 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NaH mediated isomerisation–allylation reaction of 1,3-substituted propenols†
Adam J. S. Johnston, Mark G. McLaughlin, Jolene P. Reid and Matthew J. Cook*
School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, Northern Ireland, UK. E-mail: m.cook@qub.ac.uk; Fax: +44 (0)28 90976524; Tel: +44 (0)28 90974682
First published on 3rd October 2013
Abstract
A base mediated isomerisation–allylation protocol of 1,3-disubstituted propenols has been established. The use of diaryl and aryl-silyl substrates is reported alongside the use of substituted allyl bromides. Mechanistic experiments have also been conducted to elucidate the reaction pathway.
The isomerisation of allylic alcohols to ketones and aldehydes can be a useful transformation that avoids the use of both oxidants and reductants.1 The most common strategy to achieve this operation is to utilise a transition metal catalyst that isomerises the alkene moiety into an enol (or enolate) which upon tautomerisation (or workup) affords the corresponding carbonyl functionality.2 In most cases, the reaction is driven by a thermodynamic preference for the enol (or enolate) over the allylic alcohol (or alkoxide) with up to 25 kcal mol−1 difference between the two.3
Many of these processes have also been rendered enantioselective.4 The allyl amine variant is generally superior producing significantly higher levels of enantiomeric enrichment.5 Motherwell also demonstrated that enolates derived from rhodium or nickel catalysed isomerisations of allylic alkoxides could be intercepted by electrophiles such as aldehydes or allylic bromides to form α-branched products (eqn (1)).6
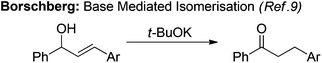
Although known for over 100 years, base mediated variants of this reaction are much less common with relatively few reported examples.7 Dimmel demonstrated the use of butyl lithium and Na/K to isomerise phenyl substituted allylic alcohols which was proposed to proceed via a double anion with both the alcohol and the allylic C–H being deprotonated followed by reprotonation to form the more conjugated product.8 Borschberg reported an analogous isomerisation of a diarylsubstituted allylic alcohol under much milder reaction conditions using t-BuOK as base (eqn (2)).9 Following deuterium labelling studies they proposed that following initial deprotonation of the alcohol, a competing [1,2] or [1,3]-hydride shift occurs as deuterium is observed at both of these positions in the product. 2-Pyridyl substituted allylic alcohols have also been shown to isomerise both thermally and under sodium hydride mediated conditions.10,11 To our knowledge there have been no reports of base mediated isomerisations of 1,3-disubstituted allylic alcohols followed by electrophilic trapping. Herein we describe the base mediated isomerisation of 1,3-diaryl and 1-aryl-3-silyl propen-1-ols and subsequent trapping with an allylic bromide (eqn (3)).
 | (1) |
 | (2) |
 | (3) |
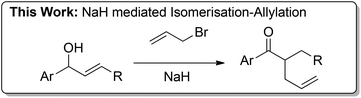
We discovered an unusual product during the course of an allylation reaction of silyl allylic alcohol 1 (Table 1).12,13 When allylic alcohol was treated with sodium hydride and allyl bromide 2 in THF with heating the major product observed was not the expected allyl ether but ketone 3.14 The mass balance of the reaction was made up of unallylated ketone product and the expected bis-allyl ether. This was further optimised and found that lowering the equivalents of NaH increased the yield to 57%. When the sodium hydride content was lowered further, a reaction was still observed (at 1.05 equivalents) however O-allylation was the major product. Other bases such as t-BuOK gave decomposition and a solvent screen showed that THF was the optimal solvent. Interestingly, when DMF was used as solvent no ketone was observed with only O-allylation being observed. Finally, the use of additives was explored with 15-crown-5 giving no ketone and HMPA affording a slightly reduced yield with several unidentified by-products being formed.

With the optimised conditions in hand, we then probed the substrate scope (Scheme 1). The reaction proceeded with both phenyl and naphthyl to afford α-allylated ketones 3a and 3b. When substitution was present on the aryl ring the reactivity was compromised with both electron withdrawing and donating groups. 3-Fluoro substituent 1c afforded allylated product 3c in low yield and 4-methoxy substituent 1d afforded no isomerisation–allylation product with the majority of the mass balance made up of O-allylated product.
 | ||
| Scheme 1 Vinyl silane substrate scope. | ||
As the substrate scope of the vinyl silanes was limited, with only three examples providing any ketone product, we next turned our attention to 1,3-diaryl propen-1-ols 4 which are readily available using a number of methods (Table 2).15 We first looked at commercially available 1,3-diphenyl propen-1-ol 4a and found the same type of reaction occurred to afford ketone 5a in a moderate yield.


We next examined the effect of changing the electronic nature of the aryl group at the 1-position. Adding electron withdrawing substituents such as 4-fluoro or 3,5-bis(trifluoromethyl) did increase the yield of the reaction affording 5b and 5c respectively. Heterocycles could be incorporated at the 1-position with little effect on reactivity. Both furyl 5d and pyridyl 5e proceeded in a similar manner to the parent phenyl compound.
We also probed the effect the 3-aryl group had on reactivity and found that changing this showed a much larger effect than the 1-position. 2-Naphthyl substituent 4f gave much improved reactivity affording 5f in 50% yield. This could be further improved by the installation of a 4-trifluoromethyl group giving 5g in good yield. Other substituents including 4-trifluoromethoxy 4h, 4-methyl 4i and 4-fluoro 4j also provided isomerised-allylated products 5h–j in around 50% yield. The installation of electron donating groups in the form of a 4-methoxy group 4k resulted in a similar yield to the phenyl group and a pyridyl group could be tolerated in the 3-position forming 5l in 42% yield. When no anion stabilising groups were present, such as alkyl substituents, no isomerisation was observed and only O-allylation was obtained.
Next we examined other substituted allyl bromides. When crotyl bromide was used a similar reaction was observed with the α-crotylated product 6 being produced in 44% yield in the same E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio as the electrophilic component (eqn (4)). This was further extended to 2,3-dimethylallyl bromide and again α-allylation was observed to form 7, albeit in reduced yields (eqn (5)).
:Z ratio as the electrophilic component (eqn (4)). This was further extended to 2,3-dimethylallyl bromide and again α-allylation was observed to form 7, albeit in reduced yields (eqn (5)).
 | (4) |
 | (5) |
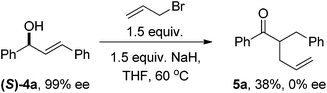
Several experiments were performed to probe the mechanism of these reactions. Firstly, in the absence of an electrophile, the allylic alcohol isomerises to the corresponding ketone 8 in excellent yield with no observed by-products (eqn (6)). Secondly, when deuterated compound 9 is used, the deuterium atom is removed from the 1-position by the base and this was observed at the 3-position in approximately 40% D-incorporation (eqn (7)). The majority of the product in this reaction was simple O-allylation product. When enantioenriched compound (S)-4a was used,16 the corresponding allylated product 5a was produced as a racemic mixture thus indicating the intermediacy of a planar achiral intermediate (eqn (8)). Finally we took the allyl ether 11 and subjected this to the reaction conditions and no reaction was observed thus ruling out the intermediacy of allyl ether 11 (eqn (9)).
 | (6) |
 | (7) |
 | (8) |
 | (9) |
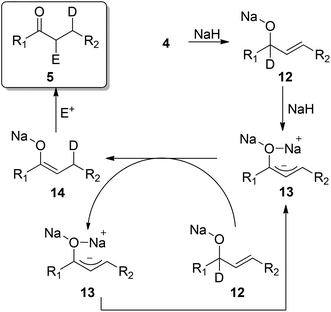
With the mechanistic data in hand we have proposed a mechanism that accounts for all observations (Scheme 2). Firstly the OH of allylic alcohol 4 is deprotonated to form the corresponding alkoxide 12. This alkoxide can undergo a second deprotonation to form allylic anion 13. The allylic anion 13 can deprotonate a second equivalent of alkoxide 12 to afford an enolate 14 incorporating deuterium in the 3-position and regenerating allylic anion 13. Finally the enolate 14 is then allylated to afford the observed isomerisation allylation product 5. As only 40%-deuterium incorporation is observed the deprotonation of 12 must be performed by both 13 and NaH at comparable rates. This indicates that the isomerisation is catalytic with respect to 13 and complete double deprotonation is not necessary before isomerisation can occur. In all cases no bis-allylated product was observed, a significant by-product in many of the other isomerisation–allylation protocols that have been reported. The position of the methyl groups when a substituted allyl bromide is used, SN2 type substitution, also rules out possibility of an O-allylation-Claisen pathway.
 | ||
| Scheme 2 Proposed mechanism. | ||
Sodium hydride has been shown to isomerise N-acyl allylic amines to their corresponding enamines with high levels of E/Z control. Clayden demonstrated the use of allylic ureas in this pathway17 and Smith used allylic acrylamides.18,19 Both of these reactions give the corresponding enamine as a single Z isomer and they both propose a sodium chelate analogous to 13. As deuterium is only observed at the 3-position, we can eliminate the possibility that a competing [1,2] and [1,3] shift is in operation as proposed by Borschberg.9
Conclusions
In conclusion, we have discovered and investigated the NaH catalyzed isomerization of allylic alcohols and α-allylation of the resultant enolate to form α-allylated ketones. We have discovered that both silyl and aryl substitution is tolerated at the γ-position. Several mechanistic probes and control reactions has shed light on the mechanism of this reaction.We thank Queen's University Belfast for support. We are also grateful to the Department of Employment and Learning, Northern Ireland for a studentship (MGM). GlaxoSmithKline and AstraZeneca are also thanked for their generous donation of laboratory equipment. We also thank Professor Andrew Smith, University of St. Andrews for useful discussions.
Notes and references
- For reviews of allylic alcohol isomerization see: (a) S. G. Davies, Organotransition Metal Chemistry. Applications to Organic Synthesis, Pergamon Press, Oxford, 1982, pp. 266–290 Search PubMed; (b) B. M. Trost, Science, 1991, 254, 1471 CAS; (c) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (d) R. C. van der Drift, E. Bouwman and E. Drent, J. Organomet. Chem., 2002, 650, 1 CrossRef CAS; (e) R. Uma, C. Crevisy and R. Grée, Chem. Rev., 2003, 103, 27 CrossRef CAS PubMed; (f) A. Gansaeuer and K. Muniz, Sci. Synth., 2007, 25, 57 Search PubMed; (g) N. Kuznik and S. Krompiec, Coord. Chem. Rev., 2007, 251, 222 CrossRef CAS PubMed; (h) V. Cadierno, P. Crochet and J. Gimeno, Synlett, 2008, 1105 CAS; (i) J. García-Álverez, S. E. García-Garrido, P. Crochet and V. Cadierno, Curr. Top. Catal., 2012, 10, 35 Search PubMed; (j) P. Lorenzo-Luis, A. Romerosa and M. Serrano-Ruiz, ACS Catal., 2012, 2, 1079 CrossRef CAS.
- For representative examples of transition-metal catalyzed isomerization of allylic alcohols see: (a) R. Damico and T. G. Logan, J. Org. Chem., 1967, 32, 2356 CrossRef CAS; (b) W. T. Hendrix, F. G. Cowherd and J. L. von Rosenberg, Chem. Commun., 1968, 97 RSC; (c) Y. Sasson and G. L. Rempel, Tetrahedron Lett., 1974, 15, 4133 CrossRef; (d) S. H. Bergens and B. Bosnich, J. Am. Chem. Soc., 1991, 113, 958 CrossRef CAS; (e) B. M. Trost and R. J. Kulawiec, J. Am. Chem. Soc., 1993, 115, 2027 CrossRef CAS; (f) H. Cherkaoui, M. Soufiaoui and R. Grée, Tetrahedron, 2001, 57, 2379 CrossRef CAS; (g) P. Crochet, M. A. Fernández-Zúmel, J. Gimeno and M. Scheele, Organometallics, 2006, 25, 4846 CrossRef CAS; (h) V. Cadierno, S. E. García-Garrido, J. Gimeno, A. Varela Álvarez and J. A. Sordo, J. Am. Chem. Soc., 2006, 128, 1360 CrossRef CAS PubMed; (i) A. Varela-Álvarez, J. A. Sordo, E. Piedra, N. Nebra, V. Cadierno and J. Gimeno, Chem.–Eur. J., 2011, 17, 10583 CrossRef PubMed; (j) N. Ahlsten, A. B. Gómez and B. Martín-Matute, Angew. Chem., Int. Ed., 2013, 52, 6273 CrossRef CAS PubMed.
- For bond energy E-values see: E. G. Lovering and K. J. Laider, Can. J. Chem., 1960, 38, 2367 CrossRef CAS.
- For representative examples of enantioselective variants see: (a) K. Tani, Pure Appl. Chem., 1985, 57, 1845 CrossRef CAS; (b) K. Tanaka, S. Qiao, M. Tobisu, M. M.-C. Lo and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 9870 CrossRef CAS; (c) K. Tanaka and G. C. Fu, J. Org. Chem., 2001, 66, 8177 CrossRef CAS PubMed; (d) M. Ito, S. Kitahara and T. Ikariya, J. Am. Chem. Soc., 2005, 127, 6172 CrossRef CAS PubMed; (e) L. Mantilli, D. Gérard, S. Torche, C. Besnard and C. Mazet, Angew. Chem., Int. Ed., 2009, 48, 5143 CrossRef CAS PubMed; (f) N. Arai, K. Sato, K. Azuma and T. Ohkuma, Angew. Chem., Int. Ed., 2013, 52, 7500 CrossRef CAS PubMed.
- (a) R. Noyori and H. Takaya, J. Am. Chem. Soc., 1990, 112, 4897 CrossRef; (b) S. Otsuka and K. Tani, Synthesis, 1991, 665 CrossRef CAS.
- (a) G. L. Edwards, W. B. Motherwell, D. M. Powell and D. A. Sandham, J. Chem. Soc., Chem. Commun., 1991, 1399 RSC; (b) L. J. Gazzard, W. B. Motherwell and D. A. Sandham, J. Chem. Soc., Perkin Trans. 1, 1999, 979 RSC; (c) W. B. Motherwell and D. A. Sandham, Tetrahedron Lett., 1992, 33, 6187 CrossRef CAS ; for photolytically activated Fe(CO)5 catalyzed variant see: ; (d) C. Crévisy, M. Wietrich, V. Le Boulaire, R. Uma and R. Grée, Tetrahedron Lett., 2001, 42, 395 CrossRef ; for reviews see: ; (e) N. Ahlsten, A. Bartoszewicz and B. Martín-Matute, Dalton Trans., 2012, 41, 1660 RSC; (f) T. D. Sheppard, Synlett, 2011, 1340 CrossRef CAS PubMed.
- (a) M. Tiffeneau, Bull. Soc. Chim. Fr., 1907, 4, 1205 Search PubMed (footnote on p. 1209); ; (b) H. Burton and C. K. Ingold, J. Chem. Soc., 1928, 904 RSC.
- D. R. Dimmel, W. Y. Fu and S. B. Gharpure, J. Org. Chem., 1976, 41, 3092 CrossRef CAS.
- G. A. Schmidt and H.-J. Borschberg, Helv. Chim. Acta, 2001, 84, 401 CrossRef.
- D. Giomi, M. Piacenti and A. Brandi, Tetrahedron Lett., 2004, 45, 2113 CrossRef CAS PubMed.
- X. Wang and D. Z. Wang, Tetrahedron, 2011, 67, 3406 CrossRef CAS PubMed.
- (a) M. G. McLaughlin and M. J. Cook, Chem. Commun., 2011, 47, 11104 RSC; (b) C. A. McAdam, M. G. McLaughlin, A. J. S. Johnston, J. Chen, M. W. Walter and M. J. Cook, Org. Biomol. Chem., 2013, 11, 4488 RSC.
- For use of silylated diallyl ethers in isomerization-Claisen reaction see: M. G. McLaughlin and M. J. Cook, J. Org. Chem., 2012, 77, 2058 CrossRef CAS PubMed.
- For Rh catalyzed isomerization of Et3Si analogue of 1 see: R. Takeuchi, S. Nitta and D. Watanabe, J. Org. Chem., 1995, 60, 3045 CrossRef CAS.
- C. Thiot, M. Schmutz, A. Wagner and C. Mioskowski, Chem.–Eur. J., 2007, 13, 8971 CrossRef CAS PubMed.
- Prepared via a Sharpless epoxidation kinetic resolution, see: M. Sasaki, H. Ikemoto, M. Kawahata, K. Yamaguchi and K. Takeda, Chem.–Eur. J., 2009, 15, 4663 CrossRef CAS PubMed.
- J. Lefranc, D. J. Tetlow, M. Donnard, A. Minassi, E. Gálvez and J. Clayden, Org. Lett., 2011, 13, 296–299 CrossRef CAS PubMed.
- P. Woods, PhD thesis, University of St. Andrews, UK, 2012 (Professor A. Smith Group).
- Organolithium and lithium amide bases have also been utilised for similar reactions: (a) J. E. Resek and P. Beak, Tetrahedron Lett., 1993, 34, 3043 CrossRef CAS; (b) P. Ribéreau, M. Delamare, S. Célanire and G. Quéguiner, Tetrahedron Lett., 2001, 42, 3571 CrossRef . For review of organolithium chemistry see: ; (c) J. Clayden, Tetrahedron Organic Chemistry, in Organlithiums: Selectivity for Synthesis, Pergamon, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob41857j |
| This journal is © The Royal Society of Chemistry 2013 |