 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A selective fluorescent chemosensor for phosphoserine†
Chad M. Cooley, Kenneth S. Hettie, Jessica L. Klockow, Shana Garrison and Timothy E. Glass*
Department of Chemistry, University of Missouri, 601 South College Avenue, Columbia, MO 65211, USA. E-mail: GlassT@missouri.edu; Fax: +1 573-882-2754; Tel: +1 573-882-3813
First published on 19th September 2013
Abstract
A fluorescent chemosensor for the detection of phosphoserine is reported. The ditopic sensor features a phosphate-coordinating zinc(II)–dipicolylamine (Zn2+–DPA) unit tethered to an amine-binding coumarin aldehyde fluorophore. With phosphoserine, the sensor demonstrates a 30-fold fluorescence enhancement under buffered aqueous conditions.
Introduction
Phospholipids constitute the dominant class of lipids composing the lipid bilayer of cellular membranes and confer structural support, organization of membrane proteins, and intercellular signaling mechanisms.1–6 Phosphatidylcholine (PC), sphingomyelin (SM), phosphatidylethanolamine (PE), and phosphatidylserine (PS) represent over 97% of the total phospholipid composition and are asymmetrically distributed between the inner and outer leaflets of a cellular membrane.7 In quiescent cells, PC and SM constitute ∼87% of the total phospholipid content of the outer leaflet, with PS being exclusively restricted to and comprising ∼30% of the total phospholipid content of the inner leaflet (Fig. 1). Cell death randomizes the asymmetric distribution of phospholipids between the inner and outer leaflets, thereby externalizing PS to the outer cell surface.8–10 In fact, exposure of PS to the outer leaflet of the cellular membrane is the hallmark molecular biomarker for all types of cell death and serves as a signal for phagocytosis by providing ∼106–109 binding sites per cell.11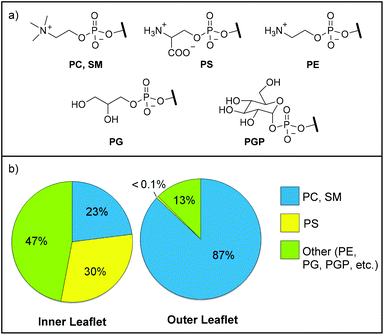 | ||
| Fig. 1 (a) Common cellular membrane phospholipid head groups. PC = phosphatidylcholine, SM = sphingomyelin, PS = phosphatidylserine, PE = phosphatidylethanolamine, PG = phosphatidylglycerol, PGP = phosphatidyl-glucopyranose. (b) Asymmetric phospholipid composition (mol%) of the outer and inner leaflets of healthy human cellular membranes.7 | ||
Several methods to tag apoptotic cells and phospho-amino acid residues have appeared in the literature.12–17 In addition to biological probes (e.g., annexin assays), the vast majority of synthetic approaches have focused on detecting anionic phospholipid head groups, which includes phosphoserine. In particular, zinc(II)–dipicolylamine (Zn2+–DPA) complexes have proven to be admirable receptors for negatively-charged lipid head groups and have been used to tag apoptotic cells as well as bacterial cells.11,12,14,15,18,19 These primarily consist of two pre-coordinated Zn2+–DPA units tethered to a synthetic fluorophore that associate with the membrane surface of apoptotic cells through strong multipoint interactions with the anionic phospholipid head groups. These molecular probes are phospholipid tags and do not operate in a turn-on fashion. By contrast, a turn-on sensor for PS could give quantitative information about the amount of lipid present with low background as well as enable new types of experiments such as the ability to monitor the progression of cell death in a cell population.
Recently, we have been exploring the use of water-soluble shape-selective molecular tubes for hydrophobic sensing of lipids.20 In order to build selective sensors for PS, we require a head-group binding unit. We chose to use the highly-water soluble head groups as a model system for the analyses to demonstrate sensor efficacy because the head groups are the exposed units at the cell surface and the phospholipids lack water solubility. The head groups lack the hydrophobic lipid, yet remain functionally equivalent to the phospholipids and are reasonable analogues for evaluating head group binding units. In the case of phosphoserine, both the head group and PS possess a primary amine and an anionic phosphoester. Herein, we present a fluorescent turn-on molecular sensor based on the coumarin aldehyde scaffold for the selective recognition and sensing of phosphoserine (Fig. 2).
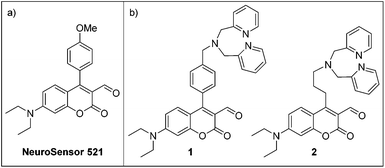 | ||
| Fig. 2 (a) Structure of NeuroSensor 521. (b) Ditopic sensors for phosphoserine. | ||
The ditopic sensors 1 and 2 consist of an aldehyde recognition element that is integrated into the coumarin fluorophore and a single Zn2+–DPA recognition element appended to the coumarin scaffold to afford reversible binding to the primary amine and anionic phosphate moieties of phosphoserine, respectively (Scheme 1). The coumarin aldehyde has been shown to effectively produce a turn-on response to amine binding. Indeed, NeuroSensor 521 (NS521, Fig. 2a) was developed as a monotopic receptor for catecholamines and provides a turn-on response in live cells.21 In this study, we modified the NS521 scaffold by appending a Zn2+–DPA unit to the C4 position of the coumarin with both a rigid and flexible spacer to afford sensors 1 and 2, respectively. We hypothesized that a more rigid linker would provide stronger binding to the analyte but developed a flexible linker for comparison. Molecular models indicated that the methyl phenyl and propyl linkers would provide the optimal spacing for phosphoserine (see ESI†).
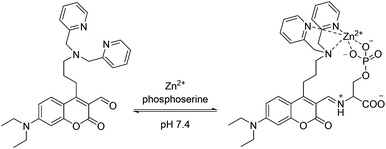 | ||
| Scheme 1 Ditopic binding of 2–Zn2+ to phosphoserine. | ||
We reasoned that only phospholipid head groups with primary amines should bind to the coumarin aldehyde and form an iminium ion which causes an ∼40 nm bathochromic shift in absorption.21–23 By exciting the fluorophore at this new, more red wavelength, we can visualize a fluorescence increase due only to the bound sensor. The Zn2+–DPA group is likely the best receptor for phosphates in water and was incorporated to achieve selectivity.
Results and discussion
Synthesis of ditopic sensors
The syntheses of sensors 1 and 2 are shown in Scheme 2. To make sensor 1, compound 3 was coupled via its tosylate to an aryl boronic acid to produce compound 4. Chlorination followed by formylation using the Vilsmeier reagent gave compound 6 which was alkylated with a dipicolylamine unit to yield sensor 1. Sensor 2 was prepared from alcohol 723 by conversion to iodide 8 followed by alkylation with dipicolylamine.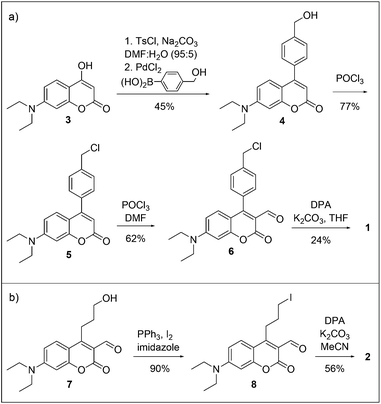 | ||
| Scheme 2 Syntheses of sensors (a) 1 and (b) 2. | ||
UV and fluorescence titrations
| Analyte | 1 | 1–Zn2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b | 2 | 2–Zn2+b | ||||
|---|---|---|---|---|---|---|---|---|
| Ka (M−1) | Isat/I0c | Ka (M−1) | Isat/I0c | Ka (M−1) | Isat/I0c | Ka (M−1) | Isat/I0c | |
| a Analytes of various concentrations were added to sensor (10 μM) in buffer (50 mM HEPES, 100 mM NaCl, pH 7.4). Sensor 1 was titrated in 97:3 buffer–DMSO and sensor 2 was titrated in 100% buffer.b Zinc acetate (10 μM) was pre-coordinated with sensor prior to addition of analyte.c Isat was taken from the theoretical maximum of the binding isotherm; λex = 488 nm; λem, 1 = 521 nm, λem, 2 = 513 nm. The phospholipid head groups phosphocholine, α-glycerolphosphate, and D-gluco-pyranose 1-phosphate did not bind or alter the fluorescence properties of either sensor. | ||||||||
| Phosphoserine | 3.1 | 7.2 | 5.4 | 6.2 | 120 | 5.8 | 310 | 30 |
| Phosphoethanolamine | 5.7 | 4.8 | 5.7 | 4.9 | 93 | 9.1 | 180 | 39 |
| Glutamate | 4.7 | 7.0 | 7.0 | 4.3 | 22 | 13 | 190 | 17 |
| Glycine | 6.1 | 6.1 | 6.2 | 4.0 | 5.3 | 16 | 6.6 | 29 |
Phosphocholine, α-glycerolphosphate, and D-gluco-pyranose 1-phosphate did not bind to the coumarin aldehyde of 1–Zn2+ and therefore did not alter the spectroscopic properties. All primary amine analytes bound to the coumarin aldehyde with low affinity and relatively low fluorescence enhancements (<7-fold) with emission maxima at 521 nm when excited at 488 nm. The binding results were similar to the control experiments conducted without Zn2+ indicating that the Zn2+–DPA had little effect on enhancing analyte binding to the coumarin aldehyde (see ESI†). Indeed, these binding constants are similar to those achieved with simple, unsubstituted coumarin aldehydes.21–23
Sensor 2 displayed similar absorption properties compared to sensor 1. The absorbance of 2–Zn2+ also shifted to the red only upon addition of phosphoserine, phosphoethanolamine, glutamate, and glycine. As seen with sensor 1, the phospholipid head groups that did not contain primary amines did not bind. The binding for phosphoserine was remarkably higher than for the other analytes tested with Ka = 310 M−1 (Fig. 3). Titrations with phosphoethanolamine and glutamate also demonstrated reasonable affinity, though not as high as with phosphoserine. The fluorescence increased over 30-fold with the addition of both phosphoserine and phosphoethanolamine, with emission at 513 nm when excited at 488 nm.
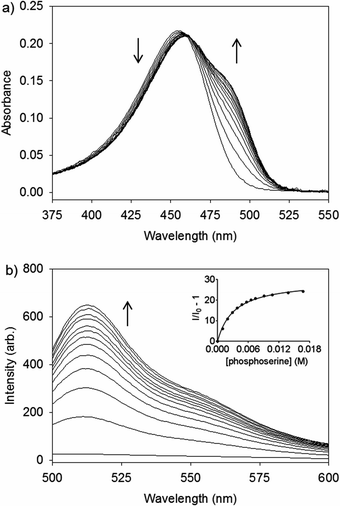 | ||
| Fig. 3 (a) UV/Vis and (b) fluorescence spectra of 2–Zn2+ (10 μM) with aliquots of 100 mM phosphoserine in buffer (50 mM HEPES, 100 mM NaCl, pH 7.4). λex = 488 nm. λem = 513 nm. Inset is the fit to a binding isotherm. | ||
For sensor 2, the spectroscopic properties with Zn2+ were much more favorable than the control experiments without Zn2+, though the binding constants of 2 with phosphoserine, phosphoethanolamine, and glutamate were elevated compared to the typical low binding constants seen with monotopic coumarin aldehyde sensors.21–23 Presumably, the protonated form of the DPA can interact favorably with the charged phosphate to enhance binding. The fluorescence response of sensor 2 was lower without Zn2+ due to photoinduced electron transfer quenching from the uncoordinated DPA.
It is clear from the spectroscopic studies of both sensors that 2–Zn2+ is the best sensor for phosphoserine both in terms of binding constant and maximum fluorescence response (Fig. 4). Because of the flexible linker, 2–Zn2+ demonstrated more than two orders of magnitude better binding than the more rigid sensor 1. Clearly, the rigid structure for 1–Zn2+ prohibits a two-point binding interaction. Even sensor 2 lacking Zn2+ was a better receptor for phosphoserine than sensor 1.
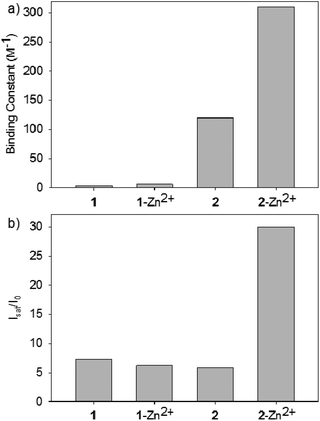 | ||
| Fig. 4 (a) Binding constants and (b) fluorescence enhancements (Isat/I0) of sensors 1 and 2 with phosphoserine. | ||
Compared to the phospholipid tagging probes, 2–Zn2+ is unique in that it can coordinate to the analyte phosphate group and primary amine to achieve selective binding to phosphoserine over other phospholipid head groups. Thus, the head groups phosphocholine, α-glycerolphosphate, and D-gluco-pyranose-1-phosphate give no response, which is extremely important since the majority of membrane phospholipids are choline-based. In addition, 2–Zn2+ is also sensitive to the charge on the head group. Phosphoethanolamine and glutamate bind less effectively, as each bears one less negative charge than phosphoserine. It should be noted that while glutamate and glycine gave enhanced binding and fluorescence output, they are not expressed at high concentrations on a cellular membrane.
Conclusions
In conclusion, 2–Zn2+ demonstrates a means to selectively sense phosphoserine over other major phospholipid head groups by forming a two-point binding interaction with the phosphate and primary amine functional groups of the analyte under buffered aqueous conditions. Future work in this area will focus on joining this head group binding unit to our lipid receptor to produce sensitive and selective sensors for bioactive lipids.Experimental section
Spectroscopic analysis
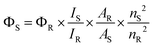 | (1) |
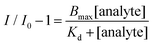 | (2) |
Synthetic procedures
:5). The solution stirred at 60 °C for 1 h. Then 4-(hydroxymethyl)phenylboronic acid (179.2 mg, 1.179 mmol) and PdCl2 (29 mg, 0.161 mmol) were added and N2 was bubbled through for 5 min. The solution stirred at 60 °C for 16 h. The inorganic solids were filtered off and rinsed with acetone. The solvent was removed in vacuo and the remaining crude material was purified by chromatography (95:5 CH2Cl2–EtOAc) to yield compound 4 (156.5 mg, 45%) as a yellow solid (mp 62 °C): 1H NMR (500 MHz, CDCl3) δ 7.50 (d, 2H, J = 7.5 Hz), 7.43 (d, 2H, J = 7.5 Hz), 7.23 (d, 1H, J = 9.0 Hz), 6.57 (d, 1H, J = 2.0 Hz), 6.51 (dd, 1H, J = 9.0, 2.0 Hz), 5.99 (s, 2H), 4.80 (s, 2H), 3.42 (q, 4H, J = 7.0), 1.21 (t, 6H, 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 162.1, 156.8, 155.8, 150.6, 142.1, 135.5, 128.6, 127.8, 127.1, 108.5, 108.2, 107.9, 97.9, 64.8, 44.8, 12.4; IR (neat, cm−1) 3412, 2970, 1695, 1605, 1589, 1417, 1352, 1115; HRMS calculated for C20H21NO3Na (M + Na+): 346.1414. Found: 346.1414.:10 CH2Cl2–EtOAc) to yield compound 5 (81 mg, 77%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.51 (d, 2H, J = 8.0 Hz), 7.43 (d, 2H, J = 8.0 Hz), 7.22 (d, 1H, J = 9.0 Hz), 6.56 (d, 1H, J = 2.5 Hz), 6.51 (dd, 1H, J = 9.0, 2.5 Hz), 5.98 (s, 1H), 3.41 (q, 4H, J = 7.0 Hz), 1.20 (t, 6H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 161.9, 156.7, 155.4, 150.6, 138.5, 136.3, 128.8, 128.7, 127.7, 108.5, 108.3, 107.6, 97.8, 45.6, 44.7, 12.4; IR (neat, cm−1) 2970, 1708, 1614, 1593, 1524, 1417, 1356, 1115; HRMS calculated for C20H20ClNO2Na (M + Na+): 364.1075. Found: 364.1077.:5 CH2Cl2–EtOAc) yielded compound 6 as a yellow oil (97 mg, 62%): 1H NMR (500 MHz, CDCl3) δ 9.89 (s, 1H), 7.54 (d, 2H, J = 8.0 Hz), 7.26 (d, 2H, J = 8.0 Hz), 6.94 (d, 1H, J = 9.5 Hz), 6.49–6.53 (m, 2H), 4.68 (s, 2H), 3.45 (q, 4H, J = 7.0 Hz), 1.23 (t, 6H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 188.2, 160.8, 160.1, 157.7, 153.1, 138.3, 133.4, 131.0, 128.6, 128.5, 112.0, 109.8, 108.9, 97.0, 45.7, 45.2, 12.4; IR (neat, cm−1) 1614, 1559, 1494, 1417, 1352; HRMS calculated for C21H20ClNO3Na (M + Na+): 392.1024. Found: 392.1022.:20 EtOAc–MeOH) to yield sensor 1 as a yellow oil (54 mg, 47%). 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H) 8.55 (d, 2H, J = 5.0 Hz), 7.69 (td, 2H, J = 7.5, 1.5 Hz), 7.61 (d, 2H, J = 8.0 Hz), 7.54 (d, 2H, J = 8.0 Hz), 7.21 (d, 2H, J = 8.0 Hz), 7.17 (m, 2H), 6.93 (d, 1H, J = 9.5 Hz), 6.45–6.51 (m, 2H), 3.88 (s, 4H), 3.80 (s, 2H), 3.43 (q, 4H, J = 7.0 Hz), 1.21 (t, 6H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 188.3, 162.2, 159.5, 159.4, 157.7, 153.0, 148.9, 140.2, 136.5, 131.5, 130.9, 128.7, 128.5, 122.8, 122.0, 112.2, 109.6, 108.9, 96.9, 60.1, 58.1, 45.1, 12.4; IR (neat, cm−1) 1746, 1611, 1495, 1422, 1356, 1133, 731; HRMS calculated for C33H32N4O3Na (M + Na+): 555.2367. Found: 555.2366.:1 CH2Cl2–EtOAc to give compound 8 (48.9 mg, 90%) as a yellow solid (decomp 138–141 °C). 1H NMR (500 MHz, CDCl3) δ 10.35 (s, 1H), 7.73 (d, 1H, J = 16.0 Hz), 6.69 (dd, 1H, J = 15.5, 4.5 Hz), 6.47 (d, 1H, J = 4.5 Hz), 3.33–3.52 (m, 8H), 2.05–2.15 (m, 2H), 1.26 (t, 6H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 190.9, 162.9, 161.5, 157.6, 152.8, 128.6, 111.8, 110.1, 108.4, 97.3, 45.1, 33.5, 28.9, 12.5, 6.8; IR (neat, cm−1) 2913, 1716, 1671, 1614, 1556, 1503, 1454, 1352; HRMS calculated for C17H20INO3Na (M + Na+): 436.0380. Found: 436.0372.:1 CH2Cl2–EtOAc gave sensor 2 (24.1 mg, 56%) as a yellow solid (mp 109 °C). 1H NMR (500 MHz, CDCl3) δ 10.34 (s, 1H), 8.55 (d, 2H, J = 7.0 Hz), 7.56–7.71 (m, 5H), 7.14–7.19 (m, 2H), 6.52 (dd, 1H, J = 9.5, 4.5 Hz), 6.44 (d, 1H, J = 4.5 Hz); 13C NMR (125 MHz, CDCl3) δ 190.7, 163.8, 163.1, 159.6, 157.5, 152.6, 149.0, 136.4, 128.7, 123.0, 121.9, 111.7, 109.8, 108.5, 97.2, 60.2, 54.3, 45.0, 27.8, 25.8, 12.4; IR (neat, cm−1) 2970, 2921, 1712, 1676, 1615, 1559, 1508, 1452, 1355, 1148, 1131; HRMS calculated for C29H32N4O3Na (M + Na+): 507.2367. Found: 507.2357.Acknowledgements
This work was supported by the National Science Foundation (CHE-1112194).Notes and references
- M. I. Gurr, J. L. Harwood and K. N. Frayn, Lipid Biochemistry: An Introduction, Oxford, UK, 5th edn, 2002, pp. 215–259 Search PubMed.
- R. O. Ryan and D. J. van der Horst, Annu. Rev. Entomol., 2000, 45, 233 CrossRef CAS PubMed.
- M. Dykstra, A. Cherukuri, H. W. Sohn, S.-J. Tzeng and S. K. Pierce, Annu. Rev. Immunol., 2003, 21, 457 CrossRef CAS PubMed.
- M. Kielian, P. K. Chatterjee, D. L. Gibbons and Y. E. Lu, Subcell. Biochem., 2000, 34, 409 CrossRef CAS.
- K. Lauber, E. Bohn, S. M. Kröber, Y.-J. Xiao, S. G. Blumenthal, R. K. Lindemann, P. Marini, C. Wiedig, A. Zobywalski, S. Baksh, Y. Xu, I. B. Autenrieth, K. Schulze-Osthoff, C. Belka, G. Stuhler and S. Wesselborg, Cell, 2003, 113, 717 CrossRef CAS.
- S. A. Islam, S. Y. Thomas, C. Hess, B. D. Medoff, T. K. Means, C. Brander, C. M. Lilly, A. M. Tager and A. D. Luster, Blood, 2006, 107, 444 CrossRef CAS PubMed.
- J. A. Virtanen, K. H. Cheng and P. Somerharju, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4964 CrossRef CAS.
- Y. Y. Tyurina, A. A. Shvedova, K. Kawai, V. A. Tyurin, C. Kommineni, P. J. Quinn, N. F. Schor, J. P. Fabisiak and V. E. Kagan, Toxicology, 2000, 148, 93 CrossRef CAS.
- G. Preta and B. Fadeel, PLoS One, 2012, 7, 1 Search PubMed.
- T. T. Tung, K. Nagaosa, Y. Fujita, A. Kita, H. Mori, R. Okada, S. Nonaka and Y. Nakanishi, J. Biochem., 2013, 153, 483 CrossRef CAS PubMed.
- B. A. Smith, S. T. Gammon, S. Xiao, W. Wang, S. Chapman, R. McDermott, M. A. Suckow, J. R. Johnson, D. Piwnica-Worms, G. W. Gokel, B. D. Smith and W. M. Leevy, Mol. Pharmaceutics, 2011, 8, 583 CrossRef CAS PubMed.
- B. A. Smith and B. D. Smith, Bioconjugate Chem., 2012, 23, 1989 CrossRef CAS PubMed.
- B. A. Smith, S. Xiao, W. Wolter, J. Wheeler, M. A. Suckow and B. D. Smith, Apoptosis, 2011, 16, 722 CrossRef PubMed.
- R. G. Hanshaw and B. D. Smith, Bioorg. Med. Chem., 2005, 13, 5035 CrossRef CAS PubMed.
- S. W. Bae, M. S. Cho, A. R. Jeong, B.-R. Choi, D.-E. Kim, W.-S. Yeo and J.-I. Hong, Small, 2010, 6, 1499 CrossRef CAS PubMed.
- P. Atkinson, B. S. Murray and D. Parker, Org. Biomol. Chem., 2006, 4, 3166 CAS.
- T. Anai, E. Nakata, Y. Koshi, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 6232 CrossRef CAS PubMed.
- A. G. White, B. D. Gray, K. Y. Pak and B. D. Smith, Bioorg. Med. Chem. Lett., 2012, 22, 2833 CrossRef CAS PubMed.
- C. Lakshmi, R. G. Hanshaw and B. D. Smith, Tetrahedron, 2004, 60, 11307 CrossRef CAS PubMed.
- C. T. Avetta, B. J. Shorthill, C. Ren and T. E. Glass, J. Org. Chem., 2012, 77, 851 CrossRef CAS PubMed.
- K. S. Hettie, X. Liu, K. D. Gillis and T. E. Glass, ACS Chem. Neurosci., 2013, 4, 918 CrossRef CAS PubMed.
- K. E. Secor and T. E. Glass, Org. Lett., 2004, 6, 3727 CrossRef CAS PubMed.
- E. K. Feuster and T. E. Glass, J. Am. Chem. Soc., 2003, 125, 16174 CrossRef CAS PubMed.
- C. A. Parker and W. T. Rees, Analyst, 1960, 85, 587 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis and fluorescence spectroscopic results and 1H and 13C NMR spectra. See DOI: 10.1039/c3ob41677a |
| This journal is © The Royal Society of Chemistry 2013 |