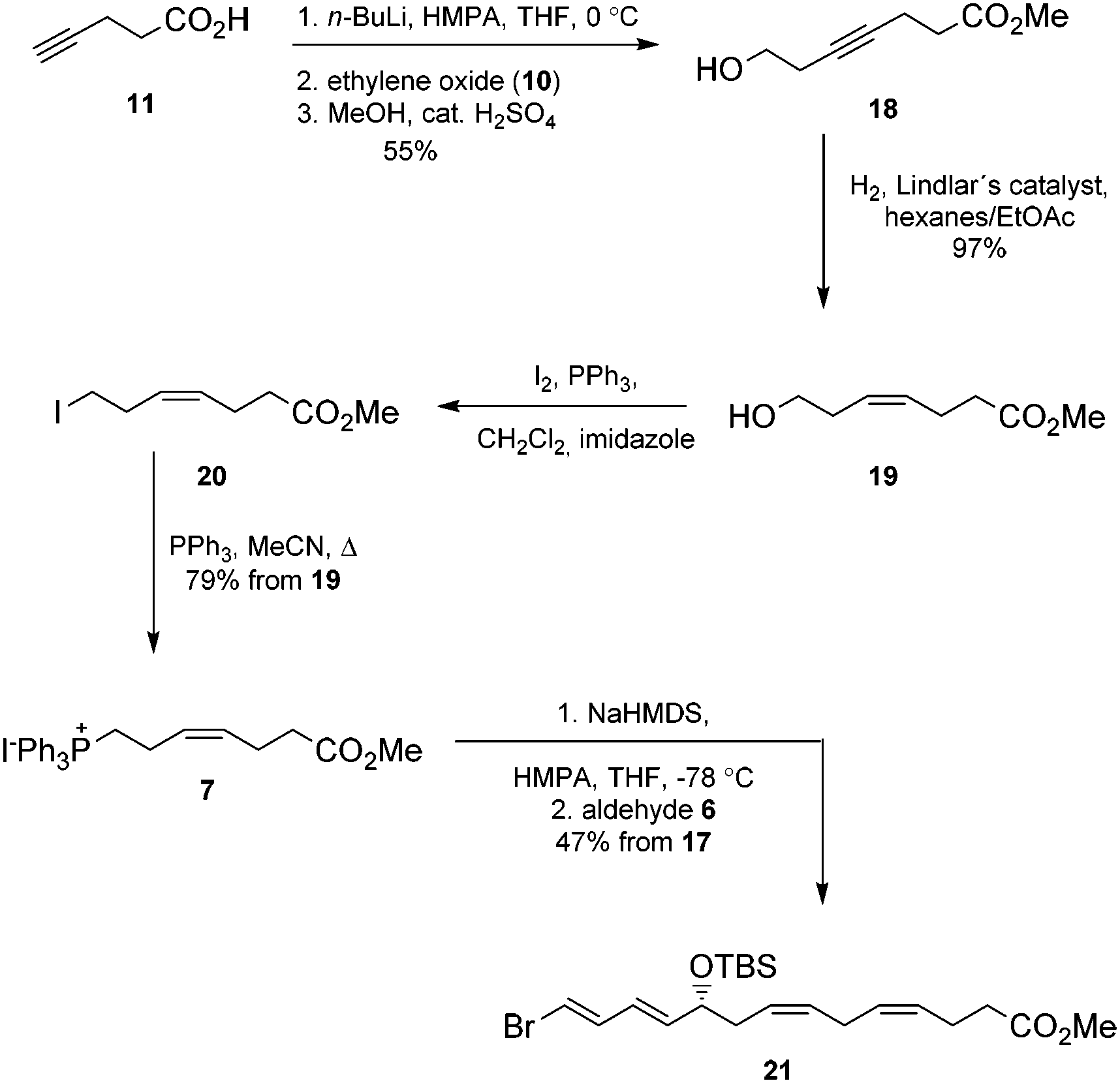 | ||
| Scheme 2 Synthesis of ester 21. | ||
Chromatographic purification on silica gel yielded stereochemically pure product 21 (HPLC, 1H-NMR) in 47% yield over two steps. Then alkyne 5 was reacted with 21 in a Sonogashira reaction23 at ambient temperature in the presence of Pd(PPh3)4 and CuI using diethyl amine as a solvent. This afforded the bis-hydroxyl-protected methyl ester 22 in 95% yield. Deprotection of the two TBS-groups in 22 was achieved with an excess of five equivalents of TBAF in THF at 0 °C to afford 81% yield of the diol 23.24 The internal conjugated alkyne in 23 was reduced to the methyl ester 24 in 65% yield after chromatographic purification on silica. A modified Lindlar hydrogenation reaction25 produced triene 24 with high stereoselectivity, while the diimide reduction26 or the standard Lindlar hydrogenation reaction27 of 23 failed to give a high conversion to 24. The Boland reduction28 gave in our hands a large amount of elimination of water from 23. Finally, lenient saponification of the methyl ester 24 at 0 °C with dilute aqueous LiOH in methanol followed by mild acidic work-up (aqueous NaH2PO4) afforded a 78% yield of protectin D1 (2) in the last step (Scheme 3).
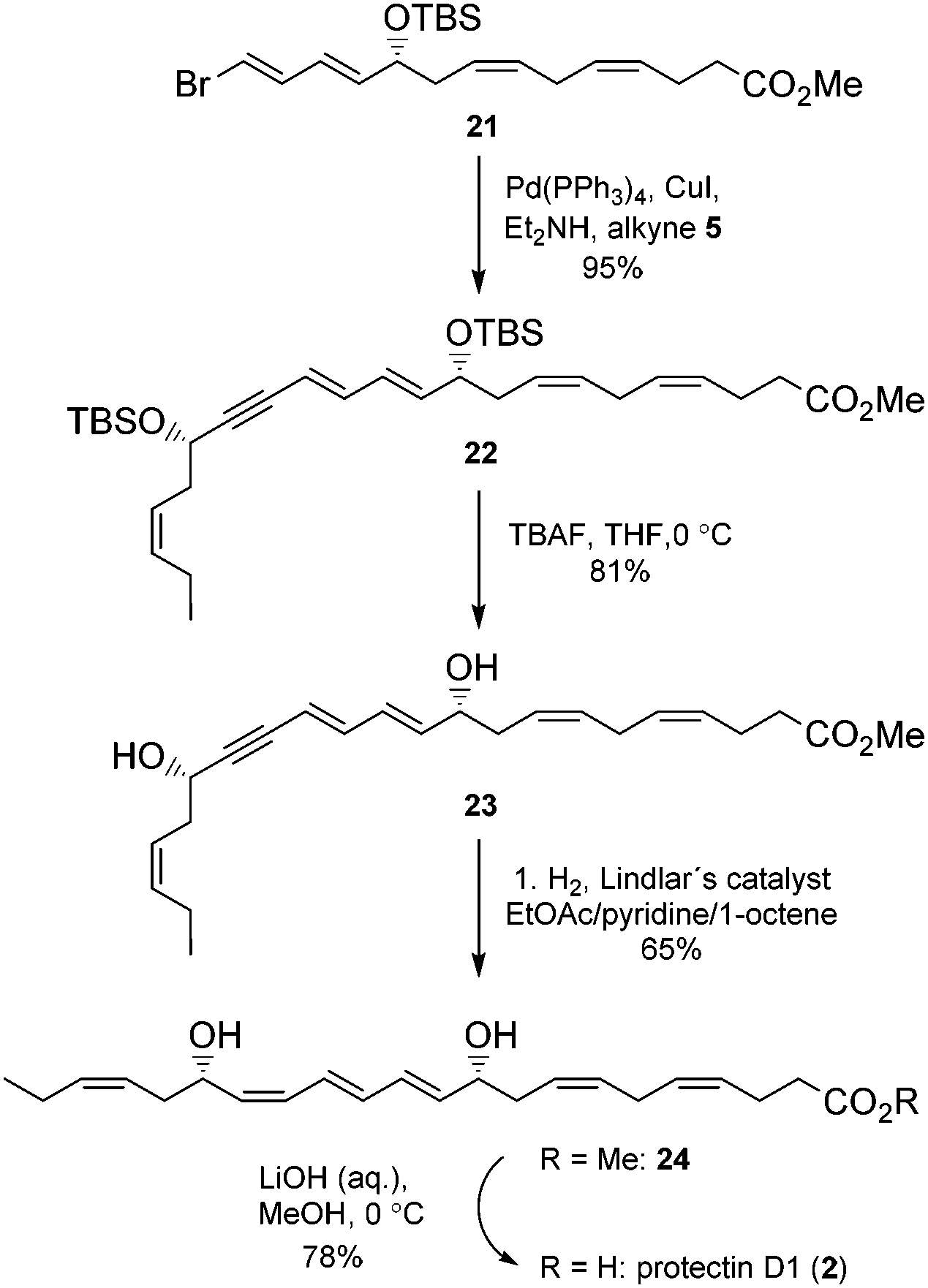 | ||
| Scheme 3 Synthesis of protectin D1 (2). | ||
The chemical purity of synthetic 2 and 24 was determined to be >95% and >98%, respectively, by HPLC analyses (see ESI†). The UV spectrum of synthetic protectin D1 (2) showed absorbance peaks (λMeOHmax) at 262, 271 and 282 nm, which is in excellent agreement with the literature.6 In order to obtain evidence that synthetic 2 and 24 matched that of authentic protectin D1 (2), protectin D1 (2) was obtained from endogenous murine self-resolving exudates.29Fig. 3 shows that the synthetic 2 was matched with endogenously produced 2.
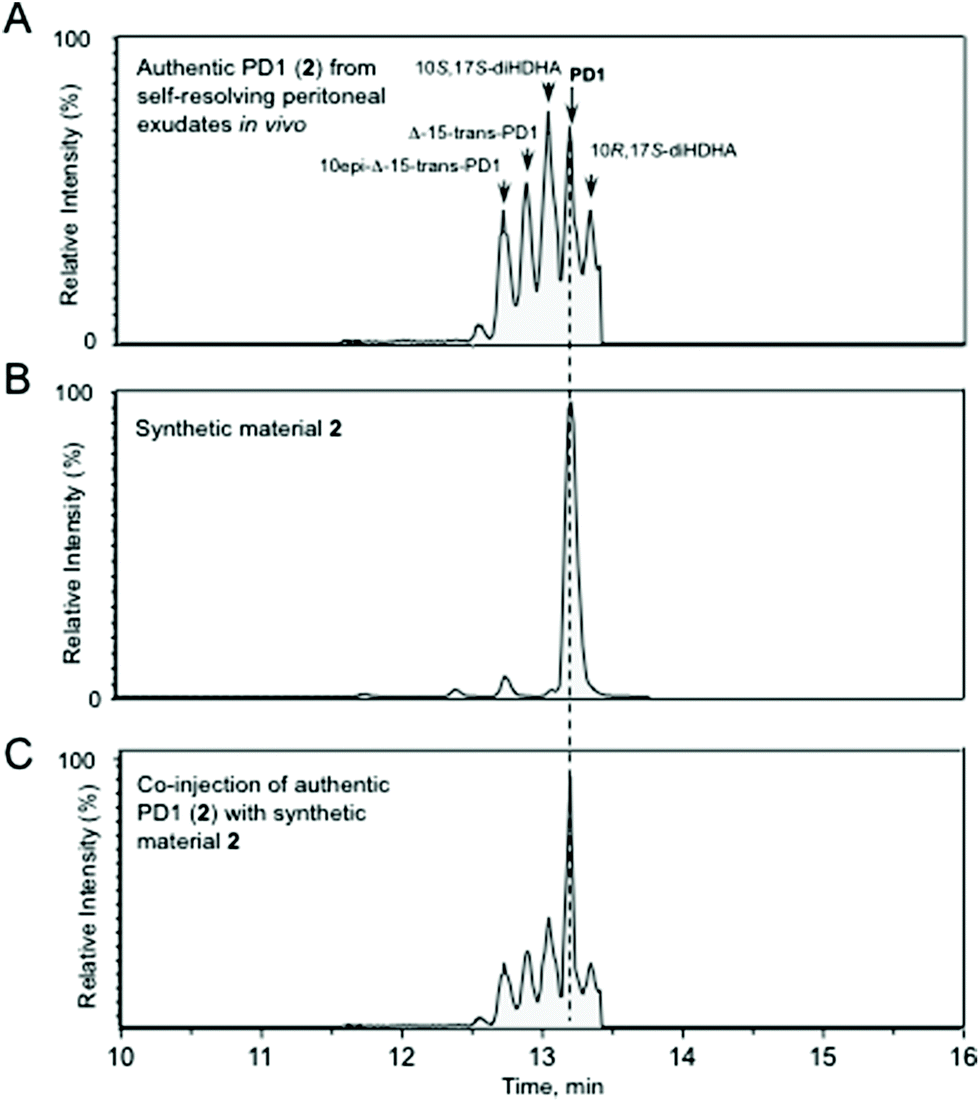 | ||
| Fig. 3 HPLC chromatograms obtained from the matching experiments. Authentic protectin D1 (2) from self-resolving peritoneal inflammatory exudates matched synthetic material protectin D1 (2). Selected ion chromatograms (m/z 359–153) depicting (A) authentic protectin D1 (2), marked as PD1, obtained from mice injected with Escherichia coli (105 CFU) and exudates collected at 12 h; (B) synthetic protectin D1 (2) and (C) coinjection of protectin D1 (2) from self-resolving inflammatory exudates with synthetic material protectin D1 (2). Figures (A)–(C) are representative HPLC chromatograms (n = 4). | ||
In Fig. 3A authentic protectin D1 (2) obtained in vivo from exudates is displayed amongst its stereoisomers.30Fig. 3B shows the chromatographic behaviour of endogenously produced 2 (TR = 13.2 min) and Fig. 3C demonstrates that synthetic 2 co-elutes with endogenous 2. In addition, the MS–MS spectra for both biosynthesized 2 and synthetic 2 displayed essentially identical MS–MS fragmentation spectra with the following fragments assigned: m/z 359 = M-H, m/z 341 = M-H-H2O, m/z 323 = M-H-2H2O, m/z 315 = M-H-CO2, m/z 297 = M-H-H2O-CO2, m/z 279 = M-H-2H2O-CO2, m/z 243 = 261-H2O, m/z 199 = 261-H2O-CO2, m/z 188 = 206-H2O, m/z 135 = 153-H2O, m/z 121 = 181-H2O-CO2, m/z 109 = 153-CO2 (see ESI†). Similar results were also obtained when synthetic ester 24 was hydrolysed to the acid 2 and compared with authentic protectin D1 (2). The chromatographic properties of synthetic 2 and the free acid of 24, the latter obtained by hydrolysis with aqueous LiOH in THF,6 were matched with data of endogenously formed protectin D1 (2). These results demonstrated that hydrolyzed 24 co-elutes with authentic 2. Furthermore, the MS–MS spectra for both the free acid obtained from 24 and biosynthesized 2 displayed essentially identical MS–MS fragmentation spectra (see ESI†). Our NMR spectral data of synthetic 2 were in accord with those published by Petasis, Serhan and co-workers,13b but not with the spectra published by others.13a
Conclusions
In summary, the potent endogenously produced lipid mediator protectin D1 (2) was prepared in eight steps and in 15% yield from the known aldehyde 8 in a convergent manner. Our synthesis of 2 compares well with those previously reported with respect to yields and simplicity, affording multi-mg quantities of this potent and biologically interesting natural product. The synthetic material displayed identical chromatographic properties with endogenously produced protectin D1 (2). Further in vivo biological studies are ongoing and will be reported elsewhere.Experimental
(R,4E,6E)-7-Bromo-3-((tert-butyldimethylsilyl)oxy)hepta-4,6-dienal (6)
Aldehyde 6 was prepared by a DIBAL-H reduction of the protected thiazolidinethione 17 according to the procedure of Olivo et al.19b All spectroscopic and physical data were in full agreement with those reported in the literature.19b [α]20D = 31.5 (c = 0.2, CHCl3); 1H NMR (300 MHz, CDCl3) δ 9.75 (t, J = 2.2 Hz, 1H), 6.69 (dd, J = 13.4, 10.8 Hz, 1H), 6.33 (d, J = 13.6 Hz, 1H), 6.16 (ddd, J = 15.2, 10.6, 1.3 Hz, 1H), 5.75 (ddd, J = 15.3, 5.9, 0.8 Hz, 1H), 4.66 (dd, J = 6.8, 5.5 Hz, 1H), 2.75–2.41 (m, 2H), 0.87 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 201.2, 136.6, 136.1, 127.6, 109.6, 68.5, 51.4, 25.9, 14.3, −4.2, −4.9.(R,4E,6E)-7-Bromo-3-hydroxy-1-((S)-4-isopropyl-2-thioxothia-zolidin-3-yl)hepta-4,6-dien-1-one (16a)
The (R)-aldol product 16a was prepared in 86% yield from dienal 8 and the auxiliary 9a according to the procedure of Olivo and coworkers.19a The diastereomeric ratio (15.3:1) on the crude product was determined by HPLC analysis (Eclipse XDB-C18, MeOH–H2O 70:30, 1.0 mL min−1, tr(minor) = 8.65 min and tr(major) = 10.85 min). All spectroscopic and physical data were in full agreement with those reported in the literature.19a [α]20D = 271.3 (c = 0.13, CHCl3); 1H NMR (300 MHz, CDCl3) δ 6.72 (dd, J = 13.5, 10.8 Hz, 1H), 6.35 (d, J = 13.6 Hz, 1H), 6.26 (ddd, J = 15.3, 10.8, 1.5 Hz, 1H), 5.79 (dd, J = 15.3, 5.4 Hz, 1H), 5.16 (dd, J = 7.8, 6.4 Hz, 1H), 4.76–4.65 (m, 1H), 3.70 (dd, J = 17.6, 3.1 Hz, 1H), 3.53 (dd, J = 11.5, 7.9 Hz, 1H), 3.29 (dd, J = 17.6, 8.6 Hz, 1H), 3.04 (dd, J = 11.6, 1.1 Hz, 1H), 2.93 (d, J = 4.5 Hz, 1H), 2.36 (dq, J = 13.6, 6.8 Hz, 1H), 1.07 (d, J = 6.7 Hz, 3H), 0.99 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 203.1, 172.3, 136.7, 134.8, 128.0, 109.6, 71.5, 68.1, 45.1, 31.0, 30.8, 19.2, 18.0.
(R,4E,6E)-7-Bromo-3-((tert-butyldimethylsilyl)oxy)-1-((S)-4-isopropyl-2-thioxothiazolidin-3-yl)hepta-4,6-dien-1-one (17)
According to the procedure of Corey and coworkers,31 the alcohol 16a was protected with a TBS-group. Yield: 4.2 g (97%); [α]20D = 263 (c = 0.5, CHCl3); 1H NMR (300 MHz, CDCl3) δ 6.69 (dd, J = 13.4, 10.7 Hz, 1H), 6.31 (d, J = 13.5 Hz, 1H), 6.15 (dd, J = 15.5, 11.1 Hz, 1H), 5.79 (dd, J = 14.9, 6.6 Hz, 1H), 5.04 (t, J = 7.0 Hz, 1H), 4.75 (q, J = 6.4 Hz, 1H), 3.64 (dd, J = 16.6, 7.8 Hz, 1H), 3.47 (dd, J = 10.9, 7.9 Hz, 1H), 3.21 (dd, J = 16.4, 4.6 Hz, 1H), 3.03 (d, J = 11.6 Hz, 1H), 2.48–2.26 (m, 1H), 1.06 (d, J = 7.2 Hz, 3H), 0.97 (d, J = 7.1 Hz, 3H), 0.86 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 202.9, 170.9, 136.8, 127.4, 109.1, 71.8, 69.8, 46.2, 31.0, 30.9, 25.9 (3C), 19.3, 18.2, 17.9, −4.2, −4.8.Methyl (R,4Z,7Z,11E,13E)-14-bromo-10-((tert-butyldimethyl-silyl)oxy)tetradeca-4,7,11,13-tetraenoate (21)
To the Wittig salt 7 (581 mg, 1.04 mmol, 1.0 equiv.) in THF (9.5 mL) was added mol. sieves and HMPA (1.5 mL) before NaHMDS (0.6 M in toluene, 1.0 equiv.) was slowly added at −78 °C and then stirred for 5 min at 0 °C. Aldehyde 6 (prepared from DIBAL-H reduction of 17 as described above) was added at −78 °C. The solution was allowed to slowly warm up to room temperature in the dry ice/acetone bath for 24 h before it was quenched with phosphate buffer (10 mL, pH = 7.2). Et2O (15 mL) was added and the phases were separated. The aqueous phase was extracted with Et2O (2 × 15 mL) and the combined organic layers were dried (Na2SO4), before it was concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes–EtOAc 95:5) to afford the title compound 21 as a yellow oil. Yield: 217 mg (47% for two steps starting from 17); TLC (hexanes–EtOAc 95:5, CAM stain): Rf = 0.29; [α]20D = −9.4 (c = 0.1, MeOH); 1H NMR (400 MHz, CDCl3) δ 6.68 (dd, J = 13.4, 10.9 Hz, 1H), 6.27 (d, J = 13.5 Hz, 1H), 6.09 (dd, J = 15.2, 10.8 Hz, 1H), 5.72 (dd, J = 15.2, 5.8 Hz, 1H), 5.48–5.32 (m, 4H), 4.16 (q, J = 6.0 Hz, 1H), 3.67 (s, 3H), 2.84–2.73 (m, 2H), 2.38–2.35 (m, 4H), 2.35–2.21 (m, 2H), 0.89 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 173.6, 138.0, 137.1, 129.9, 129.4, 128.0, 126.7, 125.6, 108.3, 72.6, 51.7, 36.3, 34.1, 26.0 (3C), 25.9, 23.0, 18.4, −4.4, −4.6. HRMS (TOF ES+): Exact mass calculated for C21H35O3Si79BrNa [M + Na]+: 465.1436, found 465.1431.
Methyl (4Z,7Z,10R,11E,13E,17S,19Z)-10,17-bis((tert-butyldi-methylsilyl)oxy)docosa-4,7,11,13,19-pentaen-15-ynoate (22)
To a solution of vinyl bromide 21 (218 mg, 0.49 mmol, 1.0 equivalent) in Et2NH (1.2 mL) and benzene (0.4 mL), Pd(PPh3)4 (17 mg, 0.02 mmol, 3 mol%) was added and the reaction was stirred for 45 min in the dark. CuI (5 mg, 0.03 mmol, 5 mol%) in a minimum amount of Et2NH was added followed by dropwise addition of alkyne 5 (117 mg, 0.49 mmol, 1.0 equiv.) in Et3N (1.0 mL). After 20 h of stirring at ambient temperature, the reaction was quenched by the addition of saturated NH4Cl (15 mL). Et2O (15 mL) was added and the phases were separated. The aqueous phase was extracted with Et2O (2 × 15 mL) and the combined organic layers were dried (Na2SO4), before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes–EtOAc 95:5) to afford the title compound 22 as a pale yellow oil. Yield: 278 mg (95%); TLC (hexanes–EtOAc 9:1, CAM stain): Rf = 0.44; [α]20D = −15.5 (c = 0.20, MeOH); 1H NMR (400 MHz, CDCl3) δ 6.51 (dd, J = 15.6, 10.9 Hz, 1H), 6.19 (dd, J = 15.2, 10.8 Hz, 1H), 5.76 (dd, J = 15.2, 6.0 Hz, 1H), 5.58 (dd, J = 15.3, 1.2 Hz, 1H), 5.55–5.47 (m, 1H), 5.45–5.33 (m, 5H), 4.47 (td, J = 6.5, 1.6 Hz, 1H), 4.19 (q, J = 6.3 Hz, 1H), 3.67 (s, 3H), 2.82–2.74 (m, 2H), 2.43 (t, J = 7.2 Hz, 2H), 2.40–2.33 (m, 4H), 2.33–2.20 (m, 2H), 2.07 (p, J = 7.4 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.89 (s, 9H), 0.12 (d, J = 8.3 Hz, 6H), 0.03 (d, J = 8.7 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 173.7, 141.2, 139.2, 134.4, 129.8, 129.5, 128.7, 128.0, 125.8, 124.1, 110.7, 93.5, 83.5, 72.8, 63.7, 51.7, 36.8, 36.4, 34.2, 26.0 (3C), 26.0 (3C), 25.9, 23.0, 20.9, 18.5, 18.4, 14.4, −4.3 (2C), −4.6, −4.8. HRMS (TOF ES+): Exact mass calculated for C35H60O4Si2Na [M + Na]+: 623.3927, found 623.3923.
Methyl (4Z,7Z,10R,11E,13E,17S,19Z)-10,17-dihydroxydocosa-4,7,11,13,19-pentaen-15-ynoate (23)
TBAF (587 mg, 2.25 mmol, 5.0 equiv., 1.0 M in THF) was added to a solution of TBS-protected alcohol 22 (270 mg, 0.45 mmol, 1.0 equiv.) in THF (6.0 mL) at 0 °C. The reaction was stirred for 20 h before it was quenched with phosphate buffer (pH = 7.2, 3.5 mL). Brine (30 mL) and EtOAc (30 mL) were added and the phases were separated. The water phase was extracted with EtOAc (2 × 30 mL) and the combined organic layer was dried (Na2SO4) before being concentrated in vacuo. The crude product was purified by column chromatography on silica (hexanes–EtOAc 7:3) to afford the title compound 23 as a pale yellow oil. Yield: 135 mg (81%); TLC (hexanes–EtOAc 7:3, CAM stain): Rf = 0.19; [α]20D = −9.2 (c = 0.3, MeOH); 1H NMR (400 MHz, MeOD-d4) δ 6.54 (dd, J = 15.6, 10.8 Hz, 1H), 6.29 (dd, J = 15.2, 10.8 Hz, 1H), 5.82 (dd, J = 15.2, 6.2 Hz, 1H), 5.66 (dd, J = 15.1, 1.8 Hz, 1H), 5.57–5.32 (m, 6H), 4.41 (t, J = 6.7 Hz, 1H), 4.14 (q, J = 6.5 Hz, 1H), 3.66 (s, 3H), 2.87–2.74 (m, 2H), 2.46–2.28 (m, 6H), 2.10 (p, J = 7.4 Hz, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, MeOD-d4) δ 175.3, 142.5, 139.8, 135.3, 131.0, 130.3, 130.3, 129.0, 126.4, 124.7, 111.8, 93.9, 84.3, 72.7, 63.3, 52.1, 36.9, 36.2, 34.8, 26.7, 23.8, 21.7, 14.6. HRMS (TOF ES+): Exact mass calculated for C23H32O4Na [M + Na]+: 395.2198, found 395.2206.
Methyl (4Z,7Z,10R,11E,13E,15Z,17S,19Z)-10,17-dihydroxydo-cosa-4,7,11,13,15,19-hexaenoate (24)
To a solution of alkyne 23 (30 mg, 0.082 mmol) in EtOAc–pyridine–1-octene (0.83 mL, 10:1:1) under argon, Lindlar's catalyst (10 mg) was added and the flask was evacuated and filled with argon. The reaction was stirred for 3.5 h at ambient temperature under a balloon of hydrogen gas until completion. The reaction mixture was loaded directly onto a silica gel column and purified by chromatography (hexanes–EtOAc 8:2) to afford the title compound 24 as a pale oil. The chemical purity (>98%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH–H2O 75:25, 1.0 mL min−1): tr(minor) = 12.62 min, and tr(major) = 9.07 min. Yield: 19.5 mg (65%); TLC (hexanes–EtOAc 6:4, CAM stain): Rf = 0.19; [α]20D = −22.2 (c = 0.4, MeOH); UV (MeOH) λmax 262, 271, 282 nm. 1H NMR (400 MHz, MeOD-d4) δ 6.52 (dd, J = 14.0, 10.7 Hz, 1H), 6.33–6.18 (m, 2H), 6.07 (t, J = 11.1 Hz, 1H), 5.76 (dd, J = 14.5, 6.5 Hz, 1H), 5.49–5.32 (m, 7H), 4.56 (dt, J = 8.9, 6.7 Hz, 1H), 4.14 (q, J = 6.5 Hz, 1H), 3.65 (s, 3H), 2.87–2.78 (m, 2H), 2.40–2.29 (m, 7H), 2.25–2.16 (m, 1H), 2.07 (p, J = 7.4 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, MeOD-d4) δ 175.3, 138.0, 134.9, 134.9, 134.7, 131.4, 130.9, 130.5, 130.3, 128.9, 128.9, 126.5, 125.3, 73.0, 68.6, 52.1, 36.4, 36.4, 34.8, 26.7, 23.8, 21.7, 14.6. HRMS (TOF ES+): Exact mass calculated for C23H34O4Na [M + Na]+: 397.2354, found 397.2365. All spectroscopic and physical data were in agreement with those reported in the literature.13b
Synthesis of protectin D1 (2)
Methyl ester 24 (18 mg, 0.032 mmol) was dissolved in methanol–water 1:1 (30 mL) and cooled to 0 °C. LiOH (1.0 M, 1.9 mL) was added dropwise. The reaction mixture was stirred at the above-mentioned temperature for 48 h, after which a saturated solution of NaH2PO4 (4.0 mL) was added. Next, NaCl (10.0 g) was added followed by EtOAc (50 ml). The organic phase was decanted, dried (Na2SO4), and concentrated in vacuo affording the title compound 2 (14 mg, 78%) as a colourless oil. The chemical purity (>95%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH–3.3 mM HCOOH in H2O, 7:3, 1.0 mL min−1): tr(minor) = 9.97 min and tr(major) = 10.68 min; TLC (hexanes–EtOAc 6:4, CAM stain): Rf = 0.03; [α]20D = −24.0 (c = 0.3, MeOH); UV (MeOH) λmax 262, 271, 282 nm. IR (neat) ν = 3316, 3012, 2961, 2930, 1713, 1557 cm−1; 1H NMR (300 MHz, MeOH-d4) δ 6.52 (dd, J = 14.1, 11.3 Hz, 1H), 6.35–6.19 (m, 2H), 6.08 (dd, J = 11.7, 10.5 Hz, 1H), 5.76 (dd, J = 14.4, 6.5 Hz, 1H), 5.52–5.31 (m, 7H), 4.56 (dt, J = 9.4, 6.8 Hz, 1H), 4.21–4.08 (m, 1H), 2.88–2.78 (m, 2H), 2.42–2.15 (m, 8H), 2.12–2.00 (m, 2H), 0.97 (t, J = 7.5 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 177.4, 137.9, 134.9, 134.8, 134.7, 131.4, 131.0, 130.6, 130.0, 129.3, 128.9, 126.5, 125.3, 73.0, 68.6, 36.4, 36.3, 35.3, 26.7, 24.0, 21.7, 14.6. HRMS (TOF ES−): Exact mass calculated for C22H31O4 [M − H]−: 359.2222, found 359.2213. All spectroscopic and physical data were in agreement with those reported in the literature.13b
Acknowledgements
The Norwegian Research Council (KOSK II) and The School of Pharmacy, University of Oslo are gratefully acknowledged for Ph.D.-scholarships to M. A. and J. E. T., respectively. T. V. H. is grateful for a Leiv Eriksson travel grant from The Norwegian Research Council. J. D. is supported by the National Institutes of Health GM Grant PO1GM095467 (awarded to Charles N. Serhan). Fruitful discussions with Professor Charles N. Serhan, Brigham and Women's Hospital and Harvard Medical School are gratefully appreciated.Notes and references
- P. C. Calder, J. Nutr., 2012, 592 CrossRef PubMed.
- (a) C. N. Serhan, S. Hong, K. Gronert, S. P. Colgan, P. R. Devchand, G. Mirick and R. L. Moussignac, J. Exp. Med., 2002, 196, 1025 CrossRef CAS PubMed; (b) P. K. Mukherjee, V. L. Marcheselli, C. N. Serhan and N. G. Bazan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8491 CrossRef CAS PubMed; (c) A. Ariel, L. Pin-Lan, W. Wang, W. X. Tang, G. Fredman, S. Hong, K. H. Gotlinger and C. N. Serhan, J. Biol. Chem., 2005, 280, 43079 CrossRef CAS PubMed.
- (a) C. N. Serhan, R. Yang, K. Martinod, K. Kasuga, P. S. Pillai, T. F. Porter, S. F. Oh and M. Spite, J. Exp. Med., 2009, 206, 15 CrossRef CAS PubMed; (b) C. N. Serhan, J. Dalli, S. Karamnov, A. Choi, C. K. Park, Z. Z. Xu, R. R. Ji, M. Zhu and N. A. Petasis, FASEB J., 2012, 26, 1755 CrossRef CAS PubMed.
- S. Hong, K. Gronert, P. R. Devchand, R. L. Moussignac and C. N. Serhan, J. Biol. Chem., 2003, 278, 14677 CrossRef CAS PubMed.
- C. N. Serhan and N. A. Petasis, Chem. Rev., 2011, 111, 5922 CrossRef CAS PubMed and references cited therein.
- C. N. Serhan, K. Gotlinger, S. Hong, Y. Lu, J. Siegelman, T. Baer, R. Yang, S. P. Colgan and N. A. Petasis, J. Immunol., 2006, 176, 1848 CrossRef CAS.
- W. J. Lukiw, J. G. Cui, V. L. Marcheselli, M. Bodker, A. Botkjaer, K. Gotlinger, C. N. Serhan and N. G. Bazan, J. Clin. Invest., 2005, 115, 2774 CAS.
- J. M. Calandria, V. L. Marcheselli, P. K. Mukherjee, J. Uddin, J. W. Winkler, N. A. Petasis and N. G. Bazan, J. Biol. Chem., 2009, 284, 17877 CrossRef CAS PubMed.
- V. L. Marcheselli, P. K. Mukherjee, M. Arita, S. Hong, R. Antony, K. Sheets, J. W. Winkler, N. A. Petasis, C. N. Serhan and N. G. Bazan, Prostaglandins Leukot. Essent. Fatty Acids, 2010, 82, 27 CrossRef CAS PubMed.
- Y. Zhao, F. Calon, C. Julien, J. W. Winkler, N. A. Petasis, W. J. Lukiw and N. G. Bazan, PLoS ONE, 2011, 6, e15816 CAS.
- (a) Z. Z. Xu, X. J. Liu, T. Berta, C. K. Park, N. Lü, C. N. Serhan and R. R. Ji, Ann Neurol., 2013, 74, 490 CrossRef CAS PubMed; (b) M. Morita, K. Kuba, A. Ichikawa, M. Nakayama, J. Katahira, R. Iwamoto, T. Watanebe, S. Sakabe, T. Daidoji, S. Nakamura, A. Kadowaki, T. Ohto, H. Nakanishi, R. Taguchi, T. Nakaya, M. Murakami, Y. Yoneda, H. Arai, Y. Kawaoka, J. M. Penninger, M. Arita and Y. Imai, Cell, 2013, 153, 112 CrossRef CAS PubMed.
- (a) C. N. Serhan and N. Chiang, Br. J. Pharmacol., 2008, 153, S200 CrossRef CAS PubMed; (b) C. N. Serhan and N. Chiang, Curr. Opin. Pharmacol., 2013, 13, 632 CrossRef CAS PubMed.
- (a) N. Ogawa and Y. Kobayashi, Tetrahedron Lett., 2011, 52, 3001 CrossRef CAS; (b) N. A. Petasis, R. Yang, J. W. Winkler, M. Zhu, J. Uddin, J. N. G. Bazan and C. N. Serhan, Tetrahedron Lett., 2012, 53, 1695 CrossRef CAS PubMed.
- (a) T. V. Hansen and Y. Stenstrøm, Synth. Commun., 2000, 30, 2549 CrossRef CAS; (b) T. V. Hansen and Y. Stenstrøm, Tetrahedron: Asymmetry, 2001, 12, 1407 CrossRef CAS; (c) T. V. Hansen and L. Skattebøl, Tetrahedron Lett., 2004, 45, 2809 CrossRef CAS; (d) H. F. Anwar and T. V. Hansen, Org. Lett., 2009, 11, 587 CrossRef CAS PubMed; (e) A. Vik, T. V. Hansen, A. K. Holmeide and L. Skattebøl, Tetrahedron Lett., 2010, 51, 2852 CrossRef CAS; (f) M. A. Yasser and T. V. Hansen, Pure Appl. Chem., 2011, 83, 489 Search PubMed; (g) M. A. Yasser and T. V. Hansen, Tetrahedron Lett., 2011, 52, 1057 CrossRef; (h) A. Vik and T. V. Hansen, Tetrahedron Lett., 2011, 52, 1060 CrossRef CAS; (i) M. G. Jakobsen, A. Vik and T. V. Hansen, Tetrahedron Lett., 2012, 53, 5837 CrossRef CAS; (j) M. A. Yasser, A. Vik, T. Hofer, J. Hammer Andersen and T. V. Hansen, Chem. Phys. Lipids, 2013, 170–171, 41 Search PubMed.
- (a) K. C. Nicolaou, S. E. Webber, J. Ramphal and Y. Abe, Angew. Chem., 1987, 99, 1077 CrossRef CAS; (b) J. S. Yadav, P. K. Deshpande and G. V. M. Sharma, Tetrahedron, 1992, 48, 4465 CrossRef CAS.
- (a) J. Becher, Org. Synth., 1979, 59, 79 CrossRef; (b) D. Soullez, G. Ple, L. Duhamel and P. Duhamel, J. Chem. Soc., Perkin Trans. 1, 1997, 1639 RSC.
- Y. Nagao, W. M. Dai, M. Ochiai, S. Tsukagoshi and E. Fujita, J. Org. Chem., 1989, 54, 5211 CrossRef CAS.
- (a) D. A. Evans, J. Barartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127 CrossRef CAS; (b) D. A. Evans, J. M. Takacs, L. R. McGee, M. D. Ennis, D. J. Mathre and J. Bartroli, Pure Appl. Chem., 1981, 53, 1109 CrossRef CAS.
- (a) M. Romero-Ortega, D. A. Colby and H. F. Olivo, Tetrahedron Lett., 2002, 43, 6439 CrossRef CAS; (b) R. Tello-Aburto, A. Ochoa-Teran and H. F. Olivo, Tetrahedron Lett., 2006, 47, 5915 CrossRef CAS.
- E. J. Corey, H. Cho, C. Rücker and D. H. Hua, Tetrahedron Lett., 1981, 22, 3455 CrossRef CAS.
- D. Seebach, G. Adam, R. Zibuck, W. Simon, M. Rouilly, W. L. Meyer, J. F. Hinton, T. A. Privett, G. E. Templeton, D. K. Heiny, U. Gisi and H. Binder, Liebigs Ann. Chem., 1989, 1233 CrossRef CAS.
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801 CrossRef.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446 CrossRef CAS.
- E. J. Corey and B. B. Snider, J. Am. Chem. Soc., 1972, 94, 6190 CrossRef CAS.
- (a) K. C. Nicolaou, T. Ladduwahetty, I. M. Taffer and R. E. Zipkin, Synthesis, 1986, 344 CrossRef CAS; (b) L. E. Overman and A. S. Thompson, J. Am. Chem. Soc., 1988, 110, 2248 CrossRef CAS.
- D. J. Pasto and R. T. Taylor, Org. React., 1991, 40, 91 CrossRef CAS.
- W. Boland, N. Schroer, C. Sieler and M. Feigel, Helv. Chim. Acta, 1987, 70, 1025 CrossRef CAS.
- N. Chiang, G. Fredman, F. Bäckhed, S. F. Oh, T. Vickery, B. A. Schmidt and C. N. Serhan, Nature, 2012, 484, 524 CrossRef CAS PubMed.
- C. N. Serhan, G. Fredman, R. Yang, S. Karamnov, L. S. Belayev, N. G. Bazan, M. Zhu, J. W. Winkler and N. A. Petasis, Chem. Biol., 2011, 18, 976 CrossRef CAS PubMed.
- E. J. Corey, H. Cho, C. Rucker and D. H. Hua, Tetrahedron Lett., 1981, 22, 3455 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and characterization data, 1H-, 13C-NMR, HRMS, LC-MS/MS and UV/VIS spectra as well as chromatograms of HPLC analyses. See DOI: 10.1039/c3ob41902a |
| This journal is © The Royal Society of Chemistry 2014 |