 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogenation effects in intramolecular furan Diels–Alder reactions: broad scope synthetic and computational studies†
Robert L. Rae, Justyna M. Żurek, Martin J. Paterson* and Magnus W. P. Bebbington*
Institute of Chemical Science, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK. E-mail: m.w.p.bebbington@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 8071
First published on 14th October 2013
Abstract
For the first time a comprehensive synthetic and computational study of the effect of halogen substitution on both furan and dienophile for the intramolecular Furan Diels–Alder (IMDAF) reaction has been undertaken. Contrary to our initial expectations, halogen substitution on the dienophile was found to have a significant effect, making the reactions slower and less thermodynamically favourable. However, careful choice of the site of furan halogenation could be used to overcome dienophile halogen substitution, leading to highly functionalised cycloadducts. These reactions are thought to be controlled by the interplay of three factors: positive charge stabilisation in the transition state and product, steric effects and a dipolar interaction term identified by high level calculations. Frontier orbital effects do not appear to make a major contribution in determining the viability of these reactions, which is consistent with our analysis of calculated transition state structural data.
Introduction
The intramolecular furan Diels–Alder (IMDAF) reaction has been the subject of numerous studies over the last 30 years.1 The reaction is very sensitive to both furan and dienophile substitution,1b which has rendered it a useful probe of more general fundamental effects on pericyclic reactions. The reaction has also been employed as a key complexity-generating step in a number of target syntheses.2Halogen substitution was first examined more than 25 years ago,3 but not until a series of elegant experimental and computational studies by Padwa and Houk3d,4 (Scheme 1) did the effect of halogen substitution on furan become clear – IMDAF reactions with halogenated furans were faster and more exergonic than those with non-halogenated furans. Although halogen substitution at the 3-position of 2-furanyl amides was found to promote the cycloaddition largely for conformational reasons, the electronic effect of halogen substitution was most profound at the 5-position (Scheme 1). This was attributed to greater stabilisation of the partial positive charge at the 5-carbon in both transition state and cycloadduct than in the starting material, as a result of the change in hybridisation from sp2 to sp3.4 The effect was calculated to be greatest for fluorine, although this is yet to be demonstrated experimentally, perhaps reflecting current difficulty in the preparation of simple α-fluorofurans.5 Chlorine or bromine substitution is, however, sufficient to have a profound promoting effect in the cases where a true comparison is available with the corresponding non-halogenated substrates.3
 | ||
| Scheme 1 Padwa's demonstration of activation by halogen substitution in the intramolecular furan Diels–Alder reaction. | ||
Although considerable progress has been made in understanding these substituent effects, there remain some unresolved issues. For example, although the change in hybridisation at carbon has been identified as one of the contributing factors in substrate activation by halogen substitution at the 5-position, significant activation is still predicted by halogen substitution at the 4-position,4 where no change in hybridisation occurs during the reaction and no significant conformational effects would be anticipated. At the beginning of our studies we also noted that very little work6 had been done on IMDAF substrates involving halogenation on the dienophile, where change in hybridisation from sp2 to sp3 at the halogen-bearing carbon also occurs. We thus undertook to investigate these fundamental issues through examination of suitable cycloaddition precursors.
Results and discussion
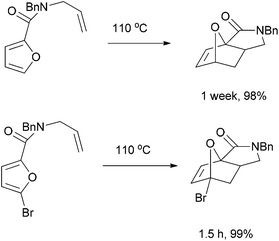
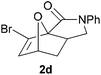
We prepared various classes of IMDAF substrates that would allow us to probe the significance of halogenation with respect to variations in electronics that would also affect the viability of the cycloaddition. Thus, we first prepared substrates closely related to Padwa's original furoic acid-derived examples that would be used essentially for control experiments and to compare directly the effect of halogenation of the diene and the dienophile in turn.Substrates 1a,b and 1d are directly analogous to those prepared by Padwa and co-workers3d,7 and show similar reactivity, consistent with the early work by that group. Bromine substitution at any of the 3-, 4- and 5-positions increases the rate and yield of the reaction compared to the parent unsubstituted system 1a (Table 1).8
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:
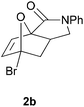
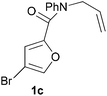
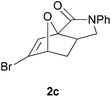
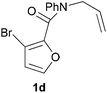
All of these reactions, and subsequent ones described herein, are optimised for maximum isolated yield of cycloadduct – longer reaction times than those listed led to some decomposition. The effect of the N-substituent on the cycloaddition rates of furanyl amides has been studied and found to vary according to the steric bulk of the substituent, rather than the relative populations of the different amide rotamers.9 Our results are consistent with this work, which suggests that the N-phenyl substituent would lead to a slightly slower forward reaction rate (by a factor of ∼3) than the N-benzyl one.
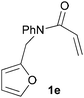
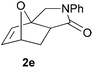
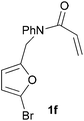
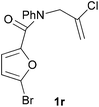
In order to determine whether the halogenation effects showed any variation with a change in the electron-demand of the IMDAF reaction, we prepared substrates 1e–1h (Table 2), bearing electron-deficient alkenes, from N-phenyl furfurylamines (see ESI† for details). In these cases, the difference between the 5-bromo and 4-bromo substrates 1f and 1g was marginal, although both showed higher reactivity than the non-halogenated substrate. The 3-bromo derivative 1h was once again the most reactive of the series, with the cycloaddition proceeding at noticeable rate even at room temperature, suggesting that conformational effects play an important part in promoting this reaction whether or not the carbonyl group is attached directly to the furan ring.
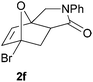
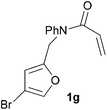
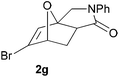
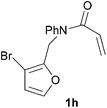
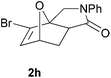
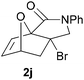
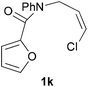
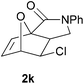
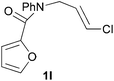
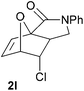
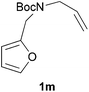
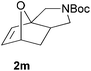
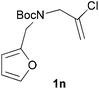
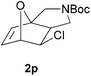
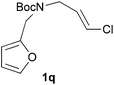
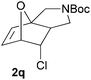
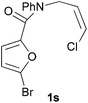
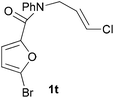
We then elected to study substrates that were halogenated on the dienophile. Chlorine was chosen in the majority of cases because the three isomeric N-chloroallyl anilines are readily prepared from the commercially available chloroallyl chlorides.10 The 2-bromoallyl derivative was also easily synthesised. Clearly, a change in hybridisation at the halogen-bearing carbon, from sp2 to sp3, is a necessary feature of the IMDAF reaction for substrates 1i–1l and 1n–1q (Table 3),11 and we initially hypothesised that this effect would promote the cycloaddition with respect to the non-halogenated substrates as was observed for the earlier work with halogenated furans.3 However, our results indicate the opposite – in all cases the reaction is slower when the halogen is present, although this effect is greater for the amides 1i–1l than for the N-Boc derivatives 1m–1q. This suggests that steric effects are more important when the dienophile is halogenated than when the furan is, since transannular interactions are expected to be more significant in these cases. The mass balance was not completely recovered in these optimised reactions, so in order to verify that the retardation in rate was genuine, substrates 1a and 1h were heated for only 3 h. The conversion to 2a was 37% and to 2h was 15%. The crude reaction mixtures were analysed by 1H NMR using a known quantity of cyclohexane as an internal standard and no significant mass loss was observed for these shorter reactions,12 supporting our initial supposition of a decrease in rate for the halogenated system. In particular, the use of a Z-chloroalkene as dienophile (in 1k and 1p) resulted in only 11–12% conversion after 24 h at 110 °C. This is somewhat surprising as the chlorine is installed on the normally less hindered face of the product. The lower reactivity of the Z-dienophiles compared to the E-dienophiles is in contrast to that observed in some other IMDA reactions using acyclic dienes.13 Indeed, reaction of the E-substrates 1l and 1q and also the 2-haloallyl systems 1i, 1j, 1n and 1o requires the halogen to be on the more hindered face of the [2.2.1]-bicyclic ring system.
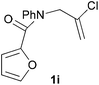
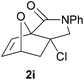
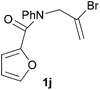
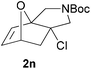
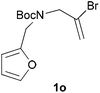
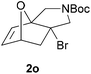
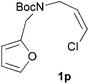
We next decided to examine frontier orbital effects using DFT (B3LYP/6-31G(d))14 calculations to attempt to explain these results. Furan is normally considered to be an electron-rich diene in Diels–Alder reactions, such that the important frontier molecular orbital interaction is HOMO(furan)–LUMO(dienophile).1b Many examples of IMDAF are consistent with this, but in a number of cases the cycloaddition occurs still occurs readily when, as here, the furan bears an electron-withdrawing carbonyl group, so there has been some debate regarding frontier orbital effects in IMDAF.3a,b,d,15 The effect of halogen substitution on frontier orbital energies is less clear cut,16 due to a mixture of electron-donating field/mesomeric and electron-withdrawing inductive effects. The similar trends observed for substrates 1i–1l and 1n–1q, which would be expected to have rather different FMO properties, suggest that frontier orbital effects are not overriding source of the observed differences in reactivity. Our calculations17 predict that both furan and 2-chlorofuran react with ethylene in a normal electron-demand sense. The presence of the Cl atom only results in a very minor change (<0.1 eV) in the HOMO–LUMO energy gap in these systems. Examination of the relevant orbitals in the more complex intramolecular reactions indicates that non-halogenated substrates 1e and 1m react with normal demand, whereas 1a, with an electron-withdrawing group on furan, reacts with inverse demand. Halogenation does not appear to change the nature of the FMO interactions, and produces only minor changes in orbital energies (0.1–0.4 eV) across most of the substrates studied.17 The exceptions to this are 1e–1h, where the FMO energy gap increases by 1.1–1.3 eV upon furan halogenation, although the halogenated systems appear to react more rapidly. Similarly, comparison of the HOMO–LUMO energy gaps in inverse demand substrates 1a, 1i and 1j suggests that halogenation decreases the energy gap (by 0.3–0.4 eV), even though 1a undergoes cycloaddition fastest of these three substrates. Intriguingly, very similar experimental and FMO trends are found in normal demand substrates 1m, 1n and 1o, where the FMO energy gap is predicted to decrease for halogenated systems 1n and 1o, even though the cycloaddition appears marginally faster in the non-halogenated substrate 1m. Therefore, dienophile halogenation decreases the FMO energy gap for both normal- and inverse-demand systems. These results therefore support the hypothesis that FMO interactions are not the most important factor in controlling these reactions. This is consistent with a less exergonic reaction than is usual for a Diels–Alder cycloaddition (e.g. with a non-aromatic diene),18 giving rise to a less reactant-like transition state that is not well approximated by examination of the orbital energies of the starting materials. An examination of the calculated transition state and product structures is also supportive of later transition states among the less exergonic reactions (vide infra).
Our results could instead be explained if a dipolar interaction between the C–X and furan C–O bonds served to raise the energy of the transition state for 1k and 1p and had the opposite effect in the other substrates. Indeed, such a possibility is supported by the calculations. Using a simple approximation for the interaction of two (weak) dipoles we have calculated the orientational dependence of the dipolar interactions of the C–O and the dienophile C–X dipoles at the transition state structures of certain reactions. Table S4 of the ESI† presents these values for reactions of 1i–1l and 1p–1q. The X–C–C–O dihedral angle differs significantly in the transition states for reactions of 1k and 1l, which leads to a significant difference in the dipolar interaction term. There is some repulsion present in the structure of the transition states for reactions of 1k and 1p thus leading to higher Diels–Alder activation energy. Transition states for reactions of 1i, 1j, 1l and 1q acquire more attraction interaction, leading to lower potential energy for the transition state, and thus lower activation energy for Diels–Alder reaction.
The 2-bromoallyl derivatives 1j and 1o undergo cycloaddition to a slightly greater extent than the corresponding 2-chloroallyl substrates 1i and 1n, and indeed any of the other chlorinated precursors, despite the fact that the bulky bromine atom is attached to a crowded tertiary centre. This indicates that the increased steric requirements of the halogenated alkene, positive charge stabilisation at halogenated carbon in the transition state, or dipolar interactions do not account for these observations, since all of these factors should be more significant for Cl-substituted systems. It is presently unclear why the brominated systems react marginally more rapidly than the chlorinated ones.
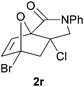
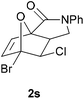
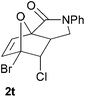
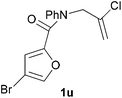
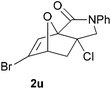
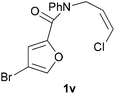
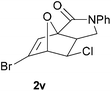
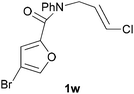
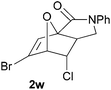
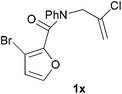
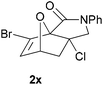
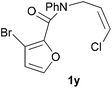
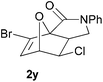
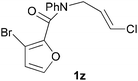
In order to compare experimentally the beneficial effect of furan halogenations versus the detrimental effect of dienophile halogenation, we finally prepared dihalogenated examples 1r–1z leading to adducts 2r–2z (Table 4). In general, greater reactivity was observed than in substrates 1i–1l. Interestingly, bromination at the 5-position of furan was not always more effective in promoting the cycloaddition than at the 4-position (substrates 1svs. 1v),4 and indeed the 5-bromo-Z-chloro substrate 1s showed little more reactivity than the analogous compound 1k lacking a brominated furan, presumably for steric reasons. The 3-bromo derivatives 1y and 1z show the highest reactivity, which is consistent with our earlier results. Substrate 1x is anomalous given the reactivity of 1r and 1u, and illustrates the complexity of the interaction between the steric and electronic factors controlling these reactions. Careful adjustment of the bromination site is thus sufficient to override the detrimental effect of the dienophile halogenations, demonstrating the power of this substitution in facilitating previously difficult cycloadditions.
Computation
All calculations were performed with the Gaussian09 program (versions a and c).19 The Complete Basis Set (CBS) set of methods of Petersson and co-workers was used to obtain accurate thermochemical data.20–23 The specific extrapolation used here (Table 5) is the CBS-QB3 one. Details of others used are given in ESI (Table S1†) for full comparison. All calculations were performed in the gas phase. Free energies with CBS models were calculated for two temperatures: 298 K and 383 K (the temperature of the experimental studies). The results for CBS-QB3 model of chosen Diels–Alder reactions for 383 K are presented in Table 5.24| Reaction | ΔrH° | ΔrG° | ΔactivH | ΔactivG |
|---|---|---|---|---|
| a All energies reported in kcal mol−1. | ||||
| 1a → 2a | −10.6 | −5.2 | 19.4 | 23.8 |
| 1b → 2b | −14.6 | −9.1 | 17.9 | 22.4 |
| 1c → 2c | −14.0 | −8.7 | 17.6 | 21.7 |
| 1d → 2d | −14.0 | −8.1 | 16.4 | 21.3 |
| 1e → 2e | −17.2 | −9.9 | 15.6 | 21.8 |
| 1f → 2f | −20.7 | −14.5 | 15.0 | 19.9 |
| 1g → 2g | −19.8 | −13.7 | 14.5 | 19.1 |
| 1h → 2h | −18.8 | −12.8 | 14.7 | 19.5 |
| 1i → 2i | −9.6 | −4.0 | 20.5 | 25.0 |
| 1j → 2j | −9.9 | −4.3 | 20.4 | 25.0 |
| 1k → 2k | −11.6 | −5.9 | 19.4 | 23.9 |
| 1l → 2l | −12.1 | −5.3 | 18.1 | 23.9 |
| 1m → 2m | −8.2 | −2.7 | 19.9 | 24.5 |
| 1n → 2n | −8.2 | −2.4 | 20.4 | 25.2 |
| 1o → 2o | −8.6 | −3.1 | 20.1 | 25.2 |
| 1p → 2p | −7.2 | −0.1 | 21.5 | 27.6 |
| 1q → 2q | −8.4 | −2.7 | 20.0 | 24.7 |
| 1r → 2r | −13.5 | −7.7 | 18.8 | 23.4 |
| 1s → 2s | −13.7 | −8.0 | 18.4 | 22.9 |
| 1t → 2t | −14.9 | −9.2 | 20.6 | 21.6 |
| 1u → 2u | −13.0 | −7.3 | 18.7 | 23.4 |
| 1v → 2v | −14.4 | −8.6 | 17.7 | 22.4 |
| 1w → 2w | −13.8 | −7.9 | 17.6 | 22.6 |
| 1x → 2x | −12.6 | −6.2 | 18.7 | 24.0 |
| 1y → 2y | −15.3 | −10.1 | 15.6 | 19.7 |
| 1z → 2z | −14.6 | −8.5 | 16.2 | 21.4 |
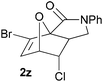
Comparison of the calculated thermodynamic and experimental data is somewhat complicated by decomposition of many of the adducts at the extended reaction times (typically a number of days) sometimes needed for the reactions to go to completion. All of the reactions studied are predicted to be exergonic in nature (Fig. 1 and Table 5), although there is significant variation in both activation and overall reaction free energies.
 | ||
| Fig. 1 Scatter plot of Gibbs free energies of reaction (in kcal mol−1) for systems a–z (black = unhalogenated; red = halofuran substrate; blue = haloalkene substrate; yellow = dihalogenated substrate). | ||
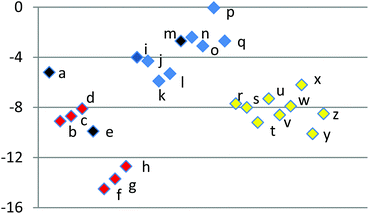
Those reactions with calculated higher activation energies predominantly show, as a general trend, lower conversions to product over the timescale of the reactions, where directly comparable. However, a quantitative correlation between activation barrier and yield was not observed. A plot of the calculated activation energies (Fig. 2 and Table 5) is colour-coded to show grouping of the different types of substrate. Thus, precursors with halogenated furans (1b–d, 1f–h) possess lower activation barriers for cycloaddition (19.1–22.4 kcal mol−1 with mean value 20.6 and standard deviation ±1.3) than those with halogenated dienophiles (1i–l, 1n–q) which lie between 23.9 and 27.5 kcal mol−1 (mean 25.1 ± 1.2). In turn, those substrates with halogens on both furan and dienophile (1r–z) have activation barriers that are intermediate in value (21.4 to 23.4 kcal mol−1, mean 22.4 ± 1.3). Substrate 1y is anomalous, with a low calculated activation energy (19.7 kcal mol−1), but experimentally it shows little reactivity.
 | ||
| Fig. 2 Scatter plot of Gibbs free energies of activation (in kcal mol−1) for systems a–z. | ||
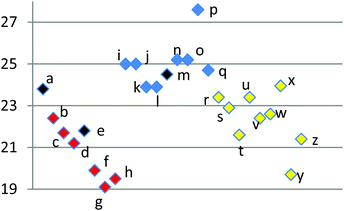
In order to probe further the validity of our conclusions from our studies on possible frontier orbital effects, we elected to quantify the “product-likeness” of the transition states in terms of the fractional shortening25 of the newly forming C–C σ-bonds (marked as a and b in Fig. 3; the corresponding calculated numerical data are in Table 6) in moving from transition state to product. A similar grouping to that seen in both activation energies and free energies of reaction is observed. Thus, the more exergonic the reactions (see Fig. 1 and 3), the earlier their transition states, so that the percentage contraction of the C–C distance in going from transition state to product is greater. Conversely, those reactions that are exhibit a lower percentage contraction are predicted to be less exergonic, consistent with later transition states. These data are thus supportive of our earlier conclusions on FMO energies which are not found to correlate with the reactivities we observe. FMO energies are thus likely to be less important contributors to transition state energies for reactions with later transition states than is usual for Diels–Alder cycloadditions, such as those discussed in our work.
 | ||
| Fig. 3 Scatter plot of percentage change in C–C bond distances from transition state to product. | ||
| System | Transition state | Product | ||
|---|---|---|---|---|
| C–C distance a | C–C distance b | C–C bond length a | C–C bond length b | |
| a All distances (a and b are defined in Fig. 3) in angstroms. | ||||
| a | 2.029 | 2.178 | 1.558 | 1.571 |
| b | 2.044 | 2.199 | 1.558 | 1.565 |
| c | 2.053 | 2.179 | 1.556 | 1.568 |
| d | 2.004 | 2.227 | 1.555 | 1.570 |
| e | 2.035 | 2.223 | 1.556 | 1.569 |
| f | 2.056 | 2.241 | 1.556 | 1.563 |
| g | 2.061 | 2.221 | 1.553 | 1.566 |
| h | 2.022 | 2.259 | 1.553 | 1.567 |
| i | 2.064 | 2.153 | 1.563 | 1.570 |
| j | 2.067 | 2.152 | 1.562 | 1.570 |
| k | 1.995 | 2.225 | 1.557 | 1.569 |
| l | 1.987 | 2.231 | 1.555 | 1.570 |
| m | 2.095 | 2.111 | 1.576 | 1.572 |
| n | 2.137 | 2.076 | 1.581 | 1.570 |
| o | 2.139 | 2.077 | 1.579 | 1.571 |
| p | 2.061 | 2.137 | 1.574 | 1.570 |
| q | 2.067 | 2.146 | 1.574 | 1.570 |
| r | 2.079 | 2.165 | 1.563 | 1.565 |
| s | 1.950 | 2.332 | 1.553 | 1.583 |
| t | 2.031 | 2.235 | 1.554 | 1.572 |
| u | 2.095 | 2.146 | 1.562 | 1.566 |
| v | 2.016 | 2.222 | 1.554 | 1.567 |
| w | 1.986 | 2.253 | 1.552 | 1.570 |
| x | 2.086 | 2.150 | 1.565 | 1.566 |
| y | 1.962 | 2.278 | 1.554 | 1.569 |
| z | 1.968 | 2.275 | 1.553 | 1.570 |
Conclusions
We have investigated the effect of both furan and dienophile halogenations in IMDAF reactions. Halogenation of the furan is found to favour the cycloaddition, irrespective of the electronic effects of the other furan and dienophile substituents. Halogenation of the dienophile was found to lead to slower reactions than those with the corresponding non-halogenated dienophiles in all cases studied. Frontier orbital effects do not appear to have a huge influence on these reactions, and the results have been attributed to a combination of positive charge stabilisation, simple steric effects, and a contribution from a dipolar interaction between the furan C–O and the dienophile C–X dipoles, supported by computation. This varies with the dihedral angle between the two bonds in the transition states, suggesting a partial explanation for the difference in reactivity between analogous systems with differing alkene geometries. The use of dihalogenated substrates can provide a means of overriding the effect of dienophile halogenations, leading to synthetically useful yields of highly functionalised cycloadducts with considerable potential for further transformation.The authors thank the ERC (for funding for M. J. P.) and the EPSRC (for a DTA for R. L. R.; for funding for J. M. Ż. and for use of the EPSRC National Mass Spectrometry Facility for mass spectra).
Notes and references
- (a) For a review of the intramolecular Diels–Alder reaction, see: E. Ciganek, Org. React., 2004, 1–371 Search PubMed , Wiley Online Library; ; (b) For a comprehensive review of the furan Diels–Alder reaction, see: C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179 CrossRef CAS.
- For a review of the use of the product oxanorbornenes in natural product synthesis, see: (a) P. Vogel, J. Cossy, J. Plumet and O. Arjona, Tetrahedron, 1999, 55, 13521 CrossRef CAS ; For selected more recent examples, see: ; (b) E. N. Pitsinos, N. Athinaios and V. P. Veroniki, Org. Lett., 2012, 14, 4666 CrossRef CAS PubMed; (c) C.-C. Wang and W.-D. Z. Li, J. Org. Chem., 2012, 77, 4217 CrossRef CAS PubMed; (d) P. Fischer, M. Gruner, A. Jaeger, O. Kataeva and P. Metz, Chem.–Eur. J., 2011, 17, 13334 CrossRef CAS PubMed; (e) K. Tanino, M. Takahashi, Y. Tomata, H. Tokura, T. Uehara, N. Takashi and M. Miyashita, Nat. Chem., 2011, 3, 484 CrossRef CAS PubMed; (f) F. R. Petronijevic and P. Wipf, J. Am. Chem. Soc., 2011, 133, 7704 CrossRef CAS PubMedY.-P. Xue and W.-D. Z. Li, J. Org. Chem., 2011, 76, 57 CrossRef CAS PubMed; (g) M. B. O'Keefe, D. M. Mans, D. E. Kaelin and S. F. Martin, J. Am. Chem. Soc., 2010, 132, 15528 CrossRef PubMed; (h) S. Mitsuru and Y. Hayashi, Eur. J. Org. Chem., 2007, 3783 Search PubMed; (i) S. M. Sparks, C.-L. Chen and S. F. Martin, Tetrahedron, 2007, 63, 8619 CrossRef CAS PubMed; (j) G. E. Morton and A. G. M. Barrett, Org. Lett., 2006, 8, 2859 CrossRef CAS PubMed; (k) A. Padwa and J. D. Ginn, J. Org. Chem., 2005, 70, 5197 CrossRef CAS PubMed; (l) M. Takadoi, T. Katoh, I. Tadashi and S. Terashima, Tetrahedron, 2002, 58, 9903 CrossRef CAS; (m) S. Mitsuru, J. Yamaguchi, H. Kakeya, H. Osada and Y. Hayashi, Angew. Chem., Int. Ed., 2002, 41, 3192 CrossRef.
- For earlier studies on IMDAF reactions with halogenated furans, see: (a) Z. Klepo and K. Jakopcic, J. Heterocycl. Chem., 1987, 24, 1787 CrossRef CAS; (b) A. D. Mance, M. Sindler-Kulyk, K. Jakopcic, A. Hergold-Brundic and A. Nagl, J. Heterocycl. Chem., 1997, 34, 1315 CrossRef CAS; (c) K. R. Crawford, S. K. Bur, C. S. Straub and A. Padwa, Org. Lett., 2003, 5, 3337 CrossRef CAS PubMed; (d) A. Padwa, K. R. Crawford, C. S. Straub, S. N. Pieniazek and K. N. Houk, J. Org. Chem., 2006, 71, 5432 CrossRef CAS PubMed.
- S. N. Pienazek and K. N. Houk, Angew. Chem., Int. Ed., 2006, 45, 1442 CrossRef PubMed.
- Procedures for the preparation of α-fluorofurans tend to be low-yielding and lacking in generality – for a review of fluorofuran synthesis, see: O. Serdyuk, A. Butin and V. Abaev, J. Fluorine Chem., 2010, 131, 296 CrossRef CAS PubMed.
- For examples of IMDAF reactions using haloalkene dienophiles, see: (a) M. Karaarslan and A. Demircan, Asian J. Chem., 2007, 19, 2999 CAS; (b) A. Demircan, M. Karaarslan and E. Turac, Heterocycl. Commun., 2006, 12, 233 CrossRef CAS PubMed . No comparison is made with non-halogenated systems herein and the halogen is always attached to a tertiary centre. For examples of IMDAF reactions with chloroacrylate dienophiles, see: ; (c) M. E. Jung and L. J. Street, J. Am. Chem. Soc., 1984, 106, 8327 CrossRef CAS; (d) M. E. Jung and L. J. Street, Heterocycles, 1988, 27, 45 CrossRef CAS . A slight retardation of the cycloaddition is observed for chloroacrylates compared with acrylates.
- No substrate analogous to 1c, with a sole Br atom in the 4-position has ever been studied prior to this work.
- The conversions of substrates 1a and 1b after only 1 h at 110 °C to the cycloadducts 2a and 2b were 19% and 63% respectively, as judged by 1H NMR spectroscopy.
- M. Nakamura, I. Takahasi, S. Yamada, Y. Dobashi and O. Kitagawa, Tetrahedron Lett., 2011, 52, 53 CrossRef CAS PubMed.
- J. Barluenga, F. Foubelo, F. J. Fañanás and M. Yus, J. Chem. Soc., Perkin Trans. 1, 1989, 553 RSC.
- Somewhat surprisingly, non-halogenated control substrate 1m has not previously been prepared.
- Some decomposition was observed (NMR) in reactions heated for 24 h or longer. Some mass loss is also thought to have occurred in chromatography – separation was difficult and some mixed fractions were obtained in a number of cases, the contents of which obviously are not included in the isolated yields.
- T. N. Cayzer, M. N. Paddon-Row, D. Moran, A. D. Payne, M. S. Sherburn and P. Turner, J. Org. Chem., 2005, 70, 5561 CrossRef CAS PubMed.
- Calibration calculations using larger bases on selected systems did not alter the conclusions from this part of the study.
- T. Takebayashi, N. Iwasawa and T. Mukaiyama, Chem. Lett., 1983, 1107 CAS.
- R. Robiette, J. Marchand-Brynaert and D. Peeters, J. Org. Chem., 2002, 67, 6823 CrossRef CAS PubMed.
- See ESI.†.
- Calculations in ref. 4 indicate that Diels–Alder reactions with non-aromatic dienes are considerably more exergonic than those with furan. Hammond's postulate would hence suggest that FMO energies of the starting materials would be less predictive of reactivity in the case of furan cycloadditions, which would be expected to have more product-like transition states.
- M. J. T. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, 2009 Search PubMed.
- M. R. Nyden and G. A. Petersson, J. Chem. Phys., 1981, 75, 1843 CrossRef CAS.
- J. W. Ochterski, G. A. Petersson and J. A. Montgomery, J. Chem. Phys., 1996, 104, 2598 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532 CrossRef CAS.
- For analogous data calculated at 298.15 K, see the ESI.†.
- For a table detailing these values, see Table S5 in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, 1H and 13C NMR spectra for all new compounds; further computational experiments on model systems and full discussion of the CBS methodologies used, results for reactions of 1a–z at 298 K, dipolar interaction, selected FMO and transition state structural data. See DOI: 10.1039/c3ob41616j |
| This journal is © The Royal Society of Chemistry 2013 |