 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversal of facial selectivity in a thia-Claisen rearrangement by incorporation of a vinylic bromine substituent†
Adam R. Ellwooda, Anne J. Price Mortimera, Jonathan M. Goodmanb and Michael J. Porter*a
aDepartment of Chemistry, University College London, Christopher Ingold Building, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: m.j.porter@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 7679 4710
bUnilever Centre for Molecular Science Informatics, Department of Chemistry, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 26th September 2013
Abstract
Thia-Claisen rearrangements have been carried out using N-benzylpyrrolidine-2-thione and chiral allylic bromides derived from D-mannitol. Introduction of a bromine atom onto the double bond of the allylic bromide reverses the sense of diastereoselectivity in the [3,3]-sigmatropic rearrangement. Density functional theory calculations lead us to rationalise the observed selectivity in terms of a Cíeplak effect.
Introduction
The Claisen rearrangement of allyl vinyl ethers and related [3,3]-sigmatropic rearrangements have been extensively studied and widely utilised in synthesis.1 In these reactions, a new carbon–carbon bond is formed, and one or two new stereogenic centres may be created.As a consequence of the ordered transition state of such rearrangements, it is possible to use existing stereogenic centres to direct the formation of new ones.2 Most of the examples of stereoselective rearrangement reported in the literature involve 1,3-chirality transfer from a stereogenic centre within the six atoms of the allyl vinyl ether system. Examples of asymmetric induction from an external stereogenic centre are less common; in particular, there are very few cases in which such a stereogenic centre on the “allyl” portion of the molecule has proven effective in inducing high levels of asymmetry in the rearrangement step.3,4
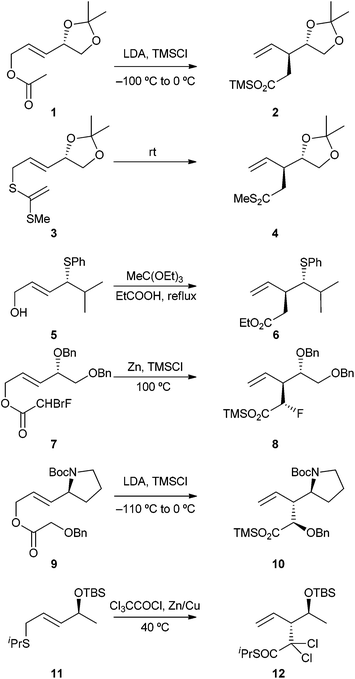
Several [3,3]-sigmatropic rearrangements of E-alkenes bearing an electronegative substituent in the allylic position are depicted in Scheme 1; the sense of asymmetric induction is the same in all cases, although its magnitude varies. For example, Ireland–Claisen rearrangement of ester 1 gives 2 as the major product with a diastereomeric ratio of 1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,5,6 while rearrangement of the related ketene dithioacetal 3 occurs at room temperature to give 4 as the major component of a 1.9:1 mixture.7 Johnson–Claisen rearrangement of allylic alcohol 5, bearing a sulfur substituent at the allylic stereogenic centre, gives ester 6 as the major component of a 1.5:1 mixture of stereoisomers.8 Reformatskii–Claisen rearrangement of allylic ester 7 gives 8 as the major compound in an 8.7:3.4:1.8:1 mixture of diastereomers.9
:1,5,6 while rearrangement of the related ketene dithioacetal 3 occurs at room temperature to give 4 as the major component of a 1.9:1 mixture.7 Johnson–Claisen rearrangement of allylic alcohol 5, bearing a sulfur substituent at the allylic stereogenic centre, gives ester 6 as the major component of a 1.5:1 mixture of stereoisomers.8 Reformatskii–Claisen rearrangement of allylic ester 7 gives 8 as the major compound in an 8.7:3.4:1.8:1 mixture of diastereomers.9
 | ||
| Scheme 1 Previously reported diastereoselective Claisen rearrangements. | ||
A higher level of asymmetric induction is observed in the Ireland–Claisen rearrangement of amino acid-derived esters such as 9, with 10 being the only product stereoisomer obtained.3 Similarly, a stereoselective zwitterionic [3,3]-rearrangement takes place on treatment of 11 with dichloroketene, leading to 12 as a single stereoisomer.4a
As part of a programme directed towards the synthesis of the sarain alkaloids, we have utilised a thia-Claisen rearrangement10 to establish the two stereogenic centres in thiolactam intermediate 14 (Scheme 2). Following S-alkylation of N-benzylpyrrolidine-2-thione (13) with the appropriate allylic bromide, deprotonation with triethylamine afforded directly the rearranged product 14 with >40:1 diastereoselectivity.11
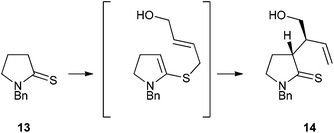 | ||
Scheme 2 Thia-Claisen reaction. Reagents and conditions: (E)-HOCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br, MeCN then Et3N, 40 °C, 67%. CHCH2Br, MeCN then Et3N, 40 °C, 67%. | ||
Herein we report our efforts to modify this diastereoselective reaction to give products such as 14 in enantiomerically enriched form. Our hope at the outset was that replacement of the achiral allylic bromide in Scheme 2 with chiral analogues such as 15 would lead to a preponderance of one of the diastereomeric products 16 and 17 (Scheme 3). Following separation of these compounds, cleavage of the protected diol unit could potentially lead to compound 14 as a single enantiomer.12
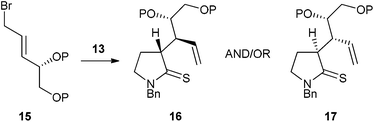 | ||
| Scheme 3 Proposed diastereoselective thia-Claisen rearrangement. | ||
Six allylic bromides were investigated (Fig. 1): E-disubstituted alkenes 18a and 19a and Z-alkene 20a, together with the analogous compounds 18b–20b in which the double bond carries an additional bromine substituent.
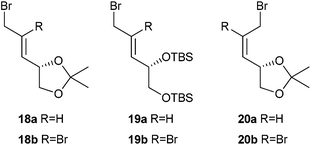 | ||
| Fig. 1 Allylic bromides used in the study. | ||
Results and discussion
Preparation of allylic bromides
Allylic bromides 18a–20b were all synthesised by standard methods from 1,2:5,6-diisopropylidene-D-mannitol. Details of these syntheses, including full spectroscopic data, can be found in the ESI.†Thia-Claisen rearrangements
Initial attempts at the key thia-Claisen rearrangement were carried out using bromide 18a under the conditions which we had previously developed.11 Thus N-benzylpyrrolidine-2-thione (13) was alkylated with allylic bromide 18a in acetonitrile. Addition of 4 Å molecular sieves was found to be helpful in suppressing undesired side reactions. Following dilution with further acetonitrile, the mixture was warmed to 40 °C and triethylamine was added. The thia-Claisen rearrangement took place to give a mixture of three stereoisomeric thiolactam products, 21a, 22a and 23a in a ratio of 69:28:3 (Scheme 4 and Table 1, entry 1). The two predominant products were those expected if the rearrangement proceeds through a chair transition state (see ESI† for details of the assignment of stereochemistry).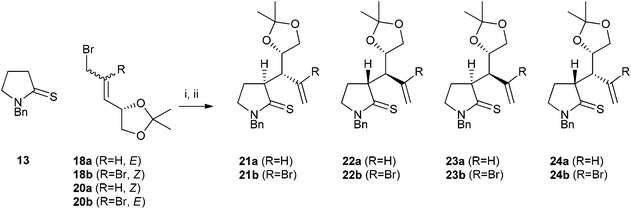 | ||
| Scheme 4 Thia-Claisen rearrangement of acetonide-containing substrates. Reagents and conditions: (i) MeCN, rt, 4 Å molecular sieves; (ii) Et3N, 40 °C. | ||
We anticipated that the use of trisubstituted alkene substrate 18b would give 21b with an enhanced level of asymmetric induction. Surprisingly, use of 18b instead led to a reversal in the stereochemical preference of the reaction, and products 21b, 22b and 23b were formed in a 7:88:5 ratio (Table 1, entry 2).
The two geometrically isomeric allylic bromides 20a and 20b were also subjected to the thia-Claisen sequence; with these substrates, diastereoselectivities were modest and, as expected, the major stereoisomer 23 from these rearrangements was one which had been a minor component in the previous reactions (Table 1, entries 3 and 4), consistent with a chair-like transition state. The same stereoisomer was the major product for both substrates.
Use of the bis-silyl ether 19a in place of the acetonide 18a gave a markedly higher ratio of thia-Claisen products (Scheme 5, Table 2, entry 1); in this case only two stereoisomers, 25a and 26a, were observed. Attempts to use bis-silyl ether 19b in thia-Claisen reactions were completely unsuccessful, yielding none of the expected products 25b and 26b.
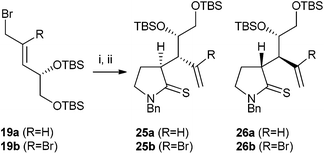 | ||
| Scheme 5 Thia-Claisen rearrangement of silyl ether-containing substrates. Reagents and conditions: (i) 13, MeCN, rt, 4 Å molecular sieves; (ii) Et3N, 40 °C. | ||
| Entry | Bromide | Ratio 25:26a | Yield 25b/% | Yield 26b/% |
|---|---|---|---|---|
| a Ratio determined from the 1H NMR spectrum of the crude reaction mixture; the reaction of 19a also generated some unidentified by-products.b Isolated yields following column chromatography.c No thia-Claisen rearrangement product was obtained from this reaction. | ||||
| 1 | 19a | >97:3 | 41 | 1 |
| 2 | 19b | —c | — | — |
Discussion
The diastereoselectivity of the thia-Claisen rearrangements ranged from poor (in the case of 18a and 20a) to excellent (18b and 19a). The choice of protecting groups was seen to have a significant effect: bis-silyl ether 19a (entry 1, Table 2) gave a much more diastereomerically enriched mixture of products than acetonide 18a.Installation of a bromine atom on the double bond had a profound effect on stereoselectivity, with the sense of asymmetric induction reversed between acetonides 18a and 18b (entries 1 and 2, Table 1).
In each of the rearrangement reactions, the relative configuration of the two newly-formed stereogenic centres in the major product is consistent with a chair-like transition state, while their absolute configuration is determined by any facial selectivity imparted by the pre-existing stereogenic centre. Analysis of the stereochemical course of the reaction indicates that the non-brominated substrates 18a and 19a show a tendency for reaction through the conformation I (Re-face selectivity at the N,S-ketene acetal, Fig. 2) while vinyl bromide 18b reacts primarily through conformation II (Si-face selectivity).
 | ||
| Fig. 2 Facial selectivity in the N,S-ketene acetal intermediates derived from 18 and 19. | ||
In trying to elucidate the reasons for this divergent stereoselectivity, it is necessary to consider six reactive conformations of the diene, corresponding to three rotamers about the bond linking the alkene to the stereogenic centre for each of the conformations I and II. These conformations are depicted in Fig. 3: in conformations Ia and IIa, the new C–C bond is formed antiperiplanar to the allylic C–O bond; in conformations Ib and IIb, it is formed antiperiplanar to the allylic C–C bond; while in conformations Ic and IIc, it is formed antiperiplanar to the allylic C–H.‡
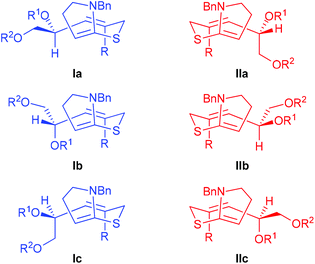 | ||
| Fig. 3 Possible reactive conformations of the N,S-ketene acetal intermediates derived from 18 and 19. | ||
The selectivity observed for the non-brominated substrates 18a and 19a is consistent with that shown by all the related literature examples (Scheme 1); such selectivity has previously been rationalised in terms of a reactive conformation corresponding to Ia. The electron-rich character of the N,S-ketene acetal component means that the rearrangement is expected to occur through a polarised transition state, with a “nucleophilic” ketene acetal fragment and an “electrophilic” allyl component.13 Attack on the electrophilic component is assumed to take place antiperiplanar to the allylic C–O bond, as this allows stabilization of the newly forming σ-bond by interaction with the low-lying σ*-orbital of the C–O bond;13,14 hence reaction should take place through either conformation Ia or IIa. The preference for the former conformation can be understood as Ia will suffer less allylic (A1,3) strain15 than IIa.
The stereochemical preferences of vinyl bromide substrate 18b are contrary to all previously reported [3,3]-sigmatropic rearrangement substrates with an electronegative allylic substituent, and this behaviour is harder to explain. Our a priori expectation had been that the increased allylic (A1,3) strain in conformation IIa when R ≠ H would lead to a stronger preference for reactive conformation Ia and hence an enhanced level of stereoselectivity in favour of product 21b. The experimental observation, conversely, was that 22b is the major product in the reaction of bromide 18b.
It seems unlikely that the reaction of bromide 18b proceeds through conformation IIa, in which allylic strain will be more severe than in conformer Ia, and so the other possible conformations must be considered. Conformations Ib or Ic would lead to product 21b, while IIb or IIc would lead to the observed major product 22b. As there was no obvious explanation for the change in stereochemical preference on bromination of the double bond, we carried out a computational study to gain further insight into the stereochemical course of these reactions.16
Computational studies
Initial attempts to locate transition states for compounds containing a dioxolane ring indicated a complicated potential energy surface on which multiple transition states, differing only in the conformation of the dioxolane, could be identified. In order to simplify the computational problem, while retaining the salient features of substrates 18 and 19, we chose to carry out the computational study on the rearrangement of allylic methyl ethers 27a and 27b (Fig. 4).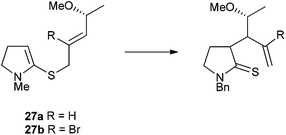 | ||
| Fig. 4 Simplified structures for computational studies. | ||
A detailed search for chair-like transition state geometries was undertaken using density functional theory: for each of the two substrates 27a and 27b, we considered (i) Re and Si-face reaction of the N,S-ketene acetal (i.e.I and II, Fig. 2); (ii) three rotamers about the bond linking the alkene to the stereogenic centre (i.e.a, b and c, Fig. 3); and (iii) three staggered rotamers about the MeO–C bond, giving a total of 18 possible transition state structures for each compound. For each of the two compounds 27a and 27b, transition states were located for 17 of the 18 possible conformations – the missing conformation in each case being one of type IIb in which the O-methyl group would be directed into the pyrroline ring.
For all six conformer types Ia–IIc for both substrates, the rotamer in which the O–CH3 bond lay antiperiplanar to the alkene portion of the molecule was markedly lower in energy than the other two rotamers around the C–OCH3 bond, and so only these rotamers were considered for the remainder of the study.
Having identified transition states corresponding to the conformers Ia–IIc, it was noted that two different conformations of the pyrroline ring, differing in the direction of pyramidalisation of the nitrogen atom, were present among the transition states identified. Further searching was therefore carried out to locate transition states for both pyrroline conformers in every case.
This exhaustive search resulted in twelve transition state structures for each of the two substrates, corresponding to the six conformations Ia–IIc each with two different pyrroline conformations. Fig. 5 shows twelve of these 24 transition state structures17 – in each case, the structure depicted is the lower-energy of the two pyrroline conformations. The relative Gibbs energies of these transition states are collected in Table 3.18 In four cases, the energetic ordering of the two pyrroline conformations was reversed between gas-phase and solution-phase calculations; these cases are marked with an asterisk.
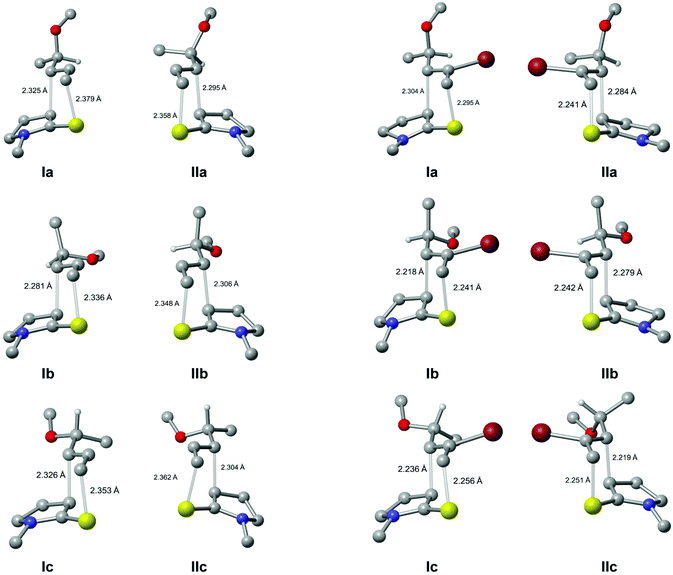 | ||
| Fig. 5 Transition state structures for thia-Claisen rearrangement of 27a (left) and 27b (right). Hydrogen atoms, other than the one at the stereogenic centre, are omitted for clarity. | ||
| Transition structure | Facial orientationa | Antiperiplanar atomb | 27a ΔΔG‡/kJ mol−1 [M06-2X/B3LYP]c,d | 27b ΔΔG‡/kJ mol−1 [M06-2X/B3LYP]c,d |
|---|---|---|---|---|
| a Face selectivity as defined by reaction of the N,S-ketene acetal moiety.b Atom bonded to the stereogenic centre which is most nearly antiperiplanar to the forming C–C bond.c Gibbs energy of transition states relative to the lowest in energy for that set. Figures in parentheses indicate solution (THF) phase energies calculated for the gas-phase optimised structures. An asterisk denotes the Gibbs energy of a structure differing in the pyrroline conformation from that depicted.d Gibbs energy calculated by a single-point energy calculation with the M06-2X functional on the B3LYP gas-phase optimised structure, followed by a free energy correction from the B3LYP values.17 | ||||
| Ia | Re | O | 21.1 (17.5) | 10.4 (8.2) |
| IIa | Si | O | 9.9 (7.2) | 27.4 (21.7) |
| Ib | Re | C | 0.0 (0.0) | 11.3 (9.9*) |
| IIb | Si | C | 3.7 (5.6*) | 0.0 (0.0) |
| Ic | Re | H | 8.3 (10.3*) | 6.0 (8.1) |
| IIc | Si | H | 22.1 (21.0) | 7.3 (8.6*) |
Contrary to our expectation, the lowest-energy transition states for the reaction of 27a did not place the C–O bond of the stereogenic centre antiperiplanar to the newly-forming C–C bond. Rather the most favourable transition states are those in which the allylic C–C bond occupies the antiperiplanar position (i.e.Ib and IIb), with structure Ib (corresponding to the observed major product) having the lowest energy of all the transition states. Only a small Gibbs energy difference was found between structures Ib and IIb (3.7 kJ mol−1 in the gas phase, 5.6 kJ mol−1 in solution). This corresponds qualitatively to the low stereoselectivity observed in the reaction.
In the case of brominated substrate 27b, the lowest energy transition state is again one in which the newly forming C–C bond is situated antiperiplanar to the C–C bond at the stereogenic centre; for this compound however, structure IIb is markedly lower in energy than structure Ib, as would be expected from the greater degree of A1,3-strain in Ib. Indeed, while IIb is the lowest-energy transition state structure for this substrate, the next-lowest is Ic, in which a hydrogen atom occupies the antiperiplanar position. The Gibbs energy difference between transition states IIb and Ic was found to be 6.0 kJ mol−1, rising to 8.1 kJ mol−1 when solvation effects were included. These increased energy differences, compared to those found for substrate 27a, again agree qualitatively with the experimental observation that substrate 18b reacts with reversed and higher levels of stereoselectivity compared to substrate 18a.
The indication from these computational studies is thus that there is an intrinsic preference for reaction to take place anti- to the carbon substituent at the stereogenic centre and not anti- to the oxygen substituent as has been suggested for related systems.3b,4a,b,5,6e,7,8,13 Intriguingly, an AM1 study of the zwitterionic Claisen reaction of 11 to give 12 (Scheme 1) also appears to show a preference for reaction anti- to the carbon substituent but no comment is made by the authors on the conformation of the transition states located.4c
A possible explanation for the preferred reactive conformation is that stabilisation of the transition state occurs most effectively not through interaction of the newly forming σ-bond with a low-lying σ* orbital but rather through a Cíeplak effect19 in which a σ-bonding orbital at the stereogenic centre interacts with the low-lying antibonding orbital of the incipient σ-bond. While the C–H bond is likely to be the best σ-donor, the conformations in which this bond is antiperiplanar to the newly forming σ-bond (Ic and IIc) are also likely to be the most sterically hindered; reaction anti- to the C–C bond is thus a compromise between σ-donor ability and steric hindrance.
For non-brominated substrate 27a, the energy of transition state Ib is slightly lower than that of IIb – possibly due to the steric interactions between the pyrroline ring and the allylic methoxy substituent in conformer IIb.
In the case of brominated substrate 27b, however, transition state Ib is greatly destabilised by the increased A1,3-strain resulting from introduction of the bromine atom; this leaves IIb as the lowest energy transition state structure.
If Z-allylic bromide 20a and its vinyl bromide congener 20b also react with Cíeplak diastereoselection, the corresponding chair conformations should be III and IV, depicted in Fig. 6. In conformation III, A1,3-strain is minimised and this should thus represent the dominant reaction pathway regardless of the identity of R. Indeed, the major product observed for both substrates is 23, corresponding to reaction through conformation III.
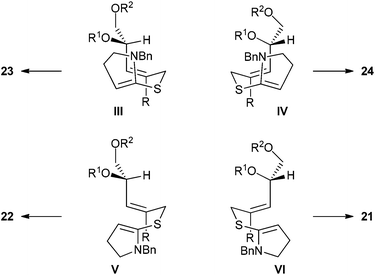 | ||
| Fig. 6 Possible chair (top) and boat (bottom) conformations of the N,S-ketene acetal intermediates derived from 20. | ||
It is clear from inspection of structures III and IV that significant destabilising interactions could exist between the allylic substituents and the pyrroline ring; these interactions would be removed in the boat conformations V and VI. Of the two possible boat conformations, we would expect V to be favoured over VI due to the minimisation of allylic strain and hence product 22 should be favoured over 21.
Experimentally, significant amounts of products arising from boat transition states are seen for both substrates. For compound 20b, as expected, product 22b predominates over 21b while for compound 20a, the products 21a and 22a are formed in equal amounts. The reasons for this lack of selectivity between the competing boat transition states when R = H are unclear.
Conclusions
Moderate to high levels of diastereoselectivity can be obtained in the thia-Claisen rearrangement of allyl vinyl sulfides derived from chiral allylic bromides and N-benzylpyrrolidine-2-thione. By installing an additional bromine atom on the alkene of the allylic bromide, the stereochemical preference of the reaction may be reversed. These results are summarised in the upper part of Scheme 7: the major isomers obtained from the reaction of bromides 18a and 18b have different relative stereochemistry, while those from the reactions of 20a and 20b have the same stereochemistry.Density functional theory calculations suggest that the preferred reactive conformation positions a C–C bond at the existing stereogenic centre antiperiplanar to the newly forming C–C bond, consistent with the operation of a Cíeplak effect. Cíeplak-type diastereoselection in [3,3]-sigmatropic rearrangements has been reported previously. Betson and Fleming20 found that Ireland–Claisen rearrangement of silyl ketene acetal 28 proceeded through the conformation shown (reaction antiperiplanar to the electropositive silyl substituent) to give 29 in a 93:7 diastereomeric ratio (Scheme 6). Similarly, Yamazaki et al.21 showed that Ireland–Claisen rearrangement of 30 proceeded with bond formation antiperiplanar to the isopropyl rather than the trifluoromethyl substituent, while Yadav et al.22 found that Johnson–Claisen rearrangement of allylic alcohol 32 occurred solely on the alkene face which is anti- to the sulfur of the oxathiolane ring, yielding 33. The preference for this orientation of reaction, together with considerations of allylic strain and other steric interactions, allow us to rationalise the observed stereoselectivities, as shown schematically in the lower part of Scheme 7. In the absence of a vinylic bromide substituent, the intermediate arising from compound 18a reacts preferentially through the conformation shown, with the allylic C–O bond approximately eclipsing the alkene and the allylic C–H bond projecting towards the pyrroline portion of the molecule. When a vinylic substituent is present that is syn- to the allylic stereocentre, allylic strain becomes the dominant factor. Hence the diene intermediate derived from vinylic bromide substrate 18b adopts the reactive conformation shown, in which the allylic C–H bond eclipses the alkene such that A1,3-strain is minimised. A similar effect is seen in the reactions of allylic bromides 20a and 20b; here, the steric interaction is with the allylic methylene group rather than the bromine atom and so both bromides react with the same sense of stereoinduction.
![Cíeplak diastereoselection in [3,3]-sigmatropic rearrangements.20–22](/image/article/2013/OB/c3ob41580e/c3ob41580e-s6.gif) | ||
| Scheme 6 Cíeplak diastereoselection in [3,3]-sigmatropic rearrangements.20–22 | ||
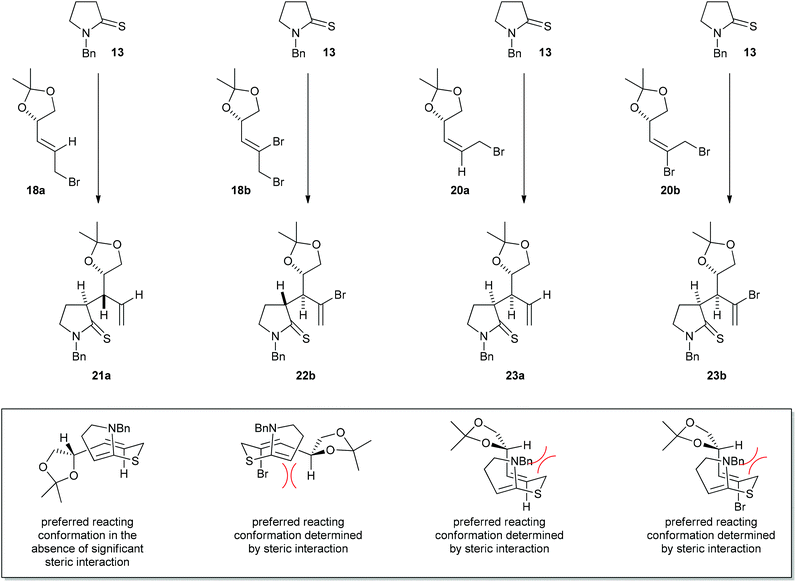 | ||
| Scheme 7 Summary of major products and reactive conformations. | ||
Experimental section
For general experimental procedures, see the ESI.†Thia-Claisen rearrangement: general procedure
A mixture of thioamide 13 (116 mg, 0.61 mmol), allylic bromide (0.67 mmol), MeCN (1 mL) and 4 Å molecular sieves (250 mg) was stirred under an argon atmosphere for 4 d. Further MeCN (2 mL) was added and the mixture warmed to 40 °C. Et3N (94 μL, 0.67 mmol) was added and the resulting solution stirred at 35 °C for 7 h. The mixture was cooled to room temperature, diluted with CH2Cl2 (30 mL) and washed with 2% citric acid (2 × 50 mL). The combined aqueous washings were extracted with CH2Cl2 (50 mL), and the combined organic layers dried (MgSO4) and concentrated in vacuo to give the crude product. Individual components were isolated by flash chromatography (SiO2; petrol–EtOAc 19:1).:3); [α]17D −5.6 (c 0.68, CHCl3); νmax/cm−1 (film) 3067, 2931 (CH), 1638 (CC), 1605, 1585; 1H NMR (CDCl3, 500 MHz) δ 1.21 (3H, s) and 1.34 (3H, s, C(CH3)2), 2.13 (1H, dtd, J 13.0, 9.2, 5.5 Hz) and 2.30 (1H, ddt, J 13.0, 9.0, 6.5 Hz, NCH2CH2), 3.05 (1H, dt, J 9.2, 3.3 Hz, CH2CHCH), 3.15 (1H, ddd, J 9.7, 6.5, 3.8 Hz, CSCH), 3.47 (1H, ddd, J 11.0, 8.9, 6.3 Hz) and 3.62 (1H, m, NCH2CH2), 3.64 (1H, t, J 7.9 Hz) and 4.05 (1H, dd, J 8.0, 6.4 Hz, OCH2), 4.29 (1H, ddd, J 7.8, 6.4, 2.8 Hz, OCH), 4.83 (1H, d, J 14.2 Hz, NCHHPh), 5.21–5.28 (3H, m, CH2CH and NCHHPh), 5.96 (1H, ddd, J 17.3, 10.2, 9.2 Hz, CH2CH), 7.30–7.36 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ 22.4 (NCH2CH2), 25.4 and 26.3 (C(CH3)2), 47.7 (CH2CHCH), 51.8 (NCH2Ph), 52.9 (NCH2CH2), 58.0 (CSCH), 68.0 (OCH2), 74.5 (OCH), 109.1 (CMe2), 118.7 (CH2CH), 128.0, 128.3 and 128.8 (aromatic CH), 135.0 (aromatic C), 135.1 (CH2CH), 205.4 (CS); m/z (CI+) 332 (MH+, 31%), 316 (24), 275 (37), 230 (80), 191 (100), 91 (40); HRMS found 332.1677, C19H26NO2S (MH+) requires 332.1684.:3); [α]22D +82.2 (c 0.60, CHCl3); νmax/cm−1 (CHCl3 cast) 2983, 2931, 2873 (CH), 1637 (CC), 1503, 1452 (CS); 1H NMR (CDCl3, 500 MHz) δ 1.38 (3H, s) and 1.42 (3H, s, C(CH3)2), 1.89 (1H, ddt, J 12.8, 8.8, 6.3 Hz) and 2.21 (1H, dtd, J 12.8, 9.1, 5.9 Hz, NCH2CH2), 2.62 (1H, m, CH2CHCH), 3.43 (1H, ddd, J 10.7, 9.0, 6.1 Hz, CSCH), 3.50–3.55 (2H, m, NCH2CH2), 3.60 (1H, dd, J 8.2, 6.6 Hz) and 3.98 (1H, dd, J 8.2, 6.1 Hz, OCH2), 4.94 (1H, d, J 14.4 Hz, NCHHPh), 5.00 (1H, m, OCH), 5.05 (1H, d, J 14.4 Hz, NCHHPh), 5.09 (1H, dd, J 10.2, 1.6 Hz) and 5.19 (1H, dd, J 17.2, 1.0 Hz, CH2CH), 5.79 (1H, dt, J 17.2, 9.9 Hz, CH2CH), 7.29–7.35 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ 24.4 (NCH2CH2), 25.7 and 27.0 (C(CH3)2), 51.5 (NCH2Ph), 52.5 (CSCH), 53.0 (CH2CHCH), 54.1 (NCH2CH2), 68.6 (OCH2), 75.0 (OCH), 109.2 (C(Me)2), 119.1 (CH2CH), 127.9, 128.3 and 128.7 (aromatic CH), 134.9 (CH2CH), 135.2 (aromatic C), 202.6 (CS); m/z (CI+) 332 (MH+, 22%), 274 (100); HRMS found 332.1676, C19H26NO2S (MH+) requires 332.1684.:3); [α]22D −27.7 (c 1.35, CHCl3); νmax/cm−1 (CDCl3 cast) 2981, 2920 (CH), 1644 (CC); 1H NMR (CDCl3, 500 MHz) δ 1.36 (3H, s) and 1.48 (3H, s, C(CH3)2), 1.97 (1H, ddt, J 13.2, 8.5, 7.5 Hz) and 2.17 (1H, dddd, J 13.2, 9.3, 7.8, 5.6 Hz, NCH2CH2), 3.35 (1H, td, J 8.9, 3.0 Hz, CH2CHCH), 3.45–3.54 (3H, m, CSCH and NCH2CH2), 3.75 (1H, dd, J 7.9, 5.7 Hz) and 4.06 (1H, dd, J 7.9, 6.0 Hz, OCH2), 4.07 (1H, dt, J 9.6, 6.0 Hz, OCH), 4.90 (1H, d, J 14.3 Hz) and 5.11 (1H, d, J 14.3 Hz, NCH2Ph), 5.13 (1H, dd, J 10.5, 1.4 Hz) and 5.24 (1H, ddd, J 17.3, 1.7, 1.0 Hz, CH2CH), 5.57 (1H, ddd, J 17.3, 10.5, 8.4 Hz, CH2CH), 7.28–7.34 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ 20.4 (NCH2CH2), 25.7 and 27.0 (C(CH3)2), 49.5 (CH2CHCH), 51.7 (NCH2Ph), 52.7 (NCH2CH2), 54.6 (CSCH), 68.6 (OCH2), 75.7 (OCH), 109.4 (C(Me)2), 120.1 (CH2CH), 127.9, 128.3 and 128.7 (aromatic CH), 132.9 (CH2CH), 135.1 (aromatic C), 203.9 (CS); m/z (EI) 331 (M+, 16%), 316 (14), 91 (12), 65 (19), 55 (21), 51 (100); HRMS found 331.1605, C19H25NO2S (M+) requires 331.1601.:15); [α]20D −33.5 (c 0.65, CHCl3); νmax/cm−1 (CHCl3 cast) 2985, 2883 (CH), 1625 (CC), 1507, 1452, 1311 (CS); 1H NMR (CDCl3, 500 MHz) δ 1.25 (3H, s) and 1.42 (3H, s, C(CH3)2), 2.20 (1H, dddd, J 13.4, 8.8, 7.6, 6.9 Hz) and 2.31 (1H, dtd, J 13.4, 8.9, 4.6 Hz, NCH2CH2), 3.37 (1H, m, CSCH), 3.49 (1H, ddd, J 11.1, 8.5, 6.9 Hz, NCHHCH2), 3.59–3.64 (2H, m, CH2C(Br)CH and NCHHCH2), 3.89 (1H, t, J 7.9 Hz) and 4.02 (1H, dd, J 8.0, 6.5 Hz, OCH2), 4.43 (1H, dt, J 7.6, 6.1 Hz, OCH), 4.88 (1H, d, J 14.3 Hz) and 5.16 (1H, d, J 14.3 Hz, NCH2Ph), 5.69 (1H, d, J 1.8 Hz) and 6.08 (1H, dd, J 1.8, 0.5 Hz, CH2CBr), 7.30–7.37 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ 22.7 (NCH2CH2), 25.1 and 26.5 (C(CH3)2), 52.1 (NCH2Ph), 52.5 (NCH2CH2), 52.8 (CH2C(Br)CH), 56.2 (CSCH), 67.2 (OCH2), 74.6 (OCH), 109.1 (C(Me)2), 120.9 (CH2CBr), 128.1, 128.4, 128.9 (aromatic CH), 131.7 (CBr), 135.0 (aromatic C), 201.6(CS); m/z (CI+) 410/412 (MH+, 24/26%), 338 (79), 141 (100); HRMS found 410.0774, C19H2579BrNO2S (MH+) requires 410.0789.:15); [α]20D +31.5 (c 1.08, CHCl3); νmax/cm−1 (CHCl3 cast) 2983, 2934, 2874 (CH), 1625 (CC), 1499, 1452, 1316 (CS); 1H NMR (CDCl3, 600 MHz) δ 1.31 (3H, s) and 1.37 (3H, s, C(CH3)2), 2.31–2.41 (2H, m, NCH2CH2), 3.22 (1H, t, J 8.6 Hz, CHCS), 3.52 (1H, ddd, J 10.8, 8.4, 7.1 Hz) and 3.66 (1H, ddd, J 10.8, 8.9, 5.2 Hz, NCH2CH2), 3.76 (1H, dd, J 8.5, 6.9 Hz, CHHO), 3.89 (1H, dd, J 10.1, 1.6 Hz, H2CC(Br)CH), 4.11 (1H, dd, J 8.5, 6.0 Hz, CHHO), 4.45 (1H, dt, J 10.1, 6.5 Hz, CHO), 4.82 (1H, d, J 14.6 Hz) and 5.19 (1H, d, J 14.6 Hz, PhCH2), 5.54 (1H, dd, J 1.9, 0.5 Hz) and 5.93 (1H, d, J 1.9 Hz, CCH2), 7.29–7.36 (5H, m, ArH); 13C NMR (CDCl3, 150 MHz) δ 22.1 (NCH2CH2), 25.9 and 26.4 (C(CH3)2), 51.8 (PhCH2), 52.7 (NCH2CH2), 55.1 (H2CC(Br)CH), 56.8 (CHCS), 68.6 (CH2O), 74.6 (CHO), 110.0 (C(CH3)2), 119.8 (CCH2), 127.8, 128.1 and 128.7 (aromatic CH), 133.3 (CCH2), 135.1 (aromatic C), 203.5 (CS); m/z (CI+, CH4) 410/412 (MH+, 16/14%), 352/354 ([MH − Me2CO]+, 46/50), 338 (63), 330 ([MH − HBr]+, 100); HRMS found 410.0799, C19H2579BrNO2S (MH+) requires 410.0789.:2); νmax/cm−1 (CHCl3 cast) 2984, 2932, 2877 (CH), 1615 (CC), 1507, 1453 (CS); 1H NMR (CDCl3, 500 MHz) δ 1.38 (3H, s) and 1.50 (3H, s, C(CH3)2), 2.19 (1H, dtd, J 13.2, 9.5, 6.0 Hz) and 2.42 (1H, ddt, J 13.2, 8.8, 6.1 Hz, NCH2CH2), 3.48–3.70 (4H, m, CSCH, CH2C(Br)CH and NCH2CH2), 3.82 (1H, dd, J 8.6, 5.1 Hz) and 4.16 (1H, dd, J 8.6, 6.0 Hz, OCH2), 4.38 (1H, dt, J 9.2, 5.6 Hz, OCH), 4.92 (1H, d, J 14.4 Hz) and 5.09 (1H, d, J 14.4 Hz, NCH2Ph), 5.56 (1H, d, J 1.6 Hz) and 6.06 (1H, d, J 1.6 Hz, CH2CBr), 7.30–7.36 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ 20.5 (NCH2CH2), 25.5 and 27.0 (C(CH3)2), 51.9 (NCH2Ph), 52.7 (CH2C(Br)CH), 53.0 (NCH2CH2), 54.9 (CSCH), 68.0 (OCH2), 76.0 (OCH), 109.9 (C(Me)2), 123.5(CH2CBr), 128.0, 128.4 (aromatic CH), 128.5 (CBr), 128.7 (aromatic CH), 135.0 (aromatic C), 202.8 (CS); m/z (CI+) 410/412 (MH+, 2/2%), 183 (100) 141 (17), 119 (18); HRMS found 410.0786, C19H2579BrNO2S (MH+) requires 410.0789.:3); 1H NMR (CDCl3, 600 MHz) δ 1.41 (3H, s) and 1.46 (3H, s, C(CH3)2), 2.00–2.48 (2H, m, NCH2CH2), 2.97 (1H, m), 3.52 (1H, m) and 3.65–3.71 (2H, m, CSCH, CH2C(Br)CH and NCH2CH2), 3.87 (1H, dd, J 8.0, 7.6 Hz) and 4.14 (1H, dd, J 8.0, 5.8 Hz, OCH2), 4.49 (1H, m, OCH), 4.92 (1H, d, J 14.3 Hz) and 5.08 (1H, d, J 14.3 Hz, NCH2Ph), 5.61 (1H, d, J 1.4 Hz) and 5.98 (1H, d, J 1.4 Hz, CH2CH(Br)), 7.30–7.36 (5H, m, aromatic CH).:1); [α]17D −14.0 (c 0.88, CHCl3); νmax/cm−1 (film) 2932 (CH), 1630 (CC); 1H NMR (CDCl3, 500 MHz) δ −0.02 (3H, s), 0.06 (3H, s), 0.07 (3H, s) and 0.07 (3H, s, 2 × Si(CH3)2), 0.86 (9H, s) and 0.91 (9H, s, 2 × C(CH3)3), 2.05–2.19 (2H, m, NCH2CH2), 2.79 (1H, dt, J 9.5, 4.8 Hz, CH2CHCH), 3.30 (1H, m, CSCH), 3.39 (1H, ddd, J 11.0, 8.6, 6.4 Hz) and 3.51 (1H, ddd, J 11.0, 8.7, 5.7 Hz, NCH2CH2), 3.63 (1H, dd, J 10.5, 4.8 Hz) and 3.82 (1H, dd, J 10.5, 4.3 Hz, OCH2), 4.30 (1H, q, J 4.7 Hz, OCH), 4.86 (1H, d, J 14.3 Hz, NCHHPh), 5.06–5.11 (2H, m, CH2CH), 5.14 (1H, J 14.3 Hz, NCHHPh), 5.95 (1H, m, CH2CH), 7.30–7.36 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ −5.4, −5.3, −4.6 and −3.7 (2 × Si(CH3)2), 18.2 and 18.3 (2 × C(CH3)3), 24.5 (NCH2CH2), 26.0 (2 × C(CH3)3), 49.6 (CH2CHCH), 51.5 (NCH2Ph), 52.2 (NCH2CH2), 56.0 (CSCH), 66.0 (OCH2), 72.8 (OCH), 117.8 (CH2CH), 127.9, 128.3 and 128.7 (aromatic CH), 135.3 (aromatic C), 136.8 (CH2CH), 203.3 (CS); m/z (CI+) 520 (MH+, 18%), 504 (25), 462 (44), 388 (100), 330 (11), 230 (54), 191 (33), 91 (34); HRMS found 520.3115, C28H50NO2SSi2 (MH+) requires 520.3101.:1); [α]20D +14.0 (c 0.3, CHCl3); νmax/cm−1 (CHCl3 cast) 2928 (CH), 1624 (CC); 1H NMR (CDCl3, 500 MHz) δ 0.01 (3H, s), 0.03 (3H, s), 0.18 (3H, s) and 0.23 (3H, s, 2 × Si(CH3)2), 0.90 (9H, s) and 0.92 (9H, s, C(CH3)3), 1.85 (1H, ddt, J 12.9, 8.9, 6.6 Hz) and 2.19 (1H, dtd, J 12.9, 9.1, 5.4 Hz, NCH2CH2), 2.43 (1H, td, J 9.9, 2.0 Hz, CH2CHCH), 3.37 (1H, ddd, J 10.9, 8.8, 6.6 Hz) and 3.45 (1H, m, NCH2CH2), 3.48 (1H, dd, J 10.7, 4.1 Hz, OCHH), 3.64 (1H, m, CSCH), 3.67 (1H, dd, J 10.7, 2.6 Hz, OCHH), 4.88 (1H, m, OCH), 4.90 (1H, d, J 14.4 Hz, NCHHPh), 5.03 (1H, dd, J 10.2, 2.3 Hz, CHHCH), 5.07 (1H, d, J 14.4 Hz, NCHHPh), 5.08 (1H, dd, J 17.2, 2.3 Hz, CHHCH), 5.91 (1H, dt, J 17.2, 10.2 Hz, CH2CH), 7.28–7.33 (5H, m, ArH); 13C NMR (CDCl3, 125 MHz) δ −5.5, −5.4, −4.1 and −4.0 (2 × Si(CH3)2), 18.3 and 18.4 (2 × C(CH3)3), 25.9 and 26.0 (2 × C(CH3)3), 26.2 (NCH2CH2), 51.2 (NCH2Ph), 52.1 (NCH2CH2), 52.3 (CSCH), 53.1 (CH2CHCH), 66.2 (OCH2), 72.8 (OCH), 118.4 (CH2CH), 127.8, 128.2 and 128.6 (aromatic CH), 135.4 (aromatic C), 135.9 (CH2CH), 202.8 (CS); m/z (CI+) 520 (MH+, 100%), 504 (44), 388 (42); HRMS found 520.3110, C28H50NO2SSi2 (MH+) requires 520.3101.Acknowledgements
We thank Dr Abil Aliev for assistance with NMR spectroscopy, and Mr John Hill and Dr Lisa Haigh for mass spectrometry. We also thank UCL Graduate School and EPSRC (grant no EP/C509153/1) for funding.Notes and references
- (a) A. M. Martin Castro, Chem. Rev., 2004, 104, 2939 CrossRef PubMed; (b) The Claisen Rearrangement, ed. M. Hiersemann and U. Nubbemeyer, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For reviews of asymmetric [3,3]-sigmatropic rearrangements, see: (a) D. Enders, M. Knopp and R. Schiffers, Tetrahedron: Asymmetry, 1996, 7, 1847 CrossRef CAS; (b) H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 29, 43 RSC; (c) U. Nubbemeyer, Synthesis, 2003, 961 CAS.
- (a) J. R. Hauske and S. M. Julin, Tetrahedron Lett., 1993, 34, 4909 CrossRef CAS; (b) J. Mulzer and M. Shanyoor, Tetrahedron Lett., 1993, 34, 6545 CrossRef CAS.
- (a) U. Nubbemeyer, R. Öhrlein, J. Gonda, B. Ernst and D. Belluš, Angew. Chem., Int. Ed. Engl., 1991, 30, 1465 CrossRef; (b) U. Nubbemeyer, J. Org. Chem., 1996, 61, 3677 CrossRef CAS PubMed; (c) J. Gonda, M. Martinková, B. Ernst and D. Belluš, Tetrahedron, 2001, 57, 5607 CrossRef CAS.
- J. K. Cha and S. C. Lewis, Tetrahedron Lett., 1984, 25, 5263 CrossRef CAS.
- For closely related Johnson–Claisen rearrangements, see: (a) T. Kametani, T. Suzuki, M. Nishimura, E. Sato and K. Unno, Heterocycles, 1982, 19, 205 CrossRef CAS; (b) T. Suzuki, E. Sato, S. Kamada, H. Tada, K. Unno and T. Kametani, J. Chem. Soc., Perkin Trans. 1, 1986, 387 RSC; (c) S. Takano, A. Kurotaki, M. Takahashi and K. Ogasawara, J. Chem. Soc., Perkin Trans. 1, 1987, 91 RSC; (d) H. Nemoto, A. Satoh, M. Ando and K. Fukumoto, J. Chem. Soc., Perkin Trans. 1, 1991, 1309 RSC; (e) S. Hatakeyama, K. Saijo and S. Takano, Tetrahedron Lett., 1985, 26, 865 CrossRef CAS; (f) J. Mulzer, K.-D. Graske and B. Kirste, Liebigs Ann. Chem., 1988, 891 CrossRef CAS; (g) K. Tadano, M. Minami and S. Ogawa, J. Org. Chem., 1990, 55, 2108 CrossRef CAS.
- S. Désert and P. Metzner, Tetrahedron, 1992, 48, 10327 CrossRef.
- D. Craig, J. W. Harvey, A. G. O'Brien and A. J. P. White, Chem. Commun., 2010, 46, 6932 RSC.
- Y. Yang, F. Zheng and F.-L. Qing, Tetrahedron, 2011, 67, 3388 CrossRef CAS PubMed.
- (a) K. C. Majumdar, S. Ghosh and M. Ghosh, Tetrahedron, 2003, 59, 7251 CrossRef CAS; (b) S. Perrio, V. Reboul, C. Alayrac and P. Metzner, in The Claisen Rearrangement, ed. M. Hiersemann and U. Nubbemeyer, Wiley-VCH, Weinheim, 2007, ch. 9, pp. 431–459 Search PubMed.
- A. J. P. Mortimer, P. S. Pang, A. E. Aliev, D. A. Tocher and M. J. Porter, Org. Biomol. Chem., 2008, 6, 2941 Search PubMed.
- A preliminary report of this research has been published: A. R. Ellwood, A. J. P. Mortimer, D. A. Tocher and M. J. Porter, Synlett, 2008, 2199 CAS [Erratum: Synlett, 2009, 3052].
- S. D. Kahn and W. J. Hehre, J. Org. Chem., 1988, 53, 301 CrossRef CAS.
- (a) M. Chérest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 2199 CrossRef; (b) N. T. Anh and O. Eisenstein, Tetrahedron Lett., 1976, 155 CrossRef; (c) N. T. Anh, Top. Curr. Chem., 1980, 88, 145 CrossRef.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841 CrossRef CAS.
- For previous computational studies on thioamide-derived thia-Claisen rearrangements, see: R. Arnaud and Y. Vallée, J. Chem. Soc., Perkin Trans. 2, 1997, 2737 RSC.
- Structure diagrams were generated using CYLView: C. Y. Legaut, CYLView v1.0.374β, Université de Sherbrooke, Quebec, Canada, 2010 Search PubMed.
- Global minima were also located for substrates 27a and 27b by a process of conformational searching and DFT geometry optimisation. These led to the following values for the free energy of activation for each substrate. 27a: 85.8 kJ mol−1 (gas phase) and 80.0 kJ mol−1 (solution phase); 27b: 94.8 kJ mol−1 (gas phase) and 100.2 kJ mol−1 (solution phase).
- A. S. Cíeplak, J. Am. Chem. Soc., 1981, 103, 4540 CrossRef.
- M. S. Betson and I. Fleming, Org. Biomol. Chem., 2003, 1, 4005 CAS.
- T. Yamazaki, N. Shinohara, T. Kitazume and S. Sato, J. Org. Chem., 1995, 60, 8140 CrossRef CAS.
- V. K. Yadav, D. A. Jeyaraj, M. Parvez and R. Yamdagni, J. Org. Chem., 1999, 64, 2928 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the synthesis of allylic bromides 18a–20b and of stereochemical assignments of thia-Claisen products; NMR spectra for all new compounds synthesised. Details of computational studies, with Cartesian coordinates and energies of transition states Ia–IIc and Ia*–IIc* for substrates 27a and 27b. See DOI: 10.1039/c3ob41580e |
| ‡ The structures in Fig. 3 are depicted for clarity with a substituent at the allylic stereocentre eclipsing the vinylic substituent R; the conformations in which the new σ-bond is formed antiperiplanar to an existing substituent are obtained by a rotation of 30° about the C(sp2)–C(sp3) bond. |
| This journal is © The Royal Society of Chemistry 2013 |