 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of absolute configuration of the phosphonic acid moiety of fosfazinomycins†
Katharina Schiessla, Alexander Rollerb and Friedrich Hammerschmidt*a
aUniversity of Vienna, Institute of Organic Chemistry, Währingerstraße 38, 1090, Vienna, Austria. E-mail: friedrich.hammerschmidt@univie.ac.at; Fax: +43 (0)1 4277 9521; Tel: +43 (0)1 4277 52105
bUniversity of Vienna, Institute of Inorganic Chemistry, Währingerstraße 42, 1090, Vienna, Austria
First published on 9th September 2013
Abstract
Fosfazinomycins A and B produced by Streptomyces lavendofoliae share the same phosphonate moiety with one chiral centre of unknown configuration which was determined by synthesising both enantiomers of 2-hydroxy-2-phosphonoacetic acid methyl ester. A chiral cyclic phosphite was reacted with methyl glyoxylate in a Pudovik reaction to give a pair of diastereomeric α-hydroxyphosphonates, which were separated by HPLC. The configurations at C-2 were assigned on the basis of single crystal X-ray structure analysis. Deprotection of these diastereomers furnished the enantiomeric α-hydroxyphosphonic acids, of which the (S)-configured had the same sign of optical rotation as the phosphonic acid moiety of the two fosfazinomycins.
Introduction
Phosphonates and phosphinates are organic compounds characterised by either one or two phosphorus–carbon bonds. Over the past few decades they have been studied extensively and are nowadays used in medicine and agriculture.1 They have interesting biological properties,2 which can be attributed to their structural similarity to phosphoric acid esters and carboxylic acids, as well as to the high stability of the incorporated P–C bonds.3,4They can be found both free and bound to structural components such as lipids or proteins. As conjugates with macromolecules they either enhance the structural rigidity of the latter or protect them against enzymatic degradation.5 There is a steadily growing, fascinating group of about a dozen small molecules of natural origin containing a P–C bond, some of which are bioactive.1,6 Their properties range from antibacterial, antiviral or antibiotic to pesticidal and enzyme inhibitory.
Fosfomycin (1) is a clinically used antibiotic,1,7 phosphinothricin (2) a commercially important and very potent herbicide,8 fosmidomycin (3) an antimalarial agent9 and 2-amino-1-hydroxyethylphosphonic acid (4)10,11 is a component of the lipophosphonoglycan of the plasma membrane of Acanthamoeba castellanii (Fig. 1).
 | ||
| Fig. 1 Naturally occurring P–C compounds. | ||
The current work deals with two other members of the group of small bioactive phosphonates, namely fosfazinomycins A (5a) and B (5b) (Scheme 1). They were first isolated in 1983 from the fermentation broth of Streptomyces lavendofoliae and are active against some filamentous fungi.12
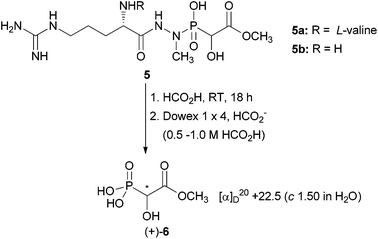 | ||
| Scheme 1 Partial hydrolysis of fosfazinomycins A and B to get phosphonic acid (+)-6 of unknown configuration. | ||
Structure elucidation revealed fosfazinomycin to be a mixture of two components, A and B. They both contain L-arginine as well as a unique phosphonohydrazine moiety.13,14 The latter somehow relates them to FR-900137, an antibacterial antibiotic particularly active against Escherichia coli.15 Fosfazinomycin A differs from B by containing L-valine attached to the α-amino group of L-arginine. Furthermore, they share the same α-hydroxyphosphonate moiety with one stereogenic centre. The corresponding free phosphonic acid has been isolated by acid hydrolysis of fosfazinomycins and purification by ion exchange chromatography. Its optical rotation was determined {[α]20D +22.5 (c 1.50 in H2O)}, but the absolute configuration remained elusive.16 The purpose of this work was to synthesise both enantiomers of 2-hydroxy-2-phosphonoacetic acid methyl ester (6) of known absolute configuration. Their specific optical rotation will allow assigning the configuration to the natural product. This information might be helpful in unravelling the biosynthesis of fosfazinomycins.
Results and discussion
Synthetic challenges
P–C bonds in phosphonates are generally chemically very stable towards cleavage by bases and acids. However, α-hydroxyphosphonates are chemically labile.17 Their formation from aldehyde and phosphite and cleavage to the same compounds are catalysed by a base. Although chiral α-hydroxyphosphonate 6 looks very simple, we anticipated some obstacles during its synthesis. First, racemisation can interfere if the chiral, nonracemic hydroxyphosphonate is treated with a base. Second, the stereogenic centre here is base-labile as the α-hydrogen is acidified by the ester and the phosphonate group, irrespective of whether it is protected or not. Third, the base can induce an α-hydroxyphosphonate–phosphate rearrangement18 assisted by the ester group, which itself can be hydrolysed. To avoid these problems, very mild reaction conditions and catalytic removal of protecting group(s) from phosphorus at the end were mandatory.Original synthetic strategy
Initially, we envisaged to generate racemic dibenzyl α-hydroxyphosphonate (±)-9, possibly separable by HPLC on a chiral stationary phase. Catalytic removal of the protecting groups would give the enantiomeric 2-hydroxy-2-phosphonoacetic acid methyl esters (R)- and (S)-(6) in the final step (Scheme 2). | ||
| Scheme 2 Initial route to prepare α-hydroxyphosphonic acids (R)- and (S)-6. | ||
Therefore, methyl glyoxylate (7) was prepared from glyoxylic acid monohydrate and methyl dimethoxyacetate by a literature procedure.19 It was reacted immediately in a Pudovik reaction20 with dibenzyl phosphite (8) at −78 °C in the presence of Et3N as a base catalyst to give the desired racemic α-hydroxyphosphonate (±)-9 in moderate yield (64%). Unfortunately, the two enantiomers could not be separated by HPLC on a Chiralcel OD-H column using various mixtures of iso-propanol–hexanes.
Revised strategy and synthesis
Therefore the synthetic strategy had to be changed. We decided to prepare a cyclic phosphite of known absolute configuration, which will yield a pair of diastereomeric cyclic α-hydroxyphosphonates upon reaction with methyl glyoxylate. Flash column chromatography and deprotection would give the free phosphonic acids (R)- and (S)-6.Thus, C2-symmetric (R,R)-1,2-diphenylethane-1,2-diol (11)21 was heated at 80 °C with commercially available bis(2,2,2-trifluoroethyl) phosphite (10)22 in dry pyridine hoping to get cyclic phosphite (R,R)-12 (Scheme 3). As the phosphorus atom in this case is a chirotopic, non-stereogenic center,23 only two diastereomers would be formed with methyl glyoxylate compared to four, when not using a C2-symmetric diol. However, only polymeric material resulted upon transesterification for unknown reasons. Increasing the reaction temperature to 120 °C or replacing phosphite 10 by diethyl phosphite did not yield cyclic five-membered phosphite (R,R)-12 either.
 | ||
| Scheme 3 Revised strategy for the synthesis of (R)- and (S)-6. | ||
Assuming that a cyclic six-membered phosphite would be more favourable, (R,R)-1,2-diphenylethane-1,2-diol was replaced by (R*,R*)-1,3-diphenylpropane-1,3-diol [(R*,R*)-14]. First trials were performed using the racemic diol (±)-14, which was readily available.24 Transesterification proceeded quantitatively within 1.5 h at room temperature in pyridine as judged by NMR spectroscopy (Scheme 4).
 | ||
| Scheme 4 Revised strategy for the synthesis of (R)- and (S)-6. | ||
As it proved to be useful, we tried to prepare diol (R,R)-14 by a literature procedure from (S)-2-chloro-1-phenylethanol via the dilithiated species, which was added to benzaldehyde. Contrary to the literature, which reported only the (R,R)-product to be formed in 79% yield, our (R,R)-14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :meso-14 ratio was 1.0:0.8 and the yield was low (35%).25 As it could not be improved, enantioselective reduction of dibenzoylmethane (17) with Ru(p-cymene)[(R,R)-Ts-DPEN] (18) seemed to be an attractive alternative to get this chiral diol (Scheme 5).26 This asymmetric transfer hydrogenation worked best with a mixture of 4.4 equiv. of HCO2H and 2.6 equiv. of Et3N as a hydrogen source and the yield of the crystallised product was 71% (ee >99% by chiral HPLC).27
:meso-14 ratio was 1.0:0.8 and the yield was low (35%).25 As it could not be improved, enantioselective reduction of dibenzoylmethane (17) with Ru(p-cymene)[(R,R)-Ts-DPEN] (18) seemed to be an attractive alternative to get this chiral diol (Scheme 5).26 This asymmetric transfer hydrogenation worked best with a mixture of 4.4 equiv. of HCO2H and 2.6 equiv. of Et3N as a hydrogen source and the yield of the crystallised product was 71% (ee >99% by chiral HPLC).27
 | ||
| Scheme 5 Highly enantioselective preparation of (R,R)-14. | ||
Having the enantiomerically pure diol (R,R)-14 in hand, it was used to generate the cyclic phosphite (R,R)-15 which was added to methyl glyoxylate (7) at −78 °C in a Pudovik reaction20 catalysed by Et3N (Scheme 4). When the reaction mixture was allowed to warm to −50 °C in the cooling bath, the two diastereomeric α-hydroxyphosphonates 16a and 16b were formed. Addition of an equimolar amount of CF3CO2H relative to Et3N, extractive workup and flash chromatography gave an inseparable mixture of the two diastereomers (in a 1:1 ratio) in low (50–55%) yield. Replacing CF3CO2H by CH3SO3H to neutralise the amine caused a lower yield. However, when the transformation was finished (as can be easily determined by NMR spectroscopy) and the reaction mixture was directly applied to the silica gel column for flash chromatography without the addition of an acid, the diastereomeric mixture of 16a and 16b was obtained in good (78%) yield. Unfortunately, the two diastereomers could not be separated by flash column chromatography, but only by preparative HPLC (CH2Cl2–EtOAc = 1:1; tR (16a) 7.8 min, tR (16b) 9.9 min). Crystallisation of both diastereomers from CH2Cl2 produced crystals suitable for single crystal X-ray structure analysis (Fig. 2 and 3). Diastereomers 16a and 16b were found to have (S)- and (R)-configuration, respectively, at the carbon atom bearing the hydroxyl group.
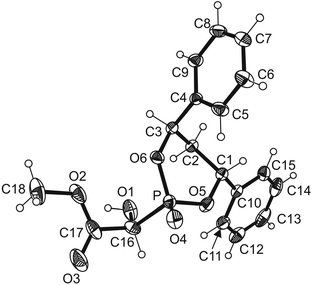 | ||
| Fig. 2 ORTEP view of the molecule 16a with thermal ellipsoids drawn at the 50% probability level. | ||
 | ||
| Fig. 3 ORTEP view of one crystallographically independent molecule of 16b with thermal ellipsoids drawn at the 50% probability level. | ||
These α-hydroxyphosphonates were used for the next and final step in the reaction sequence, removal of the diol protecting group from phosphorus by hydrogenolysis in MeOH. The 1,3-diphenylpropane formed was removed by extraction with hexanes. Concentration of the methanolic solutions furnished the α-hydroxyphosphonates (R)- and (S)-6 in sufficient purity for collecting the analytical data. Their specific optical rotations were [α]20D −28.8 (c 1.63 in H2O) and [α]20D +29.6 (c 1.40 in H2O), respectively. Surprisingly, the enantiomers 6 are configurationally stable. The optical rotation of an aqueous solution of (R)-6 did not change when left at room temperature for 8 days. However, this product racemised completely after 16 h at pH 7–8. The optical rotation measured after 1 h {[α]20D −19.7 (c 0.95 in H2O)} decreased within the next 15 h to [α]20D −0.3 (c 0.95 in H2O).
Acidic hydrolysis of fosfazinomycins, followed by ion exchange chromatography, furnished the free phosphonic acid moiety with [α]20D +22.5 (c 1.50 in H2O).16 By comparison with the synthetic samples, it was concluded that it has (S)-configuration and has evidently partly racemised during the cleavage process. The finding that the only other known α-hydroxyphosphonate of biological origin, which is 2-amino-1-hydroxyethylphosphonic acid (4), has (R)-configuration will have biosynthetic implications.11
To underpin the consistency between the structure of the α-hydroxyphosphonic acid 6 isolated from fosfazinomycins and the synthetic samples, and to determine the ee of our samples, (R)-6 was esterified with diazomethane in methanol (Scheme 6).
 | ||
| Scheme 6 Esterification of (R)-6 with ethereal diazomethane. | ||
The crude product obtained by concentration of the reaction mixture under reduced pressure was dimethyl phosphonate (R)-19 (ee 99%, by NMR spectroscopy using (R)-(+)-t-butyl(phenyl)monothiophosphinic acid as a chiral shift reagent)28,29 in admixture with a small amount of a compound tentatively assigned the structure of methyl ether (R)-20 (molar ratio of (R)-19:(R)-20 = 88:12). They were separated by flash chromatography. The NMR spectrum of homogeneous (R)-19 was identical to that of an authentic sample of (±)-19,30 prepared by base-catalysed addition of dimethyl phosphite to methyl glyoxylate in 72% yield, but its ee was only 31%. Partial racemisation evidently occurred during flash chromatography on silica gel by removal of the fairly acidic proton α to phosphorus. To prove the formation of methyl ether (R)-20 during esterification, its racemate was synthesised by etherification of racemic methyl 2-(dimethoxyphosphinyl)-2-hydroxyacetate [(±)-19] with CH2N2 in the presence of HBF4·OEt2 as a catalyst31 (Scheme 7). The product yield was poor (29%), but sufficient for collecting the necessary analytical data and proving that (R)-20 was formed as a side product during the esterification of (R)-6.
 | ||
| Scheme 7 Synthesis of (±)-20 from (±)-19. | ||
Experimental
General experimental
1H, 13C and 31P NMR spectra were recorded in CDCl3, d4-methanol and d8-toluene using a Bruker Avance DRX 400 (1H: 400.13 MHz, 13C: 100.61 MHz, 31P: 161.98 MHz), AV 400 (1H: 400.27 MHz, 13C: 100.65 MHz, 31P: 162.03 MHz) or DRX 600 (1H: 600.13 MHz) spectrometer. Chemical shifts were referenced to residual CHCl3 (δH 7.24), CHD2OD (δH 3.31) or C6D5CD2H (δH 2.09) and CDCl3 (δC 77.23), CD3OD (δC 49.15) and external H3PO4 (85%). Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. IR spectra were run using a Bruker VERTEX 70 IR spectrometer as ATR spectra. Optical rotations were measured at 20 °C using a Perkin-Elmer 341 polarimeter in a 1 dm cell. [α]D values are given in 10−1 deg cm2 g−1. Analytical HPLC for the determination of the ee of (R,R)-14 was performed on a Jasco system (PU-980 pump, UV 975 and RI 930) using a Chiralcel OD-H column, Ø 0.46 cm × 25 cm. Preparative HPLC for the separation of 16a and 16b was performed using a Dynamix Model SD-1 equipped with a Model UV-1 absorbance detector using a Nucleosil 50–5 column, Ø 3.2 cm × 25 cm. Melting points were determined using a Leica Galen III Reichert Thermovar instrument and were uncorrected.TLC was carried out on 0.25 mm thick Merck plates with silica gel 60 F254. Spots were visualised by UV and/or dipping the plate into a solution of (NH4)6Mo7O24·4H2O (25.0 g) and Ce(SO4)2·4H2O (1.0 g) in 10% aqueous H2SO4 (500 mL), followed by heating with a heat gun. Flash (column) chromatography was performed with Merck silica gel 60 (230–400 mesh).
Pyridine was dried by refluxing over powdered CaH2, followed by distillation and storage over molecular sieves (4 Å). Dichloromethane was dried by passing through aluminium oxide 90 active neutral (0.063–0.200 mm, activity I) and stored over molecular sieves (3 Å). Et2O was refluxed over LiAlH4, THF over potassium and distilled prior to use. Bis(2,2,2-trifluoroethyl) phosphite was distilled under reduced pressure (b.p. 48–50 °C/9 mm; lit.,22 43–44 °C/2 mm). All other chemicals were used as purchased from Sigma-Aldrich, Acros, Fluka or Merck.
:1, Rf 0.57). α-Hydroxyphosphonate (±)-9 (445 mg, 64%) was obtained as colourless crystals; mp 43–45 °C (CH2Cl2/hexanes); found: C, 58.28; H, 5.71; O, 27.55. Calc. for C17H20O6P: C, 58.29; H, 5.47; O, 27.40%; νmax/cm−1 3311, 2930, 2855, 1753, 1513, 1461, 1379, 1249, 1091, 1037; δH (400.27 MHz, CDCl3) 3.32–3.47 (1H, br m, OH), 3.76 (3H, s, OCH3), 4.58 (1H, dd, JHP 15.9, JHH 7.0, P–OCH3), 5.04–5.17 (4H, m, 2 × CH2), 7.27–7.38 (10H, m, Ph); δC (100.65 MHz, CDCl3) 53.58 (OCH3), 69.08 (d, JCP 6.9, CH2), 69.30 (d, JCP 155.8, CH–P), 69.31 (d, JCP 6.9, CH2), 128.21 (d, JCP 2.9, Ph), 128.77 (Ph), 128.80 (Ph), 135.96 (d, JCP 5.8, Ph), 136.02 (d, JCP 5.9, Ph), 169.96 (d, JCP 1.2, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); δP (162.03 MHz, CDCl3) 16.15; HRMS: (ESI) observed [M + H]+ 351.0990, calculated for C17H20O6P+ 351.0992.:2 (Rf 0.40) to EtOAc), followed by crystallisation from iPrOH to give diol (R,R)-14 (799 mg, 70%) as slightly reddish crystals; ee >99% before and after crystallisation (by analytical HPLC on chiral stationary phase; 10% iPrOH–hexanes, tR (S,S) 10.52 min, tR (R,R) 12.16 min). The product was directly used for the next step. A small sample was further purified by HPLC for characterisation to give colourless needles; mp 150–152 °C (from iPrOH); [α]20D +72.15 (c 0.79 in methanol) {lit.,33 for (S,S)-enantiomer: [α]20D −72 (methanol)}; νmax/cm−1 3382, 3295, 1454, 1400, 1201, 1061, 1023; δH (400.27 MHz, CDCl3) 2.16 (1H, d, J 6.1, CH2), 2.17 (1H, d, J 6.1, CH2), 2.74 (2H, br s, 2 × OH), 4.97 (2H, t, J 6.1, 2 × CH–O), 7.22–7.37 (10H, m, Ph); δC (100.65 MHz, CDCl3) 46.75 (CH2), 71.99 (2C, 2 × CH–O), 125.84 (4C, Ph), 127.72 (2C, Ph), 128.73 (4C, Ph), 144.40 (2C, Ph).:2, Rf 0.22) to obtain a mixture of the two diastereomeric α-hydroxyphosphonates 16a and 16b (1.888 g, 78%) as an oil. The ratio of 16a to 16b was 1.16:1.00 (by 31P NMR).
O); δP (162.03 MHz, CDCl3) 16.15; HRMS: (ESI) observed [M + H]+ 351.0990, calculated for C17H20O6P+ 351.0992.:2 (Rf 0.40) to EtOAc), followed by crystallisation from iPrOH to give diol (R,R)-14 (799 mg, 70%) as slightly reddish crystals; ee >99% before and after crystallisation (by analytical HPLC on chiral stationary phase; 10% iPrOH–hexanes, tR (S,S) 10.52 min, tR (R,R) 12.16 min). The product was directly used for the next step. A small sample was further purified by HPLC for characterisation to give colourless needles; mp 150–152 °C (from iPrOH); [α]20D +72.15 (c 0.79 in methanol) {lit.,33 for (S,S)-enantiomer: [α]20D −72 (methanol)}; νmax/cm−1 3382, 3295, 1454, 1400, 1201, 1061, 1023; δH (400.27 MHz, CDCl3) 2.16 (1H, d, J 6.1, CH2), 2.17 (1H, d, J 6.1, CH2), 2.74 (2H, br s, 2 × OH), 4.97 (2H, t, J 6.1, 2 × CH–O), 7.22–7.37 (10H, m, Ph); δC (100.65 MHz, CDCl3) 46.75 (CH2), 71.99 (2C, 2 × CH–O), 125.84 (4C, Ph), 127.72 (2C, Ph), 128.73 (4C, Ph), 144.40 (2C, Ph).:2, Rf 0.22) to obtain a mixture of the two diastereomeric α-hydroxyphosphonates 16a and 16b (1.888 g, 78%) as an oil. The ratio of 16a to 16b was 1.16:1.00 (by 31P NMR).The diastereomers were separated by preparative HPLC (CH2Cl2–EtOAc 1:1, tR (16a) 7.8 min, tR (16b) 9.9 min) and crystallised. A single crystal of each diastereomer was picked directly from the solution for X-ray structure analysis.
O); δP (161.98 MHz, CDCl3) 9.48; HRMS: (ESI) observed [M + Na]+ 385.0820, calculated for C18H19O6PNa+ 385.08169.O); δP (161.98 MHz, CDCl3) 9.95; HRMS: (ESI) observed [M + Na]+ 385.0817, calculated for C18H19O6PNa+ 385.08169.O); δP (161.98 MHz, d4-methanol) 15.31.Similarly, diastereomer 16b (100 mg) was converted to α-hydroxyphosphonic acid (R)-6 of opposite configuration (43 mg, 90%); [α]20D −28.8 (c 1.63 in H2O); the spectroscopic data were identical to that of the (S)-enantiomer.
:3, Rf 0.26) and crystallisation (EtOAc–Et2O) to yield colourless needles (712 mg, 72%); mp 60–63 °C (lit.,30 59–61 °C); νmax/cm−1 3269, 2961, 1754, 1440, 1247, 1185, 1108, 1028; δH (400.27 MHz, CDCl3) 3.29 (1H, br s, OH), 3.84 (3H, d, JHP 10.8, P–OCH3), 3.85 (3H, d, JHP 10.7, P–OCH3), 3.88 (3H, d, JHP 0.5, CO2CH3), 4.57 (1H, d, JHP 16.0, CH–P); δC (100.65 MHz, CDCl3) 53.81 (CO2CH3), 54.26 (d, JCP 6.8, P–OCH3), 54.60 (d, JCP 6.8, P–OCH3), 68.76 (d, JCP 156.1, CH–P), 170.06 (CO); δP (162.02 MHz, CDCl3) 17.70.:3, Rf 0.38) as a colourless solid (29 mg, 49%); ee 34% (by 31P/1H NMR using the chiral shift reagent); [α]20D −10.64 (c 1.10 in methanol). The spectroscopic data agreed with that for the racemic compound (±)-19.:3, Rf 0.49) to obtain (±)-20 (9 mg, 28%) as a colourless solid; νmax/cm−1 1744, 1443, 1259, 1190, 1119, 1027; δH (400.27 MHz, CDCl3) 3.50 (3H, d, J 0.4, CH–OCH3), 3.82 (3H, s, CO2CH3), 3.82 (3H, d, JHP 10.9, P–OCH3), 3.84 (3H, d, JHP 10.9, P–OCH3), 4.24 (1H, d, JHP 18.5, CH–P); δC (100.65 MHz, CDCl3) 53.04 (CH–OCH3), 54.32 (d, JCP 7.1, P–OCH3), 54.40 (d, JCP 7.3, P–OCH3), 60.75 (d, JCP 13.0, CO2CH3), 78.20 (d, JCP 158.2, CH–P), 167.74 (d, JCP 1.6, CO); δP (162.02 MHz, CDCl3) 16.35; HRMS: (ESI) observed [M + H]+ 213.05217, calculated for C6H14O6P+ 213.05280.Conclusions
The first chemical synthesis of both enantiomers of α-hydroxyphosphonic acid 6 was accomplished. A synthetic route taking advantage of a chirotopic, non-stereogenic phosphorus centre in a cyclic phosphite was chosen to generate two diastereomeric α-hydroxyphosphonates in a Pudovik reaction. The diastereomer with (S)-configuration at the stereogenic centre bearing the hydroxyl group was converted to the dextrorotary α-hydroxyphosphonic acid, which was also obtained by partial hydrolysis of the fosfazinomycins A and B. Therefore, the natural α-hydroxyphosphonic acid also has (S)-configuration.Acknowledgements
We greatly acknowledge the financial support by the Austrian Science Fund (FWF) (P19869-N19). We thank S. Felsinger for recording NMR spectra and J. Theiner for performing combustion analyses.Notes and references
- W. W. Metcalf and W. A. van der Donk, Annu. Rev. Biochem., 2009, 78, 65 CrossRef CAS PubMed.
- A. K. White and W. W. Metcalf, Annu. Rev. Microbiol., 2007, 61, 379 CrossRef CAS PubMed.
- S. C. Fields, Tetrahedron, 1999, 55, 12237 CrossRef CAS.
- A. Mucha, P. Kafarski and L. Berlicki, J. Med. Chem., 2011, 54, 5955 CrossRef CAS PubMed.
- F. R. McSorley, P. B. Wyatt, A. Martinez, E. F. DeLong, B. Hove-Jensen and D. L. Zechel, J. Am. Chem. Soc., 2012, 134, 8364 CrossRef CAS PubMed.
- H. Seto and T. Kuzuyama, Nat. Prod. Rep., 1999, 16, 589 RSC.
- F. M. Kahan, J. S. Kahan, P. J. Cassidy and H. Kropp, Ann. N. Y. Acad. Sci., 1974, 235, 364 CrossRef CAS.
- F. Hammerschmidt and H. Kählig, J. Org. Chem., 1991, 56, 2364 CrossRef CAS.
- J. Wiesner, S. Borrmann and H. Jomaa, Parasitol. Res., 2003, 90(Suppl. 2), 71 CrossRef PubMed.
- E. D. Korn, D. G. Dearborn, H. M. Fales and E. A. Sokoloski, J. Biol. Chem., 1973, 248, 2257 CAS.
- F. Hammerschmidt and H. Völlenkle, Liebigs Ann. Chem., 1989, 577 CrossRef CAS.
- S. Gunji, K. Arima and T. Beppu, Agric. Biol. Chem., 1983, 47, 2061 CrossRef CAS.
- T. Ogita, S. Gunji, Y. Fukazawa, A. Terahara, T. Kinoshita, H. Nagaki and T. Beppu, Tetrahedron Lett., 1983, 24, 2283 CrossRef CAS.
- I. J. Kang and Y. J. Kim, Bull. Korean Chem. Soc., 1994, 15, 595 CAS.
- (a) Y. Kuroda, T. Goto, M. Okamoto, M. Yamashita, E. Iguchi, M. Kohsaka, H. Aoki and H. Imanaka, J. Antibiot., 1980, 33, 272 CrossRef CAS; (b) Y. Kuroda, H. Tanaka, M. Okamoto, T. Goto, M. Kohsaka, H. Aoki and H. Imanka, J. Antibiot., 1980, 33, 280 CrossRef CAS.
- A. Terahara, personal communication.
- B. L. Wanner, Biodegradation, 1994, 5, 175 CrossRef CAS.
- F. Hammerschmidt, Monatsh. Chem., 1993, 124, 1063 CrossRef CAS.
- (a) M S. Foster, C. D. Oldham and S. W. May, Tetrahedron: Asymmetry, 2011, 22, 283 CrossRef CAS PubMed; (b) J. M. Hook, Synth. Commun., 1984, 14, 83 CrossRef CAS; (c) T. R. Kelly, T. E. Schmidt and J. G. Haggerty, Synthesis, 1972, 544 CrossRef CAS.
- A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81 CrossRef CAS.
- Z.-M. Wang and K. B. Sharpless, J. Org. Chem., 1994, 59, 8302 CrossRef CAS.
- D. E. Gibbs and C. Larsen, Synthesis, 1984, 410 CrossRef CAS PubMed.
- For an explanation of chirotopic non-stereogenic centers see: K. Mislow and J. Siegel, J. Am. Chem. Soc., 1984, 106, 3319 CrossRef CAS.
- J.-P. Deprés and C. Morat, J. Chem. Educ., 1992, 69, A232 CrossRef.
- J. Barluenga, J. Flórez and M. Yus, J. Chem. Soc., Perkin Trans. 1, 1983, 3019 RSC.
- J. Cossy, F. Eustache and P. I. Dalko, Tetrahedron Lett., 2001, 42, 5005 CrossRef CAS.
- T. Ikariya, S. Hashiguchi, K. Murata and R. Noyori, Org. Synth., 2005, 82, 10 CAS.
- M. Drescher, S. Felsinger, F. Hammerschmidt, H. Kählig, S. Schmidt and F. Wuggenig, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 140, 79 CrossRef CAS.
- E. Zymanczyk-Duda, M. Skwarczynski, B. Lejczak and P. Kafarski, Tetrahedron: Asymmetry, 1996, 7, 1277 CrossRef CAS.
- D. Horne, J. Gaudino and W. J. Thompson, Tetrahedron Lett., 1984, 25, 3529 CrossRef CAS.
- (a) M. Neeman, M. C. Caserio, J. D. Roberts and W. S. Johnson, Tetrahedron, 1959, 6, 36 CrossRef CAS; (b) A. B. Smith III, K. J. Hale, L. M. Laakso, K. Chen and A. Riéra, Tetrahedron Lett., 1989, 30, 6963 CrossRef.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738 CrossRef CAS.
- K. Yamamoto, H. Ando and H. Chikamatsu, J. Chem. Soc., Chem. Commun., 1987, 334 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of the compounds prepared, X-ray data. CCDC 952037 for 16a and 952038 for 16b. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41574k |
| This journal is © The Royal Society of Chemistry 2013 |