 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free, hydroacylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C and NN bonds via aerobic C–H activation of aldehydes, and reaction of the products thereof†
C and NN bonds via aerobic C–H activation of aldehydes, and reaction of the products thereof†
Vijay Chudasama, Ahmed R. Akhbar, Karim A. Bahou, Richard J. Fitzmaurice and Stephen Caddick*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H OAJ, UK. E-mail: VPEnterprise@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 3108 5071
First published on 17th September 2013
Abstract
In this report, a thorough evaluation of the use of aerobically initiated, metal-free hydroacylation of various C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and NN acceptor molecules with a wide range of aldehydes is presented. The aerobic-activation conditions that have been developed are in sharp contrast to previous conditions for hydroacylation, which tend to use transition metals, peroxides that require thermal or photochemical degradation, or N-heterocyclic carbenes. The mildness of the conditions enables a number of reactions involving sensitive reaction partners and, perhaps most significantly, allows for α-functionalised chiral aldehydes to undergo radical-based hydroacylation with complete retention of optical purity. We also demonstrate how the resulting hydroacylation products can be transformed into other useful intermediates, such as γ-keto-sulfonamides, sultams, sultones, cyclic N-sulfonyl imines and amides.
C and NN acceptor molecules with a wide range of aldehydes is presented. The aerobic-activation conditions that have been developed are in sharp contrast to previous conditions for hydroacylation, which tend to use transition metals, peroxides that require thermal or photochemical degradation, or N-heterocyclic carbenes. The mildness of the conditions enables a number of reactions involving sensitive reaction partners and, perhaps most significantly, allows for α-functionalised chiral aldehydes to undergo radical-based hydroacylation with complete retention of optical purity. We also demonstrate how the resulting hydroacylation products can be transformed into other useful intermediates, such as γ-keto-sulfonamides, sultams, sultones, cyclic N-sulfonyl imines and amides.
Introduction
The development of methods to construct new chemical bonds efficiently in a selective manner whilst minimising energy usage and production of waste has, arguably, never been of greater importance.1 Thus, the synthesis of complex molecules using environmentally benign transformations has become a major focus in organic synthesis.1 Despite recent work on the development of more efficient chemical processes, it is still the case that numerous synthetic transformations are inherently limited as they employ a multi-step mode of reactivity, i.e. where a starting material 1 is converted to a desired product 4via a number of intermediates, 2 and 3 (Fig. 1).2 Such an approach depends on: (A) the ease of introduction of a precursor functional group into a starting material (1) to give intermediate 2, (B) ease of precursor (2) conversion into an active species (3), and (C) selectivity and efficiency of the reaction of the active species to produce the desired product (4). Each step in the process introduces inefficiencies that multiply through the multi-step conversion and typically involve the use of additional reagents, which results in increased waste production. An appealing alternative to the modified substrate/reagent approach is that of C–H activation.2,3 In particular, in recent years, there have been significant developments in remote C–H activation processes using transition metal catalysis.4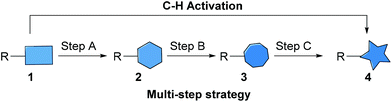 | ||
| Fig. 1 Strategies for bond formation. | ||
Although direct C–H activation has been used in the field of radical chemistry there are often significant problems with the selectivity of C–H abstraction,5–7 and the more successful protocols tend to use a modified substrate/reagent strategy. Moreover, a significant number of the most widely used methods employ expensive, toxic and environmentally damaging reagents or catalysts, and/or large excesses of noxious reagents or solvents.5–11 Nonetheless, it should still be appreciated that free radical processes have played an important role in synthetic organic chemistry and such methods can offer a complementary approach to the more vigorous conditions often required for two electron processes.5,12
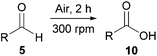
In view of the limitations of current strategies toward radical-based C–H activation, we became interested in developing an alternative. Upon examination of the generally accepted mechanism for the auto-oxidation of aldehydes to carboxylic acids, we were intrigued to note that it evokes the formation of an acyl radical 6 from an aldehyde 5via the action of molecular oxygen (Scheme 1).13 In this pathway, the acyl radical 6 reacts with molecular oxygen to form peracyl radical 7, which then abstracts an H-atom from another molecule of aldehyde regenerating acyl radical 6. The resultant peroxyacid 8 then reacts with another molecule of aldehyde to afford 9, which undergoes decomposition to yield two molecules of carboxylic acid 10.13
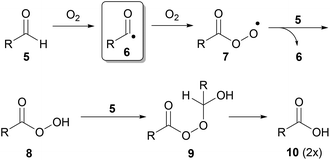 | ||
| Scheme 1 Aldehyde auto-oxidation pathway. | ||
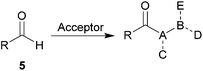
Although the synthetic applications of acyl radicals are well established8 and technologies exist for radical based hydroacylation using polarity reversal catalysis or metals,14–18 there has been limited work on the synthetic applications of aldehyde auto-oxidation.8,19–21 We have recently described the radical hydroacylation of vinyl sulfonates, sulfones and phosphonates, α,β-unsaturated esters and azodicarboxylates using acyl radicals generated via the aldehyde auto-oxidation pathway.22–26 Efficient C–C and C–N bond formation was achieved under benign conditions by the trapping of acyl radicals (6) generated from aldehydes (5) with radical acceptors (11) to initiate a chain reaction process, leading to the formation of hydroacylation product 13 (Scheme 2). We also described some of the utility of the derived hydroacylation products 13 in a range of further transformations.24,26
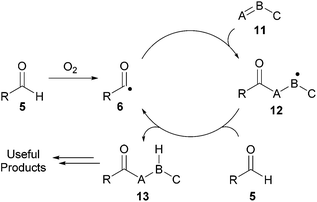 | ||
| Scheme 2 Mechanism for aerobically initiated hydroacylation. | ||
Herein, we report a thorough evaluation of the use of aerobically initiated hydroacylation with a wide range of aldehydes and double bond acceptor molecules. In our analysis, we examine aldehyde functional group tolerance with the various acceptors and the significance of the rate of aldehyde auto-oxidation on yield. Further evidence for the radical nature of the pathway is also provided. In addition, a series of methods by which the hydroacylation products can be transformed into other useful intermediates, such as γ-keto-sulfonamides, sultams, sultones, cyclic N-sulfonyl imines and amides, are also described.
Results and discussion
Aerobic auto-oxidation of aldehydes to carboxylic acids
Central to our methodology is the aerobic C–H activation of aldehydes. Thus, we sought to explore the rate at which different aldehydes auto-oxidise to their corresponding carboxylic acids (Table 1). To achieve this, a fixed volume of each aldehyde (500 μL) was stirred in the same shaped vessel at the same stirring rate (300 rpm) and the ratio of aldehyde 5 to acid 10 assessed by 1H NMR integration after 2 h. A simple set of aldehydes 5a–j, a functionally diverse series of aldehydes 5k–t, aromatic aldehydes 5u–w, and enantiopure aldehydes 5x–y were all evaluated under the reaction conditions.
| ||
|---|---|---|
| Entry | Aldehyde 5 | 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10a :10a |
| Conditions: aldehyde 5 (500 μL) stirred at 300 rpm at 21 °C for 2 h unless otherwise stated.a Ratio of aldehyde 5 to acid 10 after 2 h stirring of aldehyde 5 (500 μL) at 300 rpm determined through comparison of the 1H NMR integration of aldehyde 5 and acid 10. It should also be noted that not stirring, significantly reduced the rate of auto-oxidation. As certain aldehydes were contaminated with a minor amount of acid, the ratios quoted are relative to the initial ratio of aldehyde to acid (where applicable).b No conversion of aldehyde observed, even after 24 h.c 32% Conversion of aldehyde but no corresponding acid formation observed by integration of 1H NMR relative to pentachlorobenzene as an internal standard. | ||
| 1 |  | 1:0.37 |
| 2 | 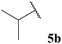 | 1:1.04 |
| 3 | 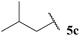 | 1:0.39 |
| 4 |  | 1:0.27 |
| 5 | 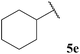 | 1:0.34 |
| 6 |  | 1:0.50 |
| 7 | 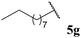 | 1:0.04 |
| 8 | 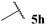 | 1:1.54 |
| 9 | 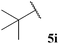 | 1:0.99 |
| 10 | 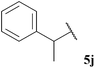 | 1:0.00b |
| 11 | 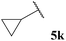 | 1:0.32 |
| 12 | 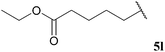 | 1:0.40 |
| 13 | 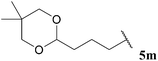 | 1:0.17 |
| 14 | 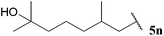 | 1:0.12 |
| 15 | 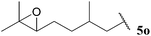 | 1:0.25 |
| 16 |  | 1:0.14 |
| 17 | 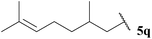 | 1:0.40 |
| 18 | 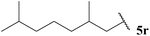 | 1:0.44 |
| 19 | 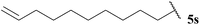 | 1:0.00b |
| 20 | 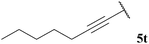 | 1:0.00b |
| 21 | 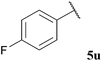 | 1:0.34 |
| 22 | 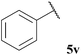 | 1:0.32 |
| 23 | 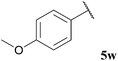 | 1:0.02 |
| 24 | 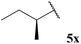 | 1:0.12 |
| 25 | 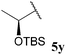 | 1:0.00c |
Although it has recently been reported that certain aldehydes (i.e. fragrant aldehydes) auto-oxidise at a rate that is partially dependent on their structure,27 there does not appear to be any obvious relationship between structure of aldehyde and rate of auto-oxidation in the series evaluated in this study. In fact, we observed that aldehydes bearing even minor structural changes brought about significant differences in the rate of acid formation (see Table 1, entries 2 and 24; and 4 and 7). Interestingly, certain aldehydes did not undergo any conversion, even after prolonged reaction times of 24 h (Table 1, entries 10, 19 and 20). Although aldehyde 5y did undergo appreciable conversion, 32%, it was converted to acetaldehyde and acetic acid rather than its corresponding acid 10y (Table 1, entry 25).
Hydroacylation of pentafluorophenyl vinyl sulfonate (PFP)
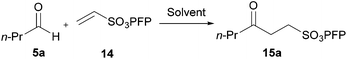
Studies toward developing an aerobically initiated hydroacylation protocol began with the hydroacylation of vinyl sulfonate 14, which has previously been shown to be an excellent radical acceptor.28 A structurally simple aldehyde that has a mid-range oxidation rate (see Table 1), n-butanal 5a, was chosen as the aldehyde component. Initially a screen of solvents was carried out to determine the optimum solvent (Table 2).
| ||
|---|---|---|
| Solvent | Conversion of 14/% | Yield 15a/% |
| Conditions: n-butanal 5a (5 mmol) and vinyl sulfonate 14 (1 mmol) were stirred at 300 rpm in solvent (1 mL) at 21 °C for 24 h. | ||
| 1,4-Dioxane | 100 | 65 |
| Et2O | 40 | 13 |
| CH2Cl2 | 82 | 39 |
| CH3OCH2CH2OCH3 | 100 | 64 |
| THF | 64 | 28 |
| PhMe | 71 | 25 |
| DMF | 82 | 30 |
| MeOH | 12 | 4 |
| H2O | 100 | 78 |
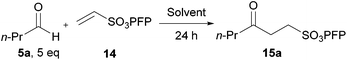
Hydroacylation was most readily affected in ethereal-based solvents such as 1,4-dioxane and ethylene glycol dimethyl ether, and perhaps most significantly, on H2O. Although the highest yield observed for the hydroacylation of 14 with 5a was on H2O, further optimisation of the reaction conditions for our hydroacylation protocol was also continued for 1,4-dioxane as solvent, since the use of H2O would preclude the use of solid aldehydes. It should also be noted that, in all solvents, addition of radical inhibitor bis(1,1-dimethylethyl)-4-methylphenol (BHT), suppressed reaction significantly.
Following our study on the effect of varying solvent we proceeded to explore the effect of aldehyde stoichiometry on yield. To do this, aldehyde stoichiometry was varied between 1 and 10 equivalent(s) for hydroacylation of 14 with 5a on H2O and in 1,4-dioxane (Table 3).
| ||||
|---|---|---|---|---|
| Solvent | 5a/eq. | Conversion 14/% | Yield 15aa/% | 15a:16ab |
| Conditions: n-butanal 5a and vinyl sulfonate 14 (1 mmol) on H2O or in 1,4-dioxane were stirred at 300 rpm for 24 h.a Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard.b Determined by 1H NMR integration. | ||||
| 1,4-Dioxane | 10 | 100 | 64 | 1:0.12 |
| 5 | 100 | 65 | 1:0.13 | |
| 4 | 100 | 57 | 1:0.16 | |
| 3 | 95 | 50 | 1:0.20 | |
| 2 | 75 | 37 | 1:0.27 | |
| 1 | 52 | 26 | 1:0.40 | |
| H2O | 10 | 100 | 80 | 1:0.04 |
| 5 | 100 | 77 | 1:0.04 | |
| 4 | 100 | 79 | 1:0.03 | |
| 3 | 100 | 78 | 1:0.04 | |
| 2 | 100 | 78 | 1:0.05 | |
| 1 | 75 | 47 | 1:0.14 | |
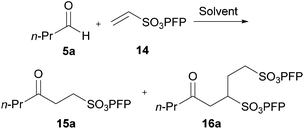
In 1,4-dioxane, maximum yield of ketone 15a was observed with 5 equivalents of aldehyde, whereas on H2O there was no vast improvement in yield when using greater than 2 equivalents of aldehyde. As such, the use of 5 equivalents of aldehyde in 1,4-dioxane, and 2 equivalents of aldehyde on H2O were chosen as the optimal stoichiometries to proceed with. The major limitation on yield where fewer equivalents of aldehyde were employed was the increased formation of double addition product 16a.
We next sought to evaluate reaction time by determining yield of ketone 15a for the hydroacylation of 14 with 5a in the H2O and 1,4-dioxane conditions at 0.5, 1, 2, 3, 4, 6, 8, 16 and 24 h (Table 4). It was determined that the optimal reaction times in the 1,4-dioxane and H2O conditions were 1 h and 3 h, respectively, with longer reactions having no significant effect on yield.
| ||
|---|---|---|
| Solvent | Time/h | Yield 15aa/% |
| Conditions for reactions in 1,4-dioxane: n-butanal 5a (5 mmol) and vinyl sulfonate 14 (1 mmol) were stirred at 300 rpm in 1,4-dioxane at 21 °C for the time specified. Conditions for reactions on H2O: n-butanal 5a (5 mmol) and vinyl sulfonate 14 (1 mmol) were stirred at 300 rpm in H2O at 21 °C for the time specified.a Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard. | ||
| 1,4-Dioxane | 0.5 | 49 |
| 1 | 65 | |
| 2 | 64 | |
| 3 | 65 | |
| 4 | 63 | |
| 6 | 65 | |
| 8 | 65 | |
| 16 | 66 | |
| 24 | 64 | |
| H2O | 0.5 | 60 |
| 1 | 70 | |
| 2 | 75 | |
| 3 | 78 | |
| 4 | 79 | |
| 6 | 82 | |
| 8 | 79 | |
| 16 | 80 | |
| 24 | 81 | |
With our optimised conditions in hand we sought to evaluate the generality of our aerobically initiated hydroacylation protocol with respect to aldehyde auto-oxidation rate. Thus, aldehydes 5a–j were specifically chosen to test this particular parameter as they exhibited a broad range of oxidation rates (see Table 1) and did not bear any complicating structural functionality. These aldehydes were also selected in view of their wide range of hydration equilibrium constants29,30 and their broad range of solubilities in water,31 in order to evaluate whether these parameters had any impact on yield of ketone in the water-based reaction conditions (Table 5).
| ||||
|---|---|---|---|---|
| Aldehyde 5 | 5:17 in D2Oa | Solubility of 5c/mass% | Yield 15 (H2O)d/% | Yield 15 (1,4-dioxane)d/% |
| Conditions for reactions in 1,4-dioxane: n-butanal 5a (5 mmol) and vinyl sulfonate 14 (1 mmol) were stirred at 300 rpm in 1,4-dioxane (1 mL) at 21 °C for the time specified in the ESI. Conditions for reactions on H2O: n-butanal 5a (2 mmol) and vinyl sulfonate 14 (1 mmol) were stirred at 300 rpm in H2O (1 mL) at 21 °C for the time specified in the ESI.a Ratio determined by 1H NMR integration in D2O.b Due to poor solubility of aldehyde in D2O, a DMSO–D2O (1:1) mixture was used.c Solubility of aldehyde in H2O at 30 °C.d All reactions proceeded with 100% conversion of 14 unless otherwise stated in parenthesis. | ||||
 | 1:1.04 | 5.48 | 78 | 65 |
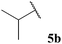 | 1:0.86 | 4.57 | 40 (60) | 58 (95) |
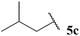 | 1:0.66 | 1.78 | 74 | 58 |
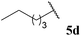 | 1:0.98 | 0.44 | 75 | 68 |
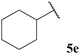 | 1:0.54 | — | 74 | 47 |
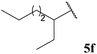 | 1:0.03b | 0.05 | 83 | 69 |
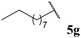 | 1:0.12b | 0.02 | 62 | 54 |
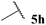 | 1:4.85 | — | 0 (0) | 37 (60) |
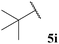 | 1:0.01b | — | 54 (75) | 5 (80) |
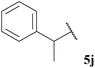 | 1:0.03b | — | 0 (0) | 0 (0) |
In both the 1,4-dioxane and water conditions, moderate to good yields of ketone were observed across the series with the exception of aldehydes 5b, 5h and 5j. The absence of any strong correlation between yield of ketone and the extent of hydration and/or solubility, except in exceptional circumstances (e.g. acetaldehyde 5h), was particularly pleasing as it showed that these parameters are fairly insignificant for efficient hydroacylation to transpire on water.
The moderate yields observed for the hydroacylation of isobutyraldehyde 5b was attributed to the propensity of this aldehyde to undergo auto-oxidation rapidly, therefore resulting in poor trapping of its corresponding acyl radical by alkene 14. This hypothesis is supported by the sub-optimal conversion of alkene 14 and complete conversion of aldehyde under the reaction conditions. A similar rationale may also be applied to explain the low yield of ketone 15h observed on application of aldehyde 5h in the 1,4-dioxane conditions. Interestingly, in the water-based conditions, the attempted hydroacylation of acetaldehyde 5h afforded no ketone and gave no alkene conversion with complete conversion of aldehyde. This result can, however, be rationalised in that acetaldehyde is completely soluble in water31 and is also known to readily react with water to form a hydrate. These characteristics of acetaldehyde thus prevent hydroacylation from ensuing when using water as solvent.
Aldehyde 5j, which did not appear to auto-oxidise in our previous study (see Table 1), underwent no conversion in both reaction conditions (even after 72 h), and therefore resulted in 0% conversion of vinyl sulfonate 14 (see Table 5). Even heating to higher temperatures, increasing aldehyde loading or increasing reaction time did not improve conversion of aldehyde and/or yield of ketone 15j.
The generally higher yields observed on water may be attributed to the higher concentration of reagents in the water-based reaction conditions, perhaps due to a hydrophobic effect. This may result in the radical intermediates under these conditions having a shorter lifetime, resulting in less unfavourable unimolecular degradation pathways ensuing. This hypothesis may also be supported by the noticeably lower yields of ketone observed when using α-substituted aldehydes 5e and 5f and α,α-disubstituted aldehyde 5i in the 1,4-dioxane conditions. Presumably, their corresponding acyl radicals, in the 1,4-dioxane conditions, have a longer lifetime and undergo more unfavourable decarbonylation. This is particularly evident for the hydroacylation of pivaldehyde 5i where 58% of decarbonylation addition product was observed and only 5% of hydroacylation product was isolated. Further support for this hypothesis is the noticeably lower yield observed in 1,4-dioxane for the hydroacylation of 14 with isovaleraldehyde 5c. The low yield of ketone 15c was due to the formation of species 18, which is almost certainly derived from acyl radical addition to 14, followed by intramolecular hydrogen atom abstraction, addition to another molecule of vinyl sulfonate 14 and intermolecular H-atom abstraction. Although we were unable to isolate ketone 18, we were able to carry out an elimination–addition sequence to afford ketone 19, and therefore indirectly confirm its structure (Scheme 3).
 | ||
| Scheme 3 Formation of by-product 18 in the 1,4-dioxane conditions and elimination–addition on ketone 18 to afford species 19. | ||
Synthetic utility of γ-keto-PFP-sulfonates
The disposition of the functional groups in a γ-keto-PFP-sulfonate motif strongly suggests that this bi-functional motif has appreciable potential for further manipulation. The carbonyl group is a versatile moiety that can be used in various synthetic transformations and, through the work pioneered by our group, PFP-sulfonates have been shown to be useful alternatives to sulfonyl chlorides for the synthesis of sulfonamides.32–35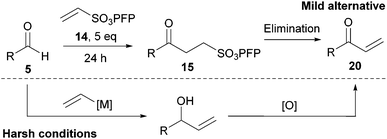 | ||
| Scheme 4 An alternative route to gain access to enones from aldehydes. | ||
Keto-sulfonate 15a was chosen as our model γ-keto-PFP-sulfonate to explore elimination to an enone. Encouragingly, elimination was achieved under basic conditions via the application of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 2 equivalents). Quantitative conversion of keto-sulfonate 15a to enone 20a was readily confirmed by 1H NMR, relative to pentachlorobenzene as an internal standard (Scheme 5).37 However isolation was not effected due to the rapid polymerisation of terminal enones upon concentration after purification. Ketone 15a was also subjected to a range of acidic conditions (acetic acid, para-toluenesulfonic acid and pyridinium para-toluenesulfonate) in various solvents (i.e. CH2Cl2, CHCl3, MeOH, Et2O, PhMe and THF), but was found to be highly stable with complete recovery of 15a being observed in all cases.
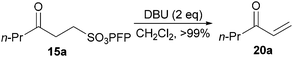 | ||
| Scheme 5 Elimination of sulfonate from 15a to form enone 20a. | ||
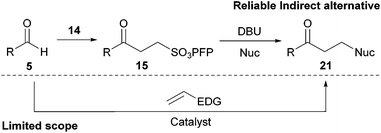 | ||
| Scheme 6 An indirect alternative to the hydroacylation of electron-rich alkenes. | ||
Through our strategy, it should also be possible to access a variety of unsymmetrical ketones, which would otherwise represent a significant challenge to current methods for the hydroacylation of electron rich alkenes, due to insurmountable polarity mis-match. A case in point is the direct hydroacylation of vinyl sulfides, a transformation that has, to the best of our knowledge, no literature precedent despite the emergence of thiol polarity reversal catalysis.38 Encouragingly, however, the indirect hydroacylation–elimination–addition strategy described above does provide access to these molecules, and in excellent yields (Table 6).
| |
|---|---|
| γ-Keto-sulfide 22 | Isolated yield/% |
| Conditions: ketone 15a, DBU (2 eq.), thiol (1.3 eq.), CH2Cl2, 21 °C. | |
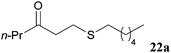 | 98 |
 | 97 |
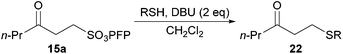
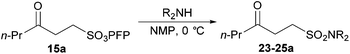
In an attempt to facilitate further formation of sulfonamide 23a the use of more polar solvents was explored. Encouragingly, application of NMP as solvent gave desired sulfonamide 23a in 45% yield, with the major side-product being enone derived. Lowering the temperature at which the amine was added, 0 °C, suppressed elimination and resulted in an improved yield of 64%. To further suppress elimination, an additional equivalent of the nucleophilic amine was used in place of triethylamine, as it did not promote elimination. To our delight, this resulted in a yield of 82%. The reaction protocol was then applied to a secondary amine and a sterically encumbered primary amine, morpholine and tert-butylamine, respectively (Table 7). Although this only afforded modest yields of sulfonamides 24a and 25a, the protocol provides reliable access to γ-keto-sulfonamides.
| |
|---|---|
| Sulfonamide | Isolated yield/% |
| Conditions: ketone 15a, amine (2 eq.), NMP, 0 °C to 21 °C. | |
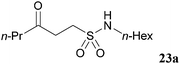 | 82 |
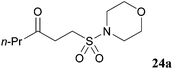 | 32 |
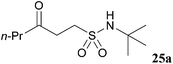 | 40 |
We also proceeded to show that secondary sulfonamide 23a could be converted into sultam 26, in a one-pot reductive-cyclisation protocol, in excellent yield (Scheme 7). It is envisaged that a wide range of similar sultams may be synthesised in an analogous manner.
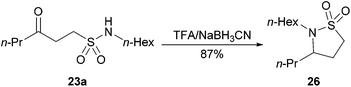 | ||
| Scheme 7 Reductive cyclisation of 23a to afford sultam 26. | ||
We next turned our attention to the synthesis of cyclic N-sulfonyl imines of the form of 27. Previously, they have been accessed via a three step protocol reported by Freitag in 63–68% overall yield.41 However, we were able to achieve direct access to N-sulfonylimine 27via the bubbling of ammonia gas into a solution of γ-keto-sulfonate 15a in CH2Cl2 in 67% yield (Scheme 8). Unlike the protocol outlined by Freitag, this method represents a simple and mild route to cyclic N-sulfonylimines in which analogue synthesis should be facile. Furthermore, through the work pioneered by Zhou, access to 3-substituted chiral sultams of the form of sultam 28 should be facile.42 The molecules generated by Zhou's asymmetric hydrogenation protocol are important organic synthetic intermediates and structural units of agricultural and pharmaceutical agents.43
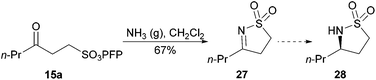 | ||
| Scheme 8 Formation of cyclic N-sulfonylimine 27 from ketone 15a. | ||
Finally, we envisaged that access to sultones could be achieved by reduction of the ketone moiety in keto-sulfonate 15a. Gratifyingly, sodium borohydride reduction of the carbonyl group in keto-sulfonate 15a gave access to sultone 29 in good yield (Scheme 9).
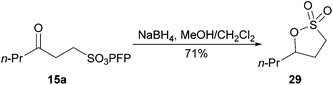 | ||
| Scheme 9 Formation of sultone 29 from ketone 15a. | ||
Hydroacylation of vinyl phosphonates
We were particularly interested in the hydroacylation of vinyl phosphonates as γ-ketophosphonates, and their corresponding phosphonic acids, have been established as useful tools in both synthetic chemistry44,45 and biology as non-hydrolysable phosphate mimetics.46–49 Thus, we applied our optimised protocols for the efficient hydroacylation of vinyl sulfonate 14 to the hydroacylation of vinyl phosphonate 30 (Scheme 10). As in the case for the optimisation of the hydroacylation of vinyl sulfonate 14, n-butanal 5a was chosen as the aldehyde component in the initial reactions.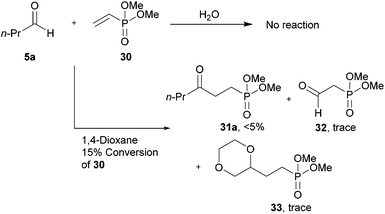 | ||
| Scheme 10 Attempted hydroacylation of vinyl phosphonate 30 with n-butanal 5a in 1,4-dioxane and water. | ||
Although no ketone was observed using the water-based conditions, γ-ketophosphonate 31a was isolated using 1,4-dioxane as solvent, albeit in very low yield (Scheme 10). In both cases, complete conversion of aldehyde 5a was observed. The lack of ketone observed in water was thought to be due to the water solubility of vinyl phosphonate 30. The low yield in 1,4-dioxane was attributed to inefficient trapping of the acyl radicals generated under the reaction conditions by alkene 30, as evidenced by the complete conversion of aldehyde and very low conversion of alkene, 15%. Careful examination of the crude 1H NMR spectrum of the reaction in the 1,4-dioxane also indicated the formation of aldehyde 32 and phosphonate 33. Phosphonate 33 is presumably formed via 1,4-dioxane radical addition to vinyl phosphonate 30. Whereas aldehyde 32 is most likely the result of peracyl radical 7a addition to vinyl phosphonate 30, to form 34, followed by aldehydic hydrogen atom abstraction to form peroxide 35, which then decomposes to aldehyde 32 and an equivalent of butanoic acid 10a (Scheme 11).
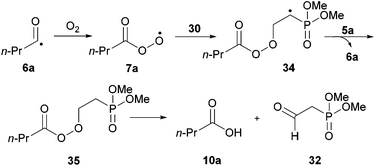 | ||
| Scheme 11 Proposed mechanism for the formation of aldehyde 32. | ||
Despite the low yield observed for the hydroacylation of vinyl phosphonate 30 with n-butanal 5a at 21 °C in 1,4-dioxane (Scheme 10) we were sufficiently intrigued to embark upon an optimisation study. As n-butanal had been completely converted to its corresponding acid under the reaction conditions and the conversion of alkene was very low, we envisaged that controlling exposure of the reaction mixture to molecular oxygen was key to achieving efficient hydroacylation. Moreover, this would also allow us, to some extent, suppress the formation of aldehyde 32. We rationalised that control of the dissolved molecular oxygen concentration and exposure of the reaction mixture to air could be readily achieved by varying reaction temperature and the surface area to volume ratio (Table 8).
| ||||||
|---|---|---|---|---|---|---|
| Entry | T/°C | [30]a/mol dm−3 | Surface area/cm2:volumeb/cm3 | Conversion 30c/% | 31a:32:33c | Isolated yield 31a/% |
| Conditions: vinyl phosphonate 30 (1 mmol), n-butanal 5a (5 mmol), 1,4-dioxane (see table), temperature (see table), 24 h.a Concentration of 30 refers to initial concentration of 30 in 1,4-dioxane before addition of 5a.b Surface area refers to surface area exposed to air.c Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard. | ||||||
| 1 | 20 | 1.00 | 1:0.32 | 15 | — | <5 |
| 2 | 0.25 | 1:1.29 | 15 | — | <5 | |
| 3 | 40 | 1.00 | 1:0.32 | 60 | — | 35 |
| 4 | 0.25 | 1:1.29 | 65 | — | 25 | |
| 5 | 60 | 5.00 | 1:0.06 | 100 | 1:0.27:0.07 | 61 |
| 6 | 2.00 | 1:0.16 | 100 | 1:0.19:0.08 | 67 | |
| 7 | 1.00 | 1:0.32 | 100 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.18:0.09 :0.18:0.09 | 70 | |
| 8 | 0.50 | 1:0.64 | 100 | 1:0.16:0.10 | 60 | |
| 9 | 0.25 | 1:1.29 | 100 | 1:0.10:0.14 | 55 | |
| 10 | 80 | 1.00 | 1:0.32 | 100 | — | 69 |
| 11 | 0.25 | 1:1.29 | 100 | — | 57 | |
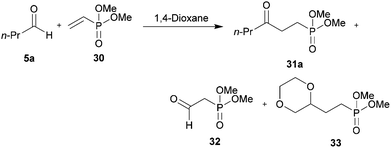
Gratifyingly, increasing the reaction temperature had a dramatic impact on yield of γ-ketophosphonate 31a with optimal yield afforded at 60 °C, 70%, at 1.00 mol dm−3 (Table 8, entry 7); heating to higher temperatures did not affect yield significantly (Table 8, entries 10 and 11). The increase in yield with increasing temperature was attributed to the lower concentration of dissolved molecular oxygen. This promotes acyl radical trapping by vinyl phosphonate 30 rather than with molecular oxygen, resulting in higher conversion of alkene and a higher yield of γ-ketophosphonate 31a. The lower yields observed at 60 °C at concentrations above and below 1.00 mol dm−3 may be rationalised by increased formation of aldehyde 32 and phosphonate 33, respectively. The higher surface area to volume ratio at higher concentrations results in an increased exposure to air and hence promotes the likelihood of an acyl radical being trapped by molecular oxygen rather than by an alkene, thus lowering the yield of γ-ketophosphonate 31a and decreasing the 31a:32 ratio (Table 8, entries 5–7). Unsurprisingly, at lower concentrations, a decreased 31a:33 ratio was observed due to the greater relative concentration of 1,4-dioxane to vinyl phosphonate 30, consequently, lowering the yield of γ-ketophosphonate 31a and increasing the yield of yield of 33 (Table 8, entries 7–9).
With optimised conditions in-hand, we then examined the scope of hydroacylation of vinyl phosphonate 30. The same set of functionally simple aldehydes that exhibit a broad range of auto-oxidation rates, 5a–j, employed in the analysis of vinyl sulfonate 14 (see Table 5) were applied to reaction with 30.
In general, moderate to good yields were observed across the aldehyde series. The major exceptions to this were the relatively low yields observed for aldehydes 5b and 5h, and the absence of ketone formed on application of aldehydes 5i and 5j. The low yields observed for aldehydes 5b and 5h may be rationalised by the propensity of these aldehydes to undergo rapid auto-oxidation, thus resulting in inefficient acyl radical trapping by alkene 30. This hypothesis is supported by the complete conversion of aldehyde and low conversion of alkene observed in both cases. The absence of the formation of γ-ketophosphonate 31i for the hydroacylation of vinyl phosphonate 30 with pivaldehyde 5i was due to the significant amount of tert-butyl radical addition that took place under the reaction conditions, resulting in the formation of dimethyl (3,3-dimethylbutyl)phosphonate in 46% yield (see Table 9 and ESI†). The tert-butyl radical is presumably derived from decarbonylation of the acyl radical formed from pivaldehyde oxidation. Consistent with our previous study on the hydroacylation of vinyl sulfonate 14 (see Table 5), aldehyde 5j gave 0% conversion of alkene since no aldehyde was oxidised.

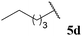
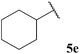
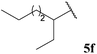
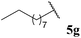
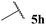
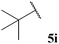
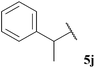
Hydroacylation of α,β-unsaturated esters
We next applied our methodology to the hydroacylation of α,β-unsaturated esters to generate 1,4-dicarbonyls, which are extremely useful intermediates. To investigate this, we initially applied our optimised protocols for the efficient hydroacylation of vinyl sulfonate 14 to the hydroacylation of 1,2-diester alkene 35 (Scheme 12), which has classically shown promise as an acyl radical acceptor.8 As in the case for the hydroacylation of vinyl sulfonate 14 and vinyl phosphonate 30, n-butanal was selected as the aldehyde component in these reactions.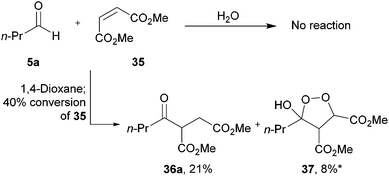 | ||
| Scheme 12 Attempted hydroacylation of alkene 35 with n-butanal 5a in 1,4-dioxane and water. *Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard by analogy with similar products. | ||
We were pleased to isolate ketone 36a in the 1,4-dioxane conditions, albeit in low yield. However, no ketone was isolated from the water-based conditions. The lack of ketone under the water conditions was thought to be due to inefficient trapping of the acyl radical with alkene 35 as complete conversion of aldehyde and low conversion of alkene was observed under the reaction conditions. This may be a consequence of the exposure to molecular oxygen in the water conditions being much higher than in 1,4-dioxane, and too high to be compatible with alkene 35. Also isolated from the crude reaction mixture in the 1,4-dioxane conditions was cyclic peroxide 37, which is presumably derived from addition of molecular oxygen to the adduct radical formed by addition of acyl radical 6a to alkene 38, to form 39, which then undergoes cyclisation and H-atom abstraction to form the cyclic peroxide (Scheme 13).
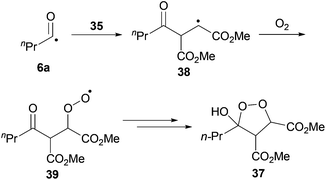 | ||
| Scheme 13 Proposed mechanism for the formation of peroxide 37. | ||
As in the case for the hydroacylation of vinyl phosphonate 30, in the 1,4-dioxane conditions, n-butanal had been completely converted under the reaction conditions and the conversion of alkene was very low. Thus, we again rationalised that controlling the exposure of the reaction mixture to molecular oxygen by manipulating reaction temperature and concentration would be vital for efficient hydroacylation.
Increasing the temperature from 20 °C to 60 °C gave an increase in the yield of ketone 36a, although heating to higher temperatures led to decomposition of the ketone product (Table 10). The increase in yield observed at higher temperature was thought to be a consequence of the lower concentration of dissolved molecular oxygen in solution. This would result in a higher conversion of alkene 35 as an acyl radical would more likely be trapped by an alkene than react with molecular oxygen; thus resulting in a higher yield of 36a. Moreover, the lower concentration of molecular oxygen also decreased the 36a:37 ratio from 1:0.38 to 1:0.15 in the 20–80 °C range. This is likely to be a consequence of adduct radical 38 in the reaction mechanism (see Scheme 13) having a greater propensity to abstract an aldehydic hydrogen atom than undergo addition to molecular oxygen; thus suppressing formation of peroxide and encouraging formation of ketone 36a.
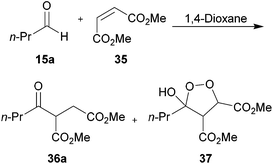
It was also reasoned that the surface area to volume ratio may have a significant impact on the exposure of the reaction medium to air, and thus, the effect of changing surface area:volume was explored (Table 11). For reasons analogous to those discussed for vinyl phosphonate 30, as the surface area to volume ratio decreased, higher conversion of alkene 35, decreased formation of cyclic peroxide 37, and a higher yield of ketone 36a was observed. Optimal yield for the hydroacylation of dimethyl maleate 35 with n-butanal 5a was observed at 0.33 mol dm−3.
| ||||
|---|---|---|---|---|
| [35]a/mol dm−3 | Surface area /cm2:volumeb/cm3 | Conversionc/% | Yield 36ac/% | 36a:37c |
| Conditions: alkene 35 (1 mmol), n-butanal 5a (5 mmol), 1,4-dioxane (see table), 60 °C, 8 days.a Concentration of 35 refers to initial concentration of 35 in 1,4-dioxane before addition of 5a.b Surface area refers to surface area exposed to air.c Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard. | ||||
| 5.00 | 1:0.06 | 50 | 37 | 1:0.34 |
| 2.00 | 1:0.16 | 60 | 35 | 1:0.22 |
| 1.00 | 1:0.32 | 85 | 50 | 1:0.07 |
| 0.50 | 1:0.64 | 100 | 64 | 1:0.05 |
| 0.33 | 1:0.96 | 100 | 77 | 1:0.04 |
| 0.25 | 1:1.29 | 100 | 74 | 1:0.03 |
| 0.20 | 1:1.61 | 100 | 70 | 1:0.03 |
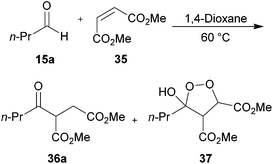
A similar trend in yields, to that obtained for the hydroacylation of dimethyl maleate 35 with n-butanal 5a with respect to changing surface area:volume, was observed for the hydroacylation of diethyl maleate 40 and dimethyl fumarate 41 with n-butanal 5a (Table 12). The good yields observed for the hydroacylation of diethyl maleate 40 and dimethyl fumarate 41 also showed that the efficiency of the hydroacylation protocol is, to some extent, independent of the nature of the ester and/or alkene geometry (E/Z).
| |||
|---|---|---|---|
| [Alkene]a/mol dm−3 | Surface area /cm2:volumeb/cm3 | Isolated yield 42/% | Isolated yield 36a/% |
| Conditions: alkene (1 mmol), n-butanal 5a (5 mmol), 1,4-dioxane (see table), 60 °C, 8 days.a Concentration of alkene refers to initial concentration of alkene in 1,4-dioxane before addition of 5a.b Surface area refers to surface area exposed to air. | |||
| 2.00 | 1:0.16 | 47 | 30 |
| 0.33 | 1:0.96 | 68 | 62 |
| 0.20 | 1:1.61 | 55 | 50 |
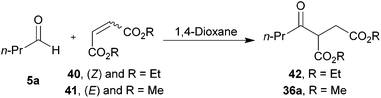
With a view to increase the scope of 1,4-dicarbonyls that can be formed by our methodology, we applied our optimised conditions to the hydroacylation of 1,1-diester alkene 43 and 2-alkoxy-1,1-diester alkene 45. Gratifyingly, very good yields of ketones 44a and 46a, respectively, were obtained (Scheme 14). We were particularly encouraged by the excellent yield of ketone 46a since it demonstrates the mild nature of our protocol as one might imagine that this species is prone to elimination under ionic conditions. Thus, our protocol gives us access to products that would otherwise be challenging to synthesise via alternative ionic-based methods.
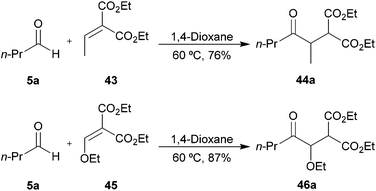 | ||
| Scheme 14 Hydroacylation of alkenes 43 and 45 with aldehyde 5a. | ||
Having established optimised conditions for the hydroacylation of a range of α,β-unsaturated esters with n-butanal 5a, we next explored aldehydes with a range of auto-oxidation rates, 5a–j (Table 13).
| ||||
|---|---|---|---|---|
| Entry | Aldehyde 5 | Ketone 36 | Ketone 44 | Ketone 46 |
| Conditions: alkene (1 mmol), aldehyde 5 (5 mmol), 1,4-dioxane (3 mL), 60 °C. | ||||
| 1 |  | 70 | 76 | 87 |
| 2 | 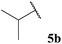 | 21 | 42 | 0 |
| 3 | 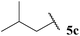 | 57 | 60 | 85 |
| 4 | 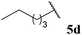 | 76 | 72 | 87 |
| 5 | 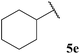 | 56 | 74 | 24 |
| 6 | 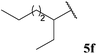 | 25 | 52 | 0 |
| 7 | 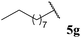 | 60 | 72 | 89 |
| 8 | 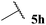 | 0 | 0 | 0 |
| 9 | 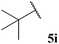 | 0 | 0 | 0 |
| 10 | 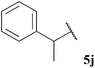 | 0 | 0 | 0 |
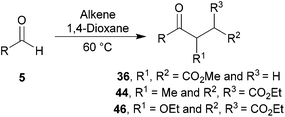
In all cases, and perhaps as expected, aldehydes that rapidly auto-oxidise to acid (i.e. aldehydes 5b, 5h and 5i, see Table 1) gave low or no yield of ketone due to inefficient trapping of their corresponding acyl radicals by alkenes compared to the rate of direct auto-oxidation. Moreover, in concert with our previous studies, aldehyde 5j, yielded no ketone and underwent no observable conversion to acid.
As expected, owing to the more electrophilic nature of 1,1-diester alkene 43 compared with 1,2-diester alkene 35 and the inherently more electrophilic adduct radical formed upon acyl radical addition, higher yields were generally observed for the hydroacylation of alkene 43 over alkene 35. It was also notable that reaction with 1,2-diester alkene 35 over 1,1-diester alkene 43 appeared to be more sensitive to steric hindrance, as evidenced by the yields observed for reaction with a variety of α-substituted aldehydes (Table 13, entries 2, 5 and 6).
Interestingly, we were able to isolate various sensitive ketones, formed by the hydroacylation of alkene 45 with linear aldehydes 5a, 5c, 5d and 5g in excellent yields (Table 13, entries 1, 3, 4 and 7). However, reaction with α-substituted aldehydes afforded no or very low yields (Table 13, entries 2, 5 and 6). These results perhaps further highlight the sensitivity of acyl radical addition to highly substituted alkenes. Nevertheless, an array of valuable ketones were synthesised using our protocol.
Hydroacylation of azodicarboxylates
Eager to extend our methodology to C–N bond formation, we applied our optimised conditions for the hydroacylation of vinyl sulfonate 14 to the hydroacylation of azodicarboxylate 47 employing n-butanal as the aldehyde component.Using either 1,4-dioxane or water as solvent, excellent yields of acyl hydrazide 48a were observed (Table 14, entries 1 and 7). Given the high yields achieved and the lack of appreciable side-products, except butanoic acid, we took the opportunity to assess whether aldehyde stoichiometry could be reduced whilst maintaining high yield (Table 14).
| ||||
|---|---|---|---|---|
| Entry | Solvent | 5a/eq. | 47/eq. | Isolated yield 48a/% |
| Conditions for 1,4-dioxane: DIAD 47, n-butanal 5a, 1,4-dioxane, 21 °C, 24 h. Conditions for H2O: DIAD 47 and n-butanal 5a, H2O, 21 °C, 24 h. | ||||
| 1 | 1,4-Dioxane | 5 | 1 | 94 |
| 2 | 2 | 1 | 80 | |
| 3 | 1.5 | 1 | 72 | |
| 4 | 1 | 1 | 64 | |
| 5 | 1 | 1.2 | 65 | |
| 6 | 1 | 2 | 67 | |
| 7 | H2O | 2 | 1 | 92 |
| 8 | 1.5 | 1 | 90 | |
| 9 | 1 | 1 | 70 | |
| 10 | 1 | 1.2 | 90 | |
| 11 | 1 | 2 | 91 | |
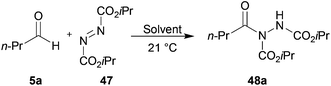
Using 1,4-dioxane as solvent we saw a significant decrease in yield on reducing the quantity of aldehyde (Table 14, entries 1–3). In contrast, reducing the 5a:47 ratio from 2:1 to 1.5:1 in the water-based conditions resulted in similarly high yields (Table 14, entries 7 and 8). However, only modest conversion (70%) was achieved with stoichiometric reaction conditions (Table 14, entry 9). The lower yield observed on reaction with a 1:1 stoichiometry of 5a:47 was attributed to conversion of diisopropyl azodicarboxylate 47 to diisopropyl hydrazinedicarboxylate. To counter this, the amount of diisopropyl azodicarboxylate 47 was increased (Table 14, entries 10 and 11), and gratifyingly, this afforded hydrazide 48a in excellent yield, 90%, based on n-butanal as the limiting reagent. A similar approach was applied to using 1,4-dioxane as solvent, however, this brought limited success (Table 14, entries 4–6).
With our optimised conditions in hand, we sought to evaluate the tolerance of azodicarboxylate 47 to aldehydes with a range of auto-oxidation rates. To appraise this, aldehydes 5a–j (1 equivalent) were reacted with azodicarboxylate 47 (1.2 equivalents) in H2O (Table 15).
| |
|---|---|
| Aldehyde 5 | Isolated yield 48/% |
| Conditions: DIAD 47 (1.2 mmol), aldehyde 5 (1 mmol), H2O (1 mL), 21 °C for the time specified in the ESI. | |
 | 91 |
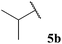 | 79 |
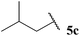 | 79 |
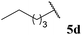 | 88 |
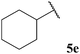 | 84 |
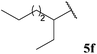 | 86 |
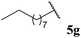 | 85 |
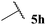 | 0 |
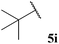 | 69 |
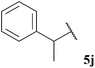 | 71 |
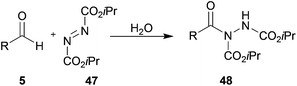
Hydroacylation of azodicarboxylate 47 showed excellent tolerance of aldehydes with a broad range of auto-oxidation rates. Previously, our protocol has shown relatively poor tolerance for aldehydes that auto-oxidise rapidly, i.e. aldehydes 5b and 5i. However, when employing azodicarboxylate 47 as the acyl radical acceptor component, these aldehydes underwent efficient hydroacylation, even when using a single equivalent of aldehyde. As in the case for the hydroacylation of vinyl sulfonate 14, under the water-based conditions, the use of acetaldehyde 5h gave no conversion of the double bond acceptor species due to its high solubility in, and reactivity with, water. Perhaps most interestingly, aldehyde 5j, which did not appear to auto-oxidise (see Table 1) and did not undergo any conversion with any of the other acceptors (see Tables 5, 9 and 13), afforded a good yield of acyl hydrazide 48j (71%). These results seem to indicate that azodicarboxylates are exceptionally good acyl radical acceptors and/or that the adduct radicals generated from acyl radical addition to azodicarboxylates are exceptionally efficient aldehyde H-atom abstractors.
Synthetic utility of acyl hydrazides
We then sought to explore the synthetic utility of acyl hydrazides as acyl donors owing to the stabilised leaving group that would be released upon an addition–elimination reaction at the amide moiety. To do this, reaction of hydrazide 48a with various amines was explored. Reaction with primary amines afforded the corresponding amides 49a–c in excellent yields with concurrent isolation of diisopropyl hydrazinedicarboxylate 50 (Table 16). Unfortunately, however, treatment of hydrazide 48a with bulkier amines such as morpholine and pyrrolidine resulted in low or no yield of amide 49. Use of more polar solvents, such as DMF and NMP, did not improve yield and/or conversion.
| |
|---|---|
| Amide 49 | Isolated yield/% |
| Conditions: acyl hydrazide 48a and amine (2.5 eq.), CH2Cl2, 21 °C. | |
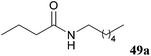 | 96 |
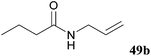 | 95 |
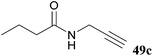 | 98 |
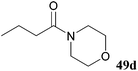 | 19 |
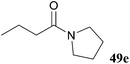 | 25 |

Upon close examination of the crude reaction mixtures for the reaction of acyl hydrazide 48a with secondary amines we were able to identify that the low yields of amides 49d–e was due to attack of the amine at the carbamate ester, resulting in the formation of 51 (Scheme 15).
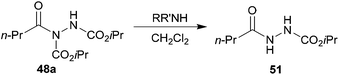 | ||
| Scheme 15 Formation of 51 derived from attack of amine on the carbamate of 48a. | ||
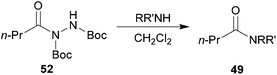
In an attempt to obviate this unfavourable side-reaction, Boc functionalised acyl hydrazide 52 was synthesised and submitted to the aminolysis reaction conditions with secondary amines morpholine and pyrrolidine. Gratifyingly, this significantly improved yield of tertiary amides 49d and 49e as it presumably suppressed attack at the carbamate (Table 17).
| |
|---|---|
| Amide 49 | Isolated yield/% |
| Conditions: acyl hydrazide 52 and amine (2.5 eq.), CH2Cl2, 21 °C. | |
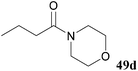 | 59 |
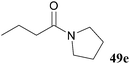 | 75 |
Application of functionalised aldehydes
Having evaluated the tolerance of our hydroacylation protocol to aldehydes with a range of auto-oxidation rates in concert with various double bond acceptor moieties, we proceeded to appraise functional group tolerance. Thus, the hydroacylation of vinyl sulfonate 14, vinyl phosphonate 30, α,β-unsaturated ester 43 and azodicarboxylate 47, under the optimised conditions that have been developed for each acceptor, with aldehydes 5k–5w were evaluated.To our delight, excellent tolerance of cyclopropyl, ester, acetal and alcohol functional groups was observed across all four double bond acceptors (Table 18, entries 1–4). Although epoxide-bearing aldehyde 5o showed excellent tolerance of acceptors 30, 43 and 47, no ketone was isolated on reaction with vinyl sulfonate 14 (Table 18, entry 5). In this case, although alkene was completely consumed under the reaction conditions, only a polymeric substance could be observed in the crude 1H NMR. Consistent with a radical mechanism was the poor tolerance of our methodology to aldehydes bearing alkenes (Table 18, entries 6 and 7), presumably due to polymerisation under the reaction conditions. The only exception to this was the moderate yields observed when using azodicarboxylate as acceptor; further testament to azodicarboxylates being excellent radical acceptors and/or their acyl radicals adducts being excellent chain propagators. To probe whether it was specifically the alkene functionality that limited yield of hydroacylation product, the corresponding reduced aldehyde of citronellal, 5r, was employed under each of the reaction conditions (Table 18, entry 8). As aldehyde 5r provided good to excellent yields of hydroacylation product in all cases, and as it has a similar auto-oxidation rate to citronellal 5q, this supports the theory of poor tolerance of alkene functional groups specifically. Consistent with our previous results, aldehydes 5s and 5t, which did not appear to undergo oxidation in our auto-oxidation study, only underwent reaction with azodicarboxylate 47 (Table 18, entries 9 and 10).
| |||||
|---|---|---|---|---|---|
| Entry | Aldehyde 5 | Vinyl sulfonate 14a | Vinyl phosphonate 30a | 1,1-Diester alkene 43a | DIAD 47a |
| Conditions for the hydroacylation of vinyl sulfonate 14: alkene 14 (1 mmol), aldehyde 5 (2 mmol), H2O (1 mL), 21 °C for the time specified in the ESI. Conditions for the hydroacylation of vinyl phosphonate 30: alkene 30 (1 mmol), aldehyde 5 (5 mmol), 1,4-dioxane (1 mL), 60 °C, 24 h. Conditions for the hydroacylation of 1,1-diester alkene 43: alkene 43 (1 mmol), aldehyde 5 (5 mmol), 1,4-dioxane (1 mL), 60 °C for the time specified in the ESI. Conditions for the hydroacylation of DIAD 47: DIAD 47 (1.2 mmol), aldehyde 5 (1 mmol), H2O (1 mL), 21 °C for the time specified in the ESI.a All reactions proceeded with 100% conversion of acceptor unless otherwise stated in parenthesis.b Determined by integration of 1H NMR relative to pentachlorobenzene as an internal standard by analogy with similar products. | |||||
| 1 |  | 64 | 57 | 62 | 87 |
| 2 | 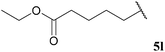 | 68 | 67 | 71 | 80 |
| 3 | 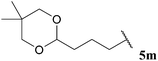 | 62 | 71 | 67 | 70 |
| 4 | 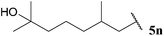 | 81 | 74 | 68 | 82 |
| 5 | 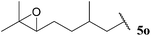 | 0 | 62 | 60 | 74 |
| 6 |  | 0 (37) | 20b | 0 | 42 |
| 7 | 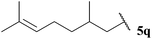 | 0 (32) | 0 | 0 | 54 |
| 8 | 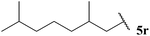 | 66 | 68 | 61 | 87 |
| 9 | 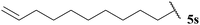 | 0 (0) | 0 (0) | 0 (0) | 75 |
| 10 | 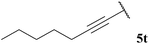 | 0 (0) | 0 (0) | 0 (0) | 55 |
| 11 | 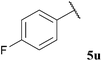 | 10 (35) | 27 (55) | <5b (10) | 75 |
| 12 | 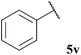 | 8b (32) | <5b (40) | <5b (10) | 79 |
| 13 | 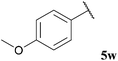 | <5b (10) | <5b (35) | <5b (10) | 44 |
The tolerance of aromatic aldehydes under our optimised conditions was evaluated with electron-poor, -neutral and -rich aldehydes 5u–w (Table 18, entries 11–13). In general, our hydroacylation protocol showed poor tolerance to aromatic aldehydes, with the only exception being observed for the hydroacylation of azodicarboxylate 47; further evidence of the exceptional propensity of azodicarboxylates to undergo efficient hydroacylation. The major obstacle to efficient hydroacylation with the alkene based double bond acceptors was centred on low conversion of alkene, unfavourable polymerisation and 1,4-dioxane addition products. These side-reactions are likely to be a consequence of inefficient aromatic acyl radical trapping by alkenes, perhaps due to unfavourable steric interactions and/or inefficient aldehyde H-atom abstraction by the resultant acyl radical-alkene adduct radical species.
Application of enantiopure aldehydes
Due to acyl radicals being σ-type radicals we envisaged that aldehydes bearing α-stereocentres would undergo hydroacylation with preservation of enantiopurity. Thus we evaluated the use of chiral non-racemic aldehydes 5x and 5y in our hydroacylation protocol. As valuable enantiopure aldehydes would almost certainly not be employed when a vast excess of aldehyde is required (e.g. 5 equivalents), hydroacylation with chiral aldehydes 5x and 5y was only analysed on reaction with vinyl sulfonate 14 and azodicarboxylate 47 where only 2 and 1 equivalent(s) is/are employed, respectively (Table 19).
| ||
|---|---|---|
| Aldehyde 5 | Vinyl sulfonate 14a | DIAD 47a |
| Conditions for the hydroacylation of vinyl sulfonate 14: alkene 14 (1 mmol), aldehyde 5 (2 mmol), H2O (1 mL), 21 °C for the time specified in the ESI. Conditions for the hydroacylation of DIAD 47: DIAD 47 (1.2 mmol), aldehyde 5 (1 mmol), H2O (1 mL), 21 °C for the time specified in the ESI.a All reactions proceeded with 100% conversion of acceptor unless otherwise stated in parenthesis. | ||
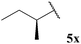 | 77, 97% ee | 88, 98% ee |
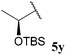 | 0 (25) | 61, 99% ee |

To our delight, aldehyde 5x underwent efficient hydroacylation with both vinyl sulfonate 14 and DIAD 47, and most importantly, with exceptional retained enantiomeric excesses (ree). Although no ketone could be isolated for reaction of aldehyde 5y with vinyl sulfonate 14, acyl hydrazide 47y could be isolated in good yield, and again, with excellent retained enantiomeric excess. These reactions represent to the best of our knowledge the first examples of hydroacylation achieved with a chiral aldehyde with retention of enantiomeric excess.
In concert with our auto-oxidation study, although all of aldehyde 5y was consumed on reaction with vinyl sulfonate 14, none of the corresponding acid was observed. The only species observed in the crude 1H NMR were acetaldehyde 5h and unreacted vinyl sulfonate 14. One possible explanation for the formation of acetaldehyde 5h is via decarbonylation of the corresponding acyl radical of aldehyde 5y, followed by β-TBS-elimination. In the presence of azodicarboxylate, however, such a pathway may have been precluded, to some extent, by the rapid reaction of acyl radicals with azodicarboxylates.
Having developed an effective protocol for the hydroacylation of acceptors using aldehydes bearing α-enantioenriched stereocentres in excellent ree, we proceeded to explore whether acyl hydrazide 47x may be converted into an amide with high ree. To our delight, reaction with benzylamine afforded known amide 53 in good yield, and most significantly, with retention of stereochemical information (Scheme 16). We envisage that a range of enantioenriched amides may be prepared in similarly high enantiomeric excess.
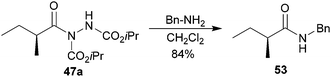 | ||
| Scheme 16 Conversion of hydrazide 47a to amide 53. | ||
Conclusions
In conclusion, we have described the use of aerobic aldehyde C–H activation for the construction of C–C and C–N bonds through the hydroacylation of vinyl sulfonates and phosphonates, α,β-unsaturated esters and azodicarboxylates. These reactions require no metal(s) and rely on only molecular oxygen in air for activation, therefore providing a clean, green route to hydroacylation. Of particular note is the hydroacylation of azodicarboxylates, which proceeded with aldehyde as limiting reagent, a stoichiometry not previously observed in the literature. Hydroacylation, in all acceptor cases, was shown to proceed in good yields for a range of aldehydes with respect to oxidation rate, as well as being tolerant of aldehydes bearing various functional groups. Moreover, the use of chiral aldehydes for hydroacylation, which has not been previously reported, was shown to be applicable to our aerobic activation protocol with exceptional retention of enantiomeric excesses observed in all cases. Throughout, we have observed products and patterns of reactivity that are most readily explained though a radical mechanism. In addition to using aerobic aldehyde C–H activation to affect hydroacylation of a range of acyl radical acceptors, the reactivity of the resultant hydroacylation products has also been demonstrated. The γ-keto-sulfonate motif may act as a precursor for the formation of γ-keto-sulfonamides, sultams, N-sulfonylimines and sultones. Perhaps most significantly, the γ-keto-sulfonate motif may undergo quantitative elimination to generate enones, providing a mild alternative route for the overall conversion of an aldehyde to an enone when taken in conjunction with the hydroacylation chemistry. Moreover, the hydroacylation–elimination–addition chemistry represents a powerful indirect alternative for the hydroacylation of electron rich alkenes, and the acyl hydrazide motif has also been highlighted as an intermediate for the construction of amides.Experimental
General
All reagents were purchased from Aldrich or AlfaAesar and were used as received without further purification. All hydroacylation reactions were carried out in stoppered carousel tubes (15 cm × 2 cm) equipped with an octagon-shaped magnetic stirrer bar (12.7 mm × 3 mm). Where described below petrol refers to petroleum ether (40–60 °C). All reactions were monitored by thin-layer chromatography (TLC) on pre-coated silica gel plates (254 μm). Flash column chromatography was carried out with Kiesegel 60M 0.04/0.063 mm (200–400 mesh) silica gel. 1H NMR spectra were recorded at 300 MHz, 400 MHz, 500 MHz and 600 MHz and 13C NMR at 75 MHz, 100 MHz, 125 MHz and 150 MHz on a Bruker AMX300, AMX400, AMX500 and AMX600 at 21 °C temperature. The chemical shifts (δ) for 1H and 13C are quoted relative to residual signals of the solvent on the ppm scale. Coupling constants (J values) are reported in hertz (Hz). Mass spectra were obtained on a VG70-SE mass spectrometer. Infrared spectra were obtained on a Perkin Elmer Spectrum 100 FTIR Spectrometer operating in ATR mode. Optical rotations were measured using a Perkin Elmer 343 polarimeter. Chiral high performance liquid chromatography (HPLC) was performed on a Varian HPLC instrument equipped with a manual injector, binary pump, and a UV detector (214 nm) using CHIRALCEL® OD column (4.6 mm × 250 mm, 10 μm) from Chiral Technologies (West Chester, PA) eluting with hexane:i-PrOH.Typical procedure for the hydroacylation of vinyl sulfonate 14 in 1,4-dioxane
To a solution of ethenesulfonic acid pentafluorophenyl ester 14 (1 mmol) in 1,4-dioxane (1 mL) was added aldehyde (5 mmol) and the reaction mixture stirred at 300 rpm at 21 °C until reaction was complete by TLC. PhMe (2 mL) was added and the solvent removed in vacuo and the crude residue purified as described in the ESI† to afford the desired ketone sulfonate ester.Typical procedure for the hydroacylation of vinyl sulfonate 14 in H2O
To a solution of ethenesulfonic acid pentafluorophenyl ester 14 (1 mmol) in H2O (1 mL) was added aldehyde (2 mmol) and the reaction mixture stirred at 300 rpm at 21 °C until reaction was complete by TLC. The solvent removed in vacuo and the crude residue purified as described in the ESI† to afford the desired ketone sulfonate ester.Typical procedure for the synthesis of γ-keto-sulfides 22
To a solution of pentafluorophenyl 3-oxohexane-1-sulfonate 15a (1 mmol) in CH2Cl2 (3 mL) was added thiol (1.3 mmol) and DBU (2 mmol), and the reaction mixture stirred at 300 rpm at 21 °C for 1 h. The solvent was removed in vacuo and the crude residue purified as described in the ESI† to afford the desired γ-keto-sulfide.Typical procedure for the synthesis of sulfonamides 23–25a
To a solution of pentafluorophenyl 3-oxohexane-1-sulfonate 15a (0.29 mmol) in NMP (2.5 mL) was added dropwise a solution of amine (0.58 mmol) in NMP (1 mL) at 0 °C. After addition was complete, the reaction mixture was warmed to 21 °C and stirred for 4 h. To work-up, the reaction mixture was diluted with Et2O (20 mL), washed with sat. LiCl (3 × 20 mL), sat. NaHCO3 (3 × 20 mL), 2 M HCl (3 × 20 mL), dried (MgSO4) and the solvent removed in vacuo to afford the desired sulfonamide.Typical procedure for hydroacylation of vinyl phosphonate 30
To a solution of vinyl phosphonate 30 (1 mmol) in 1,4-dioxane (1 mL) was added aldehyde (5 mmol) and the reaction mixture stirred at 300 rpm at 60 °C for 24 h unless otherwise stated in the ESI.† The reaction mixture was concentrated in vacuo and the crude residue purified as described in the ESI† to afford the desired γ-ketophosphonate.Typical procedure for the hydroacylation of alkenes 35, 40, 41, 43 and 45
To a solution of alkene (1 mmol) in 1,4-dioxane (3 mL) was added aldehyde (5 mmol) and the reaction mixture stirred at 300 rpm at 60 °C for the time specified in the ESI.† The solvent removed in vacuo and the crude residue purified as described in the ESI† to afford the desired hydroacylation product.Typical procedure for the hydroacylation of DIAD 47
To a mixture of azodicarboxylate (1.2 mmol) and H2O (500 μL) was added aldehyde (1.0 mmol) and the reaction mixture stirred at 300 rpm at 21 °C for the time specified in the ESI.† The solvent was removed in vacuo and the crude residue purified as described in the ESI† to afford the desired hydroacylation product.Typical procedure for the synthesis of amides 49a–c from acyl hydrazide 48a
To a solution of dipropan-2-yl 1-butanoylhydrazine-1,2-dicarboxylate 48a (1 mmol) in CH2Cl2 (2 mL) was added amine (2.5 mmol) and the reaction mixture stirred at 300 rpm at 21 °C for 16 h. The solvent was removed in vacuo and the crude residue purified as described in the ESI† to afford the desired amide.Typical procedure for the synthesis of amides 49d–e from acyl hydrazide 52
To a solution of di-tert-butyl 1-butanoylhydrazine-1,2-dicarboxylate 52 (1 mmol) in CH2Cl2 (2 mL) was added amine (2.5 mmol) and the reaction mixture stirred at 300 rpm at 21 °C for 16 h. The solvent was removed in vacuo and the crude residue purified as described in the ESI† to afford the desired amide.Acknowledgements
We gratefully acknowledge UCL for support of our programme. We also thank A. G. Davies and K. U. Ingold for helpful discussions.Notes and references
- (a) C.-J. Li and P. T. Anastas, Chem. Soc. Rev., 2012, 41, 1413–1414 RSC and references therein; ; (b) P. T. Anastas and J. C. Warner, Green Chemistry, Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed; (c) R. Noyori, Adv. Synth. Catal., 2001, 343, 1 CrossRef CAS.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (b) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC and references therein.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (e) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- W. B. Motherwell and D. Crich, Free Radical Chain Reactions in Organic Synthesis, Elsevier, 1992 Search PubMed.
- L. Grossi, Chem.–Eur. J., 2005, 11, 5419–5425 CrossRef CAS PubMed.
- D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1961, 83, 4076–4083 CrossRef CAS.
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2069 CrossRef CAS PubMed.
- D. P. Curran, Synthesis, 1988, 417–439 CrossRef CAS.
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed.
- R. Kundu and Z. T. Ball, Org. Lett., 2010, 12, 2460–2463 CrossRef CAS PubMed.
- (a) G. J. Rowlands, Tetrahedron, 2009, 65, 8603–8655 CrossRef CAS; (b) G. J. Rowlands, Tetrahedron, 2010, 66, 1593–1636 CrossRef CAS.
- J. R. McNesby and C. A. Heller, Chem. Rev., 1954, 54, 325–346 CrossRef CAS.
- S. Esposti, D. Dondi, M. Fagoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531–2534 CrossRef CAS PubMed.
- F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800–3842 CrossRef CAS PubMed.
- S. Tsujimoto, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2003, 44, 5601–5604 CrossRef CAS.
- G. Cauquis, B. Sillion and L. Verdet, Tetrahedron Lett., 1977, 18, 27–30 CrossRef.
- S. Protti, D. Ravelli, M. Fagoni and A. Albini, Chem. Commun., 2009, 7351–7353 RSC.
- N. Shapiro and A. Vigalok, Angew. Chem., Int. Ed., 2008, 47, 2849–2852 CrossRef CAS PubMed.
- S. G. Jarboe and P. Beak, Org. Lett., 2000, 2, 357–360 CrossRef CAS PubMed.
- N. Tada, H. Okubo, T. Miura and A. Itoh, Synlett, 2009, 3024–3026 CAS.
- R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Org. Biomol. Chem., 2009, 7, 235–237 CAS.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed.
- V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133–135 RSC.
- V. Chudasama, J. M. Ahern, R. J. Fitzmaurice and S. Caddick, Tetrahedron Lett., 2011, 52, 1067–1069 CrossRef CAS.
- V. Chudasama, J. M. Ahern, D. V. Dhokia, R. J. Fitzmaurice and S. Caddick, Chem. Commun., 2011, 47, 3269–3271 RSC.
- C. Marteau, F. Ruyffelaere, J.-M. Aubry, C. Penverne, D. Favier and V. Naredllo-Rataj, Tetrahedron, 2013, 69, 2268–2275 CrossRef CAS.
- O. Edetanlen-Elliot, R. J. Fitzmaurice, J. D. Wilden and S. Caddick, Tetrahedron Lett., 2007, 48, 8926–8929 CrossRef CAS.
- R. P. Bell, Adv. Phys. Org. Chem., 1966, 4, 1–29 CrossRef CAS.
- S. H. Hilal, L. L. Bornander and L. A. Carreira, QSAR Comb. Sci., 2005, 24, 631–638 CAS.
- R. M. J. Stephenson, Chem. Eng. Data, 1993, 38, 630–633 CrossRef CAS.
- S. Caddick, J. D. Wilden, H. D. Bush, S. N. Wadman and D. B. Judd, Org. Lett., 2002, 4, 2549–2551 CrossRef CAS PubMed.
- S. Caddick, J. D. Wilden and D. B. Judd, Chem. Commun., 2005, 2727–2728 RSC.
- J. D. Wilden, D. B. Judd and S. Caddick, Tetrahedron Lett., 2005, 46, 7637–7640 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024–1025 CrossRef CAS PubMed.
- (a) E. Negishi, S. M. Ma, T. Sugihara and Y. Noda, J. Org. Chem., 1997, 62, 1922–1923 CrossRef CAS; (b) R. T. Lewis, W. B. Motherwell and M. Shipman, J. Chem. Soc., Chem. Commun., 1988, 948–950 RSC.
- T. Tsuda, T. Yoshida, T. Kawamoto and T. Saegusa, J. Org. Chem., 1987, 52, 1624–1627 CrossRef CAS.
- S. H. Dang and B. P. Roberts, J. Chem. Soc., Perkin Trans. 1, 1998, 67–75 RSC.
- S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352–2353 RSC.
- P. Perlmutter, Conjugate Addition Reactions in Synthesis, Pergamon, Oxford, 1992 Search PubMed.
- D. Freitag and P. Metz, Tetrahedron, 2006, 62, 1799–1805 CrossRef CAS.
- Y. Q. Wang, S. M. Lu and Y. G. Zhou, J. Org. Chem., 2007, 72, 3729–3734 CrossRef CAS PubMed.
- (a) A. R. Katritzky, J. Wu, S. Rachwal, B. Rachwal, D. W. Macomber and T. P. Smith, Org. Prep. Proced. Int., 1992, 24, 463–467 CrossRef CAS; (b) R. A. Miller, G. R. Humphrey, D. R. Lieberman, S. S. Celiga, D. J. Kennedy, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 2000, 65, 1399–1406 CrossRef CAS PubMed; (c) M. Inagaki, T. Tsuri, H. Jyoyama, T. Ono, K. Yamada, M. Kobayashi, Y. Hori, A. Arimura, K. Yasui, K. Ohno, S. Kakudo, K. Koizumi, R. Suzuki, S. Kawai, M. Kato and S. Matsumoto, J. Med. Chem., 2000, 43, 2040–2048 CrossRef CAS PubMed; (d) L. Brunetti, I. Cacciatore, A. Di Stefano, S. Dupre, A. Giorgi, G. Luisi, B. Michelotto, G. Orlando, F. Pinnen, L. Recinella, P. Sozio and A. Spirito, Farmaco, 2002, 57, 479–486 CrossRef CAS PubMed; (e) R. J. Cherney, R. Mo, D. T. Meyer, K. D. Hardman, R.-Q. Liu, M. B. Covington, M. Qian, Z. R. Wasserman, D. D. Christ, J. M. Trzaskos, R. C. Newton and C. P. Decicco, J. Med. Chem., 2004, 47, 2981–2983 CrossRef CAS PubMed.
- (a) H. Krawczyk, K. Wasek, J. Kedzia, J. Wojciechowski and W. M. Wolf, Org. Biomol. Chem., 2008, 6, 308–318 RSC; (b) D. Appleton, A. B. Duguid, S. K. Lee, Y. J. Ha, H. J. Ha and F. J. Leeper, J. Chem. Soc., Perkin Trans. 1, 1998, 89–101 RSC; (c) R. D. Norcross, P. von Matt, H. C. Kolb and D. Bellus, Tetrahedron, 1997, 53, 10289–10312 CrossRef CAS; (d) D. R. Marshall, P. J. Thomas and C. J. M. Stirling, J. Chem. Soc., Perkin Trans. 2, 1977, 1898–1909 RSC; (e) P. Page, C. Blonski and J. Perie, Bioorg. Med. Chem., 1999, 7, 1403–1412 CrossRef CAS PubMed.
- C. Meier and W. H. G. Laux, Tetrahedron, 1996, 52, 589–598 CrossRef CAS.
- (a) J. M. Cox, T. R. Hawkes, P. Bellini, R. M. Ellis, R. Barrett, J. J. Swanborough, S. E. Russell, P. A. Walker, N. J. Barnes, A. J. Knee, T. Lewis and P. R. Davies, Pestic. Sci., 1997, 50, 297–311 CrossRef CAS; (b) C. C. Lin, F. Moris-Varas, G. Weitz-Schmidt and C. H. Wong, Bioorg. Med. Chem., 1999, 7, 425–433 CrossRef CAS PubMed.
- P. Page, C. Blonski and J. Perie, Eur. J. Org. Chem., 1999, 2853–2857 CrossRef CAS.
- D. L. Jakeman, A. J. Ivory, M. P. Williamson and G. M. Blackburn, J. Med. Chem., 1998, 41, 4439–4452 CrossRef CAS PubMed.
- S. K. Perumal and R. F. Pratt, J. Org. Chem., 2006, 71, 4778–4785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and chiral HPLC spectra (where applicable) of all novel compounds and compounds made by novel procedures. See DOI: 10.1039/c3ob41632a |
| This journal is © The Royal Society of Chemistry 2013 |