 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalyzed stereospecific C–C bond formation of β-lactams†
Sachin A.
Pawar
a,
Saba
Alapour
a,
Sibusiso
Khanyase
a,
Zamani E. D.
Cele
a,
Srinivas
Chitti
a,
Hendrik G.
Kruger
a,
Thavendran
Govender
*a and
Per I.
Arvidsson
*ab
aCatalysis and Peptide Research Unit, University of KwaZulu Natal, Durban, South Africa. E-mail: govenderthav@ukzn.ac.za; Tel: +27 312601845
bScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden. E-mail: per.arvidsson@scilifelab.se; Tel: +46 852481398
First published on 4th November 2013
Abstract
Herein, we report the development of mild, organocatalyzed routes to novel carbapenam derivatives through aldol, Mannich and Michael C–C bond forming reactions.
Compounds containing β-lactams (Fig. 1) are amongst the most important molecules in clinical use today.1–3 Most notable is their wide utility as antibacterial agents and as related β-lactamase inhibitors; however, β-lactams are also being explored in other therapeutic areas.4,5 Given the global challenge of antibiotic resistance,6 there is an urgent need for increased focus on the discovery and development of antibacterial agents. Bacterial resistance may occur through a number of pathways, e.g. production of β-lactamases,7 efflux pumps, and mutations that alters expression and function of transpeptidase enzymes – the targets of most β-lactam antibiotics.8,9 As β-lactams function as both transpeptidase- and β-lactamase inhibitors, much work is being devoted to accessing novel analogs of these critical molecular frameworks.10 However, the commercially viable synthesis of many β-lactams remains challenging due to a high degree of functionalization and chirality combined with the reactive nature of the core bicyclic ring-structures. Furthermore, most β-lactam antibiotics, except carbapenems and aztreonam, are being produced by biosynthetic routes rather than through chemical synthesis. Considering the challenges associated with synthetic modifications of the β-lactam framework, we envisioned that the mild conditions offered by organocatalysis might help overcome some of the limitations of current methodologies and open en route to hitherto unexplored β-lactams.
 | ||
| Fig. 1 Examples of β-lactam antibiotics: generic structure of penicillins with a saturated penam core and of synthetic carbapenems (e.g. imipenem, thienamycin, and panipenem). | ||
During the past decade, asymmetric organocatalysis11–13 has grown extensively as a powerful tool in the construction of complex molecular skeletons in synthetic chemistry.14–19 Aldol,15,20–24 Mannich15,25,26 and Michael15,27,28 reactions are some of the most powerful strategies in synthetic organic chemistry, since it allows the formation of new C–C bonds.29
We envisaged that (2S,5R,6S)-4-nitrobenzyl 6-((R)-1-hydroxyethyl)-3,7-dioxo-1-azabicyclo[3.2.0]heptane-2-carboxylate (1), the common intermediate for the preparation of clinically used antibiotics imipenem,30 thienamycin31,32 and panipenem,33 could be further substituted via HOMO-rising amine catalysis,34 thereby promoting reactions with electrophilic substrates (Scheme 1).
 | ||
| Scheme 1 Novel HOMO rising strategies offering a mild and facile route to carbapenam derivatives. | ||
In order to test our hypothesis, we subjected the “carbapenam ketone” intermediate 1 to a reaction with the benchmark substrate formaldehyde as the electrophile and proline as the catalyst, Table 1. Various solvents such as DMF, DCM and THF were evaluated but no conversion was observed via LC-MS except when DMSO was employed (Table 1, entry 1). When the reactions were conducted with reagent grade DMSO as the solvent, we observed the presence of the product and a hydrolyzed form of the starting materials (+18 m/z on LC-MS). The use of dry DMSO resulted in no detection of the hydrolyzed starting material but also resulted in a slower and low yielding reaction. Acid additives are common additives in the organocatalyzed aldol reactions,35 so we next investigated the effect of formic and acetic acids. It was observed that there was no difference in the reactivity or yields when acetic acid was used (Table 1, entry 2) whereas formic acid enhanced the hydrolysis side reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Catalyst/additive | Time (h) | Solvent | Yieldb (%) |
| a Reactions were performed at excess amount of aldehyde to serve as a solvent (see ESI). b Isolated yields. c Diastereomeric ratio determined by 1H NMR. d Observed yields from NMR of the crude reaction mixture. | |||||
| 1 | R = H | L-Proline | 24 | DMSO | 73 |
| 2 | R = H | L-Proline/AcOH | 24 | DMSO | 74 |
| 3 | R = H | L-Proline/AcOH | 8 | Neat | 76 |
| 4 | R = H | D-Proline | 24 | DMSO | 70 |
| 5 | R = H | Pyrrolidine | 24 | DMSO | NR |
| 6 | R = H | Pyrrolidine/AcOH | 2 | DMSO | 60 |
| 7 | R = H | NEt3 | 24 | DMSO | NR |
| 8 | R = H | NEt3/AcOH | 24 | DMSO | NR |
| 9 | R = Ph | L-Proline | 24 | DMSO | NR |
| 10 | R = Ph | L-Proline/AcOH | 24 | DMSO | NR |
| 11 | R = Ph | L-Proline/AcOH | 6 | Neata | 60 |
| 12 | R = Ph | L-Proline/AcOH | 24 | DMF | 25d |
| 13 | R = Ph | Pyrrolidine/AcOH | 24 | DMF | 55d |
| 14 | R = 4-NO2 Ph | Pyrrolidine/AcOH | 24 | DMF | 54d |
| 15 | R = 4-Me Ph | Pyrrolidine/AcOH | 24 | DMF | 29d |
| 16 | R = 4-OMe Ph | Pyrrolidine/AcOH | 24 | DMF | 51d |
| 17 | R = 2,4-OMe Ph | Pyrrolidine/AcOH | 24 | DMF | 26d |
| 18 | R = 4-F Ph | Pyrrolidine/AcOH | 24 | DMF | 46d |
| 19 | R = Et | Pyrrolidine/AcOH | 48 | DMF | 70d |

Under solvent free conditions, the rate of the reaction was improved significantly (Table 1, entry 3). D-Proline gave similar results as L-proline with respect to reaction times and yields (Table 1, entries 1 and 4), but also with respect to the diastereomeric ratio of the product formed. This result shows that the stereochemical outcome of the reaction is dictated by the chiral ketone 1 rather than the catalyst, as might be expected when considering the unique bent conformation of the cyclobutanone ring at the bicyclic core. This prompted us to evaluate simple pyrrolidine as a catalyst; pyrrolidine on its own did not result in any conversion but when one equivalent of acetic acid was added we obtained the product at a high conversion rate (Table 1, entries 5, 6).
In order to prove that the reaction was indeed operating through the postulated enamine intermediate, and did not simply involve the enol tautomer of ketone 1, we performed the reaction with triethylamine as a catalyst, with and without acetic acid; however, no reaction was observed in any of these cases (Table 1, entries 7, 8), thus supporting the need for HOMO rising catalysis of the reaction. Aromatic aldehydes, i.e. benzaldehyde as the electrophile, did not result in any conversion in DMSO (Table 1, entries 9, 10). However, benzaldehyde and other liquid aldehydes (e.g. 4-methyl and 4-fluoro benzaldehyde) gave the corresponding aldol product under neat reaction conditions (Table 1, entry 11) as detected by LC-MS. Unfortunately, purification via chromatography for all analogs except the benzaldehyde product 3 proved difficult since the β-lactam ring is prone to hydrolysis during prolonged exposure to silica gel. However, aromatic aldehydes, i.e. 4-nitro, 4-methyl, 4-methoxy, 4-fluoro benzaldehyde and propionaldehyde, gave the product upon changing the solvent from DMSO to DMF, utilizing pyrrolidine/AcOH as a catalyst. Again purification proved difficult; crude NMR yield for the reaction of these electrophiles with carbapenem ketone 1 was in the range 26–70% as determined by crude NMR (Table 1, entries 12–19).
The formation of the new C–C bond in product 2 was confirmed by observing the shift of C8 from 64.0 ppm (1) to 131.6 ppm (2) in 13C NMR and disappearance of H8 as a singlet at 4.76 ppm (1) in H-NMR, Fig. 2. In addition, protons at C17 showed an HMBC correlation with C8. The 2D NMR investigation proved the excellent stereoselectivity (d.r. >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) seen in the crude LC-MS trace and 1D spectra, and through a correlation between H17 and H4 in the NOESY spectra, we could establish the expected exo configuration of the newly attached group in 2. Compound 3 showed similar HMBC correlations and excellent diastereoselectivity as observed with 2 for the formation of the C–C bond at C8. The newly formed chiral centre C17 in compound 3 was determined to be created with a diastereomeric ratio of 90:10.
:1) seen in the crude LC-MS trace and 1D spectra, and through a correlation between H17 and H4 in the NOESY spectra, we could establish the expected exo configuration of the newly attached group in 2. Compound 3 showed similar HMBC correlations and excellent diastereoselectivity as observed with 2 for the formation of the C–C bond at C8. The newly formed chiral centre C17 in compound 3 was determined to be created with a diastereomeric ratio of 90:10.
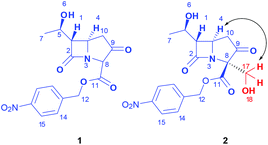 | ||
| Fig. 2 Aldol product 2, resulting from L-proline catalyzed transformation of “carbapenam ketone” 1 and Formaldehyde. | ||
Given the high potential for using organocatalysis for accessing hitherto unexplored derivatives of carbapenam and carbapenem β-lactams, we decided to explore other organocatalyzed processes, i.e. Mannich and Michael reactions.
First, we decided to perform the direct asymmetric three-component Mannich reaction of carbapenam intermediate 1 with different amines and aldehydes in DMSO (Scheme 2). In the presence of 30% L-proline, aldehydes and amines were reacted with 1 to give products 9, 10 and 11 in moderate yields (50% to 55%) respectively. These yields are typical of the one pot Mannich reaction and are attributed to the formation of the competitive aldol reaction side products as noticed by LC-MS. Various aromatic aldehydes were tested as electrophiles, but, similarly to the aldol reaction described above, only formaldehydes resulted in the formation of the Mannich adducts. A complete NMR assignment of Mannich product 9 proved that the exo-product was formed with complete diastereoselectivity, in analogy to the aldol product 2 above. In compound 9, the absolute configuration at C8 was confirmed by NOE correlation (see ESI†).
 | ||
| Scheme 2 Mannich reactions on carbapenam intermediate 1. | ||
Next, we explored the organocatalyzed Michael reaction to carbapenam intermediate 1 (Scheme 3). The most commonly studied Michael acceptors with enamine catalyzed reactions are nitrostyrenes28,36 and enones;37 hence it was decided to test these substrates in this first report. From the optimized conditions reported above, we initiated the study by examining the addition of the carbapenam intermediate 1 to trans-4-methoxy-β-nitrostyrene in DMSO catalyzed by L-proline. The reaction offered the product 12 in modest 41% yield in 24 hours. The modest yield was due to low catalytic turnover, as confirmed through LC-MS analysis of the crude mixture, where we noticed a peak that corresponded to the Michael product still bound to the catalyst. To release the product, the adduct had to be stirred with water and monitored by LC-MS until only a minor amount of the trapped product could be detected. Similarly, Michael reaction of carbapenam intermediate 1 with neat cyclopentenone using L-proline as a catalyst produced compound 7 in 67% yield. The observed HMBC correlation between H17 and C8 in both compounds (12 and 13) proved the formation of the new C–C bond at this position. From the 1H NMR shifts of H17 in compounds 12 and 13, the diastereomeric ratio was established to be 88:12 and 90:10 respectively. The configuration at C8 for 12 was also established by NOE correlation as for the aldol and Mannich reactions above.
 | ||
| Scheme 3 Michael reaction on carbapenam intermediate 1. | ||
In summary, the mild reaction conditions that characterize enamine-based organocatalysis have been shown to offer a new route to chiral β-lactam derivatives. The reaction scope has so far been shown to include aldol, Mannich and Michael reactions. High distereoselectivity was observed in all of the reactions, as would be expected considering the inherent chirality of the starting carbapenam intermediate. This methodology has the potential to offer a widely sought after, new synthetic route to novel and potentially medically useful β-lactam antibiotics. The full substrate scope for the Mannich and Michael reactions reported here are ongoing in our laboratories.
Acknowledgements
We thank NRF, UKZN and Asphen Pharmacare for financial support.Notes and references
- A. C. Rodloff, E. J. C. Goldstein and A. Torres, J. Antimicrob. Chemother., 2006, 58, 916–929 CrossRef CAS PubMed.
- R. P. Elander, Appl. Microbiol. Biotechnol., 2003, 61, 385–392 CAS.
- F.-R. Schmidt, in Industrial Applications, ed. M. Hofrichter, Springer, Berlin, Heidelberg, 2010, vol. 10, pp. 101–121 Search PubMed.
- J. M. T. Hamilton-Miller, J. Antimicrob. Chemother., 1999, 44, 729–734 CrossRef CAS PubMed.
- T. Sperka, J. Pitlik, P. Bagossi and J. Tozser, Bioorg. Med. Chem. Lett., 2005, 15, 3086–3090 CrossRef CAS PubMed.
- F. Rossi, Clin. Infect. Dis., 2011, 52, 1138–1143 CrossRef PubMed.
- M. Baroud, I. Dandache, G. F. Araj, R. Wakim, S. Kanj, Z. Kanafani, M. Khairallah, A. Sabra, M. Shehab, G. Dbaibo and G. M. Matar, Int. J. Antimicrob. Agents, 2013, 41, 75–79 CrossRef CAS PubMed.
- B. G. Spratt, J. Antimicrob. Chemother., 2012, 67, 1578–1588 CrossRef CAS PubMed.
- A. Zervosen, E. Sauvage, J.-M. Frere, P. Charlier and A. Luxen, Molecules, 2012, 17, 12478–12505 CrossRef CAS PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, C. Ducho, M. A. McDonough and C. J. Schofield, Nat. Prod. Rep., 2013, 30, 21–107 RSC.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- H. Pellissier, Tetrahedron, 2007, 63, 9267–9331 CrossRef CAS PubMed.
- B. List, Acc. Chem. Res., 2004, 37, 548–557 CrossRef CAS PubMed.
- W. Notz, F. Tanaka and C. F. Barbas, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS PubMed.
- P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS PubMed.
- M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8–27 CrossRef CAS PubMed.
- S. G. Zlotin, A. S. Kucherenko and I. P. Beletskaya, Russ. Chem. Rev., 2009, 78, 737–784 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386–7387 CrossRef CAS.
- A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 6798–6799 CrossRef CAS PubMed.
- G. Guillena, M. del Carmen Hita, C. Najera and S. F. Viozquez, J. Org. Chem., 2008, 73, 5933–5943 CrossRef CAS PubMed.
- B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827–833 CrossRef CAS PubMed.
- B. List, J. Am. Chem. Soc., 2000, 122, 9336–9337 CrossRef CAS.
- S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123–3135 RSC.
- B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425 CrossRef CAS PubMed.
- J. Franzen, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjaersgaard and K. A. Jorgensen, J. Am. Chem. Soc., 2005, 127, 18296–18304 CrossRef CAS PubMed.
- US 2002/0095034 A1, US2002/0095034 A1 Search PubMed.
- T. N. Salzmann, R. W. Ratcliffe, B. G. Christensen and F. A. Bouffard, J. Am. Chem. Soc., 1980, 102, 6161–6163 CrossRef CAS.
- Y. Nagao, Y. Nagase, T. Kumagai, Y. Kuramoto, S. Kobayashi, Y. Inoue, T. Taga and H. Ikeda, J. Org. Chem., 1992, 57, 4238–4242 CrossRef CAS.
- CN20111233337 20110816.
- L. Dell'Amico, L. Albrecht, T. Naicker, P. H. Poulsen and K. A. Jorgensen, J. Am. Chem. Soc., 2013, 135, 8063–8070 CrossRef CAS PubMed.
- D. Gryko, M. Zimnicka and R. Lipinski, J. Org. Chem., 2007, 72, 964–970 CrossRef CAS PubMed.
- H. Chen, Y. Wang, S. Wei and J. Sun, Tetrahedron: Asymmetry, 2007, 18, 1308–1312 CrossRef CAS PubMed.
- Y. G. Chi and S. H. Gellman, Org. Lett., 2005, 7, 4253–4256 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and copies of NMR spectra. See DOI: 10.1039/c3ob41858h |
| This journal is © The Royal Society of Chemistry 2013 |