 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triazole biotin: a tight-binding biotinidase-resistant conjugate†‡
Anne I. Germerotha, Jill R. Hannaa, Rehana Karima, Franziska Kundela, Jonathan Lowthera, Peter G. N. Neatea, Elizabeth A. Blackburnb, Martin A. Wearb, Dominic J. Campopianoa and Alison N. Hulme*a
aEaStCHEM School of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK. E-mail: Alison.Hulme@ed.ac.uk; Fax: +44 (0)131 650 4743; Tel: +44 (0)131 650 4711
bCentre for Translational and Chemical Biology, University of Edinburgh, Mayfield Road, Edinburgh, EH9 3JR, UK
First published on 1st October 2013
Abstract
The natural amide bond found in all biotinylated proteins has been replaced with a triazole through CuAAC reaction of an alkynyl biotin derivative. The resultant triazole-linked adducts are shown to be highly resistant to the ubiquitous hydrolytic enzyme biotinidase and to bind avidin with dissociation constants in the low pM range. Application of this strategy to the production of a series of biotinidase-resistant biotin-Gd-DOTA contrast agents is demonstrated.
Introduction
The use of the triazole as an amide bond isostere was highlighted by Sharpless in his seminal review of the impact of “click” chemistry in 2003.1 However, the application of this strategy to confer hydrolytic stability in amide linkages to the essential cofactor biotin 1a, such as found in the biological carrier biocytin 2 (biotinyl-ε-lysine),2 and recently suggested as a rare epigenetic modification of histone lysines,3 has not been explored. Functionalisation of (bio)molecules with biotin is employed across a wide array of separation, immobilisation, pre-targeted therapeutic and medical imaging technologies due to its extremely strong affinity for the proteins avidin4 and streptavidin.5 This “avidity” results from unusually strong hydrogen-bonding interactions within the binding site for the ureido moiety of biotin,6 and improved protein packing on binding the ligand.7Regulation of histone biotinylation through formation and hydrolysis of the histone lysine-biotin amide bond is only partly understood,3,8 but maintenance of levels of biotin from dietary sources is known to be achieved through the biotinidase-induced cleavage of biotin from both biocytin 2, and short biotinylated peptides.9 Indeed, biotinidase deficiency is an inherited genetic disorder that if left untreated can result in major disabilities.10 However, biotinidase has also been shown to catalyze the hydrolysis of other esters and amides,11–13 and engineering in biotinidase resistance is important in the design of pre-targeted therapies and imaging agents.14–18 Although not yet explored, hydrolysis-resistant biotinylation agents for histone modification might allow recent advances in site-specific histone labelling using near-native labels to be exploited,19 enabling understanding of the epigenetic role of biotinylation. Additionally, the development of biotinylated, hydrolytically stable, drug conjugates which exploit the over-expression of the sodium-dependent multivitamin transporter,20 which mediates the cell uptake of biotin, in several aggressive cancer cell lines is attractive.
Results and discussion
Design of biotinidase-resistant conjugates
Some resistance to cleavage by biotinidase has been achieved by blocking,14,15 reversing the sense,16 or even removing the carbonyl of the amide bond to biotin;15,17 however, each of these approaches has an impact on binding to the target protein avidin or streptavidin.17 We sought to design a biotin label which would be resistant to cleavage by biotinidase, but would retain a high binding affinity for avidin. Our hypothesis was that triazole 3, prepared by copper(I)-catalysed azide alkyne cycloaddition (CuAAC) coupling of alkyne 4 to an azide (RN3), would provide an optimum non-hydrolysable isostere of the amide bond (Fig. 1b). Correspondingly, triazole 5, derived from coupling known azide 6 to a terminal alkyne (RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), would provide a non-hydrolysable isostere of the reversed amide bond (Fig. 1b).§ Novel alkyne 4 was readily prepared from D-biotin methyl ester via reduction to the corresponding aldehyde and reaction with the Ohira–Bestmann reagent (47% overall yield);‡ whilst azide 6 was prepared in 3 steps (62% overall yield) from the same starting material.21
CH), would provide a non-hydrolysable isostere of the reversed amide bond (Fig. 1b).§ Novel alkyne 4 was readily prepared from D-biotin methyl ester via reduction to the corresponding aldehyde and reaction with the Ohira–Bestmann reagent (47% overall yield);‡ whilst azide 6 was prepared in 3 steps (62% overall yield) from the same starting material.21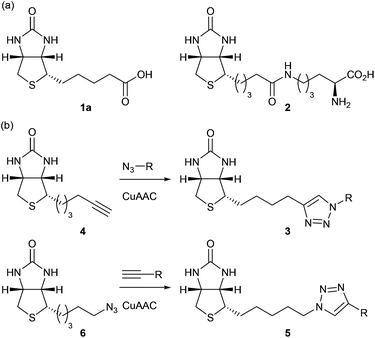 | ||
| Fig. 1 (a) Biotin 1a and biocytin 2. (b) Non-hydrolysable triazoles (3, 5) arising from CuAAC coupling of biotin-alkyne 4 and biotin-azide 6 (respectively). | ||
To determine the efficacy of the triazole-linked biotin species, in terms of biotinidase resistance and avidin binding, a series of biotinylated Gd-DOTA complexes appropriate for pre-targeted magnetic resonance imaging were generated.22 Synthesis of MR imaging agents 7–9 was achieved through the CuAAC coupling of 4 and 6 with the Gd-complexes of three known DOTA “click” ligands (Fig. 2).23–25 These complexes allowed us to investigate both modes of CuAAC coupling (7vs. 8), as well as to probe the effects of linker length between the triazole-linked biotin and bulky Gd-DOTA complex (8vs. 9). The precursor Gd-complexes were readily prepared from the DOTA “click” ligands,¶ and were coupled (CuSO4, NaASc, TBTA, 4 or 6, tBuOH–H2O, rt, 16 h) to give triazoles 7–9 which were purified by HPLC prior to biological assays.
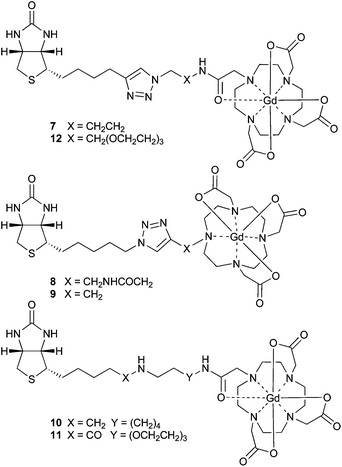 | ||
| Fig. 2 Biotin Gd-DOTA complexes for pre-targeted MRI. | ||
Biotinidase resistance
Human biotinidase was fractionated from human serum,26 and its activity was assessed by measuring the rate of hydrolysis of the commercially available substrate, N-biotinyl-p-aminobenzoic acid (N-biotinyl-PABA).27 HPLC analysis revealed that all three triazole complexes (7–9) were resistant to cleavage by biotinidase up to 126 h, in marked contrast to N-biotinyl-PABA which was fully hydrolyzed under the same conditions within 6 h (Fig. 3).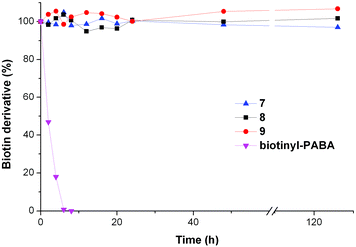 | ||
| Fig. 3 Stability of 7–9 to cleavage by partially-purified biotinidase contrasted with cleavage of N-biotinyl-PABA under the same conditions. Samples were incubated with biotinidase (0.57 AU) in phosphate buffer (0.05 M, pH 6) for up to 126 h; the percentage of the complex remaining was determined by HPLC against an internal standard (resorcinol). | ||
Binding affinity for avidin
The binding affinity of complexes 7–9 to avidin was probed using spectrophotometric competition experiments against the known ligand 4-hydroxy-azobenzene-2-carboxylic acid (HABA, KD = 5.8 × 10−6 M).28 All of the triazoles 7–9 were shown to inhibit the formation of the HABA:avidin complex in a dose-dependent manner (Fig. 4), and comparison with three additional ligands of known binding affinity (biotin 1aKD = 10−15 M; desthiobiotin 1bKD = 10−13 M; and diaminobiotin 1cKD = 1.2 × 10−7 M)4,29 suggested binding affinities for complexes 7–9 in the nM to pM range. Both the amide bond and reversed amide bond triazole isosteres bound to avidin strongly; but complex 9 bound least strongly, indicating that longer linker lengths to the bulky DOTA complex were more favourable.![Binding of MRI agents 7–9 to avidin assessed spectrophotometrically by displacement of HABA from the HABA–avidin complex at pH 7.5. [Diaminobiotin displacement assay conducted at pH 10.]](/image/article/2013/OB/c3ob41837e/c3ob41837e-f4.gif) | ||
| Fig. 4 Binding of MRI agents 7–9 to avidin assessed spectrophotometrically by displacement of HABA from the HABA–avidin complex at pH 7.5. [Diaminobiotin displacement assay conducted at pH 10.] | ||
Comparison with other biotinidase-resistant conjugates
To compare this isostere-based strategy against other biotinidase-resistant approaches we took arguably the most studied DOTA ligand, the reduced biotinamidohexylamine–DOTA conjugate referred to as r-BHD17b or ST221030 (Gd-complex 10, Fig. 2); for which an avidin dissociation constant (KD = 2.9 × 10−7 M) has recently been determined by isothermal calorimetry (ITC).31 Ligand ST2210 was synthesised as previously described,17b and both it and the conjugate of biotin-PEG3-amine with the mono-activated DOTA NHS ester (DO3A-NHS) were treated with GdCl3 to effect the formation of amine-linked complex 10 and amide-linked complex 11 respectively. Synthesis of the isostere-based, biotinidase-resistant complex 12 was achieved through CuAAC coupling of alkyne 4 with a commercial bi-functional PEG3-aminoazide; followed by coupling to DO3A-NHS and Gd-complex formation as for complex 11. Each of the complexes (10–12) was purified by HPLC prior to the determination of avidin dissociation constants by ITC.32Since the binding of complexes 10–12 to avidin was predicted to be very strong, we determined KD values in a series of competition experiments using amine-linked complex 10 as a reference.33 Calorimetric titration of 10 was determined to be close to the limit of the experimental range (KD = 4.1 ± 1.5 × 10−9 M); the binding was assumed to be non-cooperative across the avidin tetramer and the ligand was shown to saturate each of the four binding sites. This measured binding affinity is ∼102 fold greater than previously reported,31 but we attribute this to the rigorous purification of both the complex (by HPLC) and protein (by size exclusion) prior to measurement. To determine the binding affinities of amide-linked complex 11 and the natural biotin carrier biocytin 2, and compare them with the triazole-linked complex 12, the amine-linked complex 10 was added to bring the apparent equilibrium dissociation constant (K′D) closer to the range of the instrument (Fig. 5). The binding affinities of these amide- and triazole-linked ligands were determined to be in the low picomolar to femtomolar range (KD ≤ 10 × 10−12 M), and of equivalent strength to each other suggesting that the triazole-linked biotin mimics the natural biotin amide bond very closely in its binding to avidin.
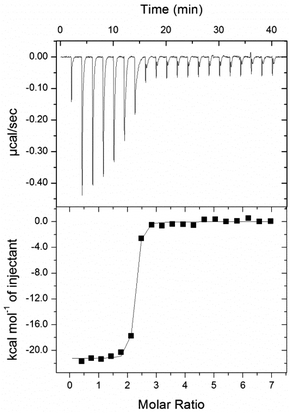 | ||
| Fig. 5 Calorimetric titration of 12 into avidin in competition with 10 in sodium acetate buffer (100 mM; pH 5.5). Top panel: individual injections of a solution of 12 (and 10) into a solution of avidin (and 10). Bottom panel: titration curve, enthalpy/(mole of 12 injected) versus12/(avidin) molar ratio. | ||
Conclusions
We have demonstrated that a triazole-linked biotin conjugate derived either from alkynyl biotin 4, or from the related biotin azide 6, provides an excellent biotinidase-resistant isostere of the biotin-amide bond. Derivatives of biotin alkyne 4 linked to appropriate spacers have equilibrium dissociation constants which are equivalent to their amide counterparts, in contrast to other biotinidase-resistant linkages. Hydrolase-resistant biotin conjugates based on the triazole isostere will lead to efficient pre-targeting agents for therapeutic and imaging applications, and an enhanced understanding of the role of protein biotinylation where hydrolytic cleavage (e.g. by biotinidase, holocarboxylase synthetase etc.) might make this otherwise difficult to address.Experimental
Preparation of biotin alkyne 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) to give terminal biotin alkyne 4 as a colourless solid (0.13 g, 0.58 mmol, 47%). Rf (DCM–MeOH, 90:10) = 0.5; mp 158–162 °C; IR 3242 (NH), 2108 (CCH), 1703 (C
:10) to give terminal biotin alkyne 4 as a colourless solid (0.13 g, 0.58 mmol, 47%). Rf (DCM–MeOH, 90:10) = 0.5; mp 158–162 °C; IR 3242 (NH), 2108 (CCH), 1703 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1H NMR δ (500 MHz, CDCl3) 4.97 (1H, s, NHCHCH), 4.89 (1H, s, NHCHCH2), 4.55 (1H, dd, J = 7.7, 4.6 Hz, NHCHCH), 4.37 (1H, dd, J = 7.7, 4.6 Hz, NHCHCH2), 3.21 (1H, ddd, J = 8.0, 6.6, 4.6 Hz, SCH), 2.97 (1H, dd, J = 12.8, 5.0 Hz, CHAHBS), 2.77 (1H, d, J = 12.8 Hz, CHAHBS), 2.26 (2H, td, J = 6.5, 2.5 Hz, CH2CCH), 2.00 (1H, t, J = 2.5 Hz, CCH), 1.75–1.65 (2H, m, CH2), 1.62–1.55 (4H, m, 2 × CH2); 13C NMR δ (126 MHz, CDCl3) 162.9 (CO), 84.1 (C), 68.8 (CH), 61.9 (CH), 60.1 (CH), 55.3 (CH), 40.6 (CH2), 28.1 (CH2), 28.0 (CH2), 27.9 (CH2), 18.1 (CH2); m/z (ESI+, MeOH) 471 ([2M + Na]+, 100%), 247 ([M + Na]+, 21), 225 ([M + H]+, 11); HRMS (ESI+, MeOH) [2M + Na]+ found 471.1864, C11H16N2OS requires 471.1859.
O); 1H NMR δ (500 MHz, CDCl3) 4.97 (1H, s, NHCHCH), 4.89 (1H, s, NHCHCH2), 4.55 (1H, dd, J = 7.7, 4.6 Hz, NHCHCH), 4.37 (1H, dd, J = 7.7, 4.6 Hz, NHCHCH2), 3.21 (1H, ddd, J = 8.0, 6.6, 4.6 Hz, SCH), 2.97 (1H, dd, J = 12.8, 5.0 Hz, CHAHBS), 2.77 (1H, d, J = 12.8 Hz, CHAHBS), 2.26 (2H, td, J = 6.5, 2.5 Hz, CH2CCH), 2.00 (1H, t, J = 2.5 Hz, CCH), 1.75–1.65 (2H, m, CH2), 1.62–1.55 (4H, m, 2 × CH2); 13C NMR δ (126 MHz, CDCl3) 162.9 (CO), 84.1 (C), 68.8 (CH), 61.9 (CH), 60.1 (CH), 55.3 (CH), 40.6 (CH2), 28.1 (CH2), 28.0 (CH2), 27.9 (CH2), 18.1 (CH2); m/z (ESI+, MeOH) 471 ([2M + Na]+, 100%), 247 ([M + Na]+, 21), 225 ([M + H]+, 11); HRMS (ESI+, MeOH) [2M + Na]+ found 471.1864, C11H16N2OS requires 471.1859.Preparation of biotinidase-resistant complex 12
:1, 30 mL) and allowed to stir for 15 min. Sodium ascorbate (0.009 g, 0.044 mmol) was then added and allowed to stir for 15 min followed by copper(II) sulfate (0.006 g, 0.022 mmol). After a further 15 min 1-amino-11-azido-3,6,9-trioxaundecane (2.19 mL, 0.219 mmol; 0.1 M in H2O) was added and the solution stirred under nitrogen at room temperature for 16 h. QuadraPure-IDA metal scavenger resin was added and the reaction mixture was gently shaken overnight. The resin was removed by filtration, and the aqueous solution extracted with EtOAc (2 × 20 mL). The aqueous solution was then lyophilised to afford crude triazole as a colourless solid. The crude product was purified by preparative RP-HPLC using a C18 column and H2O–acetonitrile + 0.1% TFA as mobile phase to give triazole-linked biotin amine 13 as a colourless solid (76 mg, 65%). HPLC Rt = 38 min (SI Method 2); 1H NMR δ (500 MHz, D2O) 7.92 (1H, NCCHN), 4.66–4.59 (3H, m, NHCHCH, CHNCH2), 4.42 (1H, dd, J = 7.9, 4.5 Hz, NHCHCH2), 3.99 (2H, t, J = 4.9 Hz, CH2O), 3.79–3.73 (2H, m, CH2O), 3.70–3.60 (8H, m, 4 × OCH2), 3.34 (1H, dt, J = 9.7, 5.1 Hz, SCH), 3.27–3.17 (2H, m, CH2NH2), 2.99 (1H, dd, J = 13.0, 5.0 Hz, CHAHBS), 2.79 (1H, d, J = 13.0 Hz, CHAHBS), 2.77 (2H, t, J = 7.3 Hz, CH2CCH), 1.82–1.52 (4H, m, 2 × CH2), 1.49–1.36 (2H, m, CH2); 13C NMR δ (126 MHz, D2O) 165.33 (CO), 148.13 (C), 123.57 (CH), 69.64 (CH2), 69.58 (CH2), 69.50 (CH2), 69.45 (CH2), 68.83 (CH2), 66.35 (CH2), 62.08 (CH), 60.23 (CH), 55.44 (CH), 49.90 (CH2), 39.64 (CH2), 39.09 (CH2), 28.23 (CH2), 27.65 (CH2), 27.63 (CH2), 24.13 (CH2); m/z (ESI+, MeOH) 465 ([M + Na]+, 5%), 443 ([M + H]+, 100); HRMS (ESI+, MeOH) [M + H]+ C19H35O4N6S requires 443.2435, found 443.2438.Acknowledgements
We thank the Wellcome Trust, University of Edinburgh (IKTF) and EC FP6 (Marie Curie Fellowship to JRH; contract MEST-CT-2005-020744) for financial support of this work. JL and DJC thank the BBSRC for funding (BB/I013687/1).Notes and references
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- J. Hymes and B. Wolf, Clin. Chim. Acta, 1996, 255, 1–11 CrossRef CAS.
- (a) T. Kuroishi, L. Rios-Avila, V. Pestinger, S. S. K. Wijeratne and J. Zempleni, Mol. Genet. Metab., 2011, 104, 537–545 CrossRef CAS PubMed; (b) Y. I. Hassan and J. Zempleni, J. Nutr., 2006, 136, 1763–1765 CAS; (c) M. A. Narang, R. Dumas, L. M. Ayer and R. A. Gravel, Hum. Mol. Genet., 2004, 13, 15–23 CrossRef CAS PubMed.
- N. M. Green, Biochem. J., 1966, 101, 774–780 CAS.
- L. Chaiet and F. Wolf, J. Arch. Biochem. Biophys., 1964, 106, 1–5 CrossRef CAS.
- J. DeChancie and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 5419–5429 CrossRef CAS PubMed.
- D. H. Williams, E. Stephens and M. Zhou, J. Mol. Biol., 2003, 329, 389–399 CrossRef CAS.
- Y. C. Chew, G. Sarath and J. Zempleni, J. Nutr. Biochem., 2007, 18, 475–481 CrossRef CAS PubMed.
- J. Hymes and B. Wolf, J. Nutr., 1999, 129, 485S–489S CAS.
- B. Wolf, Genet. Med., 2012, 14, 565–575 CrossRef CAS PubMed.
- (a) J. Knappe, W. Bruemmer and K. Biederbick, Biochem. Z., 1963, 338, 599–613 CAS; (b) M. Koivusalo, C. Elorriaga, Y. Kaziro and S. Ochoa, J. Biol. Chem., 1963, 238, 1038–1042 CAS.
- J. Oizumi and K. Hayakawa, Biochim. Biophys. Acta, 1991, 1074, 433–438 CrossRef CAS.
- L. Nilsson and B. Kågedal, Biochem. J., 1993, 291, 545–551 CAS.
- D. S. Wilbur, D. K. Hamlin, M.-K. Chyan, B. B. Kegley and P. M. Pathare, Bioconjugate Chem., 2001, 12, 616–623 CrossRef CAS PubMed.
- (a) A. Pratesi, F. Bucelli, I. Mori, M. Chinol, A. Vedoliva, G. Paganelli, V. Rivieccio, L. Gariboldi and M. Ginanneschi, J. Med. Chem., 2010, 53, 432–440 CrossRef CAS PubMed; (b) J. Hainsworth, P. Harrison and S. J. Mather, Bioconjugate Chem., 2005, 16, 1468–1474 CrossRef CAS PubMed.
- C. F. Foulon, K. L. Alston and M. R. Zalutsky, Bioconjugate Chem., 1997, 8, 179–186 CrossRef CAS PubMed.
- (a) P. A. Turhanen, J. Weisell, P. Lehtolainen-Dalkilic, A.-M. Määttä, J. Vepsäläinen and A. Närvänen, Med. Chem. Commun., 2011, 2, 886–888 RSC; (b) G. Sabatino, M. Chinol, G. Paganelli, S. Papi, M. Chelli, G. Leone, A. M. Papini, A. De Luca and M. Ginanneschi, J. Med. Chem., 2003, 46, 3170–3173 CrossRef CAS PubMed; (c) D. S. Wilbur, M.-K. Chyan, P. M. Pathare, D. K. Hamlin, M. B. Frownfelter and B. B. Kegley, Bioconjugate Chem., 2000, 11, 569–583 CrossRef CAS PubMed.
- J. Hymes and B. Wolf, J. Neurosci. Methods, 2000, 98, 171–173 CrossRef CAS.
- (a) C. David Allis and Tom W. Muir, ChemBioChem, 2011, 12, 264–279 CrossRef PubMed; (b) J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- L. Bildstein, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2011, 63, 3–23 CrossRef CAS PubMed.
- Based on: (a) N. Umeda, T. Ueno, C. Pohlmeyer, T. Nagano and T. Inoue, J. Am. Chem. Soc., 2011, 133, 12–14 CrossRef CAS PubMed; (b) C. Corona, B. K. Bryant and J. B. Arterburn, Org. Lett., 2006, 8, 1883–1886 CrossRef CAS PubMed.
- (a) K. Sano, T. Temma, T. Azuma, R. Nakai and M. Narazaki, Mol. Imaging Biol., 2011, 13, 1196–1203 CrossRef PubMed; (b) A. Mishra, J. Pfeuffer, R. Mishra, J. Engelmann, A. K. Mishra, K. Ugurbil and N. K. Logothetis, Bioconjugate Chem., 2006, 17, 773–780 CrossRef CAS PubMed.
- DOTA azide ligand required for 7: M. K. Schultz, S. G. Parameswarappa and F. C. Pigge, Org. Lett., 2010, 12, 2398–2401 CrossRef CAS PubMed.
- DOTA alkyne ligand required for 8: R. F. H. Viguier and A. N. Hulme, J. Am. Chem. Soc., 2006, 128, 11370–11371 CrossRef CAS PubMed.
- DOTA alkyne ligand required for 9: M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283–6285 RSC.
- ∼10-fold enrichment using method adapted from: J. Chauhan and K. Dakshinamurti, J. Biol. Chem., 1986, 261, 4268–4275 CAS.
- K. A. Kobza, K. Chaiseeda, G. Sarath, J. M. Takacs and J. Zempleni, J. Nutr. Biochem., 2008, 19, 826–832 CrossRef CAS PubMed.
- N. M. Green, Methods Enzymol., 1970, 18(Pt. A), 418–424 CrossRef.
- N. M. Green, Adv. Protein Chem., 1975, 29, 85–133 CrossRef CAS.
- G. Paganelli, et al., Eur. J. Nucl. Med. Mol. Imaging, 2010, 37, 203–211 CrossRef CAS PubMed.
- A. Verdoliva, P. Bellofiore, V. Rivieccio, S. Catello, M. Colombo, C. Albertoni, A. Rosi, B. Leoni, A. M. Anastasi and R. De Santis, J. Biol. Chem., 2010, 285, 9090–9099 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L. N. Lin, Anal. Biochem., 1989, 179, 131–137 CrossRef CAS.
- B. W. Sigurskjold, Anal. Biochem., 2000, 277, 260–266 CrossRef CAS PubMed.
- R. W. Dixon, R. J. Radmer, B. Kuhn and P. A. Kollman, J. Org. Chem., 2002, 67, 1827–1837 CrossRef CAS PubMed.
Footnotes |
| † Celebrating 300 years of Chemistry at Edinburgh. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization of azide 6, complexes 7–9, biotinidase activity assay, enrichment of biotinidase from serum, biotinidase stability assay, HABA displacement and ITC binding assays. See DOI: 10.1039/c3ob41837e |
| § For synthetic ease the azide 6 corresponding to the amide bond reversal of an amide of homobiotin was chosen for this investigation. |
| ¶ Attempted coupling of DOTA ligands in the absence of the lanthanide metal was found to be unsuccessful unless excess Cu(I) was employed. |
| This journal is © The Royal Society of Chemistry 2013 |