 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkaloid inspired spirocyclic oxindoles from 1,3-dipolar cycloaddition of pyridinium ylides†
Jonathan Daya,
Maliha Uroosa,
Richard A. Castledinea,
William Lewisa,
Ben McKeever-Abbasb and
James Dowden*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: james.dowden@nottingham.ac.uk; Fax: +44 (0)115 9513565; Tel: +44 (0)115 9513566
bPharmaceutical Development, AstraZeneca, Silk Road Business Park, Macclesfield, UK
First published on 15th August 2013
Abstract
Cycloaddition reactions between pyridinium ylides and 3-alkenyl oxindoles that proceed in high yield and with very good regio- and diastereoselectivity are reported. The resulting cycloadducts have the same stereochemistry of biologically active oxindole alkaloids, such as strychnofoline.
Introduction
Natural products and their signature fragments are an enduring resource for identifying biological modulators.1 A number of biologically active alkaloids, such as strychnofoline and isorhynchophylline (Fig. 1), feature spiroindolizidine oxindoles.2,3 This fragment is considered ‘privileged’ for potential therapeutic investigation and there is much interest in developing expedient synthesis of such structures.4 | ||
| Fig. 1 Example spirooxindole alkaloids. | ||
Target spiroindolizidineoxindole structures may be efficiently accessed by 1,3-dipolar cycloaddition of 3-alkylideneindoline-2-ones.5 Serov et al. described reactions between N-phenylacylquinolinium ylides with 3-alkylidene oxindoles,6 while extended studies of this type of reaction have recently been reported.7 The latter cycloadducts have a relatively high molecular weight due to additional aromatic rings and it is not easy to envisage strategies for their transformation into natural products or drug-like scaffolds.
There are no literature descriptions of corresponding 1,3-dipolar cycloadditions of pyridinium ylides to 3-alkylidene oxindoles, yet such cycloadducts would be attractive for access to spirooxindole alkaloids and possible therapeutics. Cycloaddition reactions of pyridinium ylides have previously been reported but in situ oxidation is commonly used, leading to valuable unsaturated indolizines.8 Of course, such oxidations destroy the rich stereochemical information accumulated during the cycloaddition. Early investigation of general pyridinium ylide cycloadditions noted limited stability of the tetrahydroindolizine cycloadducts and this may have discouraged further investigation of these products.9 The 1,2-dihydropyridine motif embedded within tetrahydroindolizine cycloadducts is generally regarded as unstable with few exceptions,10 although there have been exciting developments in unlocking their synthetic potential.11
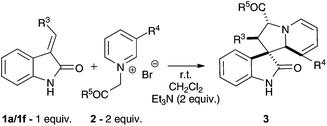
This report demonstrates that cycloaddition reactions between pyridinium ylides 2 (Table 1) and 3-alkylidene oxindoles 1 proceed with good selectivity to produce generally isolable spirotetrahydroindolizineoxindole cycloadducts 3 in good yield.
|
|||
|---|---|---|---|
| Solvent | Base | Time (h) | Yielda (%) |
| a Yield after chromatography. | |||
| Toluene | Et3N | 3 | 65 |
| EtOAc | Et3N | 4 | 64 |
| CH2Cl2 | Et3N | 2 | 88 |
| DMF | Et3N | 1 | 61 |
| EtOH | Et3N | 4 | 40 |
| CH2Cl2 | DBU | 3 | 73 |
| CH2Cl2 | tBuOK | 3 | 71 |
| THF | NaH | 2 | 82 |

Results and discussion
Starting materials for this route are readily accessible by olefination of isatin to give 3-alkylidene-2-oxindoles;12† pyridinium salts were obtained by alkylation.13† Pyridinium salts featuring N-methylene groups attached to an electron withdrawing group can be readily deprotonated using mild base to give the ylide.14,15Initially, we set out to briefly investigate the role of solvent and base on yield. An excess of pyridinium salt 2a relative to dipolarophiles 1a was used to suppress further cycloaddition onto the initial tetrahydroindolizine product.9
Generally, high yields of cycloadduct were obtained using a variety of solvents and base at room temperature, although suspending the reagents in dichloromethane, then initiating the reaction by addition of triethylamine is convenient (Table 1). The cycloadduct 3a is stable to chromatography and could be stored under argon in the freezer for no more than a week.
Cycloaddition between the ylide derived from 3-bromopyridinium salt 2a and oxindole 1a appears to be highly regio- and diastereoselective, giving a single product 3a (Table 1). 1H-NMR spectroscopy of the cycloadduct revealed an apparent triplet for the 5′ proton (J = 7.0 Hz) indicative of reaction at the C-2 position of the pyridinium salt. Initial NOESY analysis revealed enhancements between protons corresponding to 1′ ring junction and 2′ position adjacent to the spirocentre, but not between the 1′ and 3′ positions adjacent to nitrogen.
Ultimately, single crystal X-ray diffraction of this material gave unequivocal evidence of the diastereoselectivity of the reaction (Fig. 2).‡ The relative stereochemistry of the spirocentre (position 8a′) and the 1′ position is diagnostic of exo or endo selectivity; co-location of the oxindole carbonyl and 1′ proton on the same face of the cycloadduct is suggestive of an attractive interaction between the electron rich oxindole aromatic ring and the electron deficient pyridinium in the transition state. The trans arrangement of the 1′ and 3′ protons is also confirmed, indicating that the ylide is S-shaped in the transition state (Scheme 1). This relative stereochemistry is the same as that required for alkaloids related to strychnofoline and isorhynchophylline.
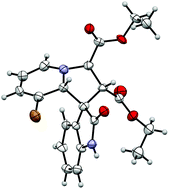 | ||
| Fig. 2 Crystal structure of cycloadduct 3a (CCDC 927103). Ellipsoids are drawn at the 50% probability level. | ||
 | ||
| Scheme 1 Proposed origin of observed diastereoselectivity. | ||
We next carried out investigation into the generality of the reaction using various 3-alkylideneoxindoles 1b–f (Table 2).11† Cycloaddition with the ylide derived from 3-bromopyridinium salt 2a generally proceeded to give good yields (80–90%) of cycloadducts 3b–f as single diastereoisomers with relative stereochemistry similar to cycloadduct 3a as judged by 1H NMR spectroscopy. 3-Methylene oxindole 1f is a stable solid that readily undergoes cycloaddition reactions in good yield (entry 5). Unfortunately, N-substituted 3-methylene oxindoles are highly reactive and generally met with polymerisation before cycloaddition could be attempted.16 On the other hand, N-propargyl and N-acetyl substituted oxindoles (entries 3 and 4, 1d and 1e respectively) bearing an ethyl ester gave good yields of cycloadducts (3d and 3e respectively).
|
|||
|---|---|---|---|
| Entry | Oxindole | Cycloadduct | Yielda (%) |
| a Yield after chromatography. | |||
| 1 |  |
 |
93 |
| 2 |  |
 |
91 |
| 3 |  |
 |
82 |
| 4 |  |
 |
83 |
| 5 |  |
 |
81 |

A selection of pyridinium salts (2g–l) were also evaluated (Table 3). Cycloadducts arising from reaction with unsubstituted pyridinium salt (2l) could be detected by mass spectrometry of the reaction mixture but were not stable to purification by chromatography, however the saturated indolizidine 3l could be obtained by in situ reduction using RANEY® nickel in good yield.
|
|||
|---|---|---|---|
| Entry | Pyridinium salt | Cycloadduct | Yielda (%) [ratio] |
| a Yield after chromatography.b Inseparable mixture of two diastereoisomers, ratio in parenthesis from relative integration of 1H NMR spectrum.c Crude cycloadduct subject to H2/RANEY® nickel (∼10 mol%) in EtOAc. | |||
| 1 |  |
 |
77 [2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1]b :1]b |
| 2 |  |
 |
95 [7:3]b |
| 3 |  |
 |
81 [1:1]b |
| 4 |  |
 |
76 |
| 5 |  |
 |
87 |
| 6 | 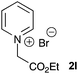 |
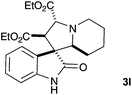 |
63c |
Pyridinium salts featuring methyl, or phenyl substituents at the 3- or 4-pyridinium position (not shown) did not give observable products in their respective cycloaddition reactions using these condition, which we presume to be due to their instability.
Pleasingly, less acidic pyridinium salts featuring either nitrile (2m) or phenyl (2n) ylide stabilising groups could be deprotonated with sodium hydride, leading to good yields of the corresponding cycloadducts 3m and 3n respectively as single diastereoisomers (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: a, NaH (2 equiv.), THF, rt. | ||
Pyridinium salts featuring resonance stabilising groups (2g–i) in the 3-position gave good overall yields of spiroindolizidine oxindoles arising from reaction at the C-6 position of the pyridinium ring, i.e. opposite to the electron withdrawing substituent. These products (3g–i) were obtained as a mixture of two diastereoisomers in varying ratios.
Crystals were obtained from the 3-cyanopyridine derived cycloadduct 3g that X-ray diffraction revealed to feature the 1′ proton on the opposite face to the oxindole carbonyl (Fig. 3).‡ This product was later confirmed as the minor diastereoisomer 3g′ by 1H NMR spectroscopy.
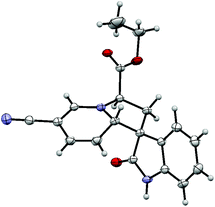 | ||
| Fig. 3 Crystal structure of the minor diastereoisomer of cycloadduct 3g′ (CCDC 927102). Ellipsoids are drawn at the 50% probability level. | ||
There are two possible explanations for these observed results. One is that pyridinium resonance stabilising groups influence the orientation of the transition state. Alternatively, interconversion of cycloadducts via ring opening to restore the pyridinium ring, then rotation about the former 2′–8′a bond and rejoining of the enolate with the pyridinium may proceed.17 An analogous reversible Mannich reaction mechanism is well known to occur in either acidic or basic media, leading to interchange between related spirocyclic oxindole alkaloid diastereoisomers, such as isorhynchophylline and rhynchophylline.18 Diastereoisomer 3g′ remained unchanged when exposed to the same cycloaddition conditions for 14 hours, however.19
A recent paper described reactions of very closely related pyridinium ylides and 3-alkylideneoxindoles in ethanol with 20 mol% triethylamine at 50 °C, but alkene products arising from elimination of pyridine were the exclusive reported products.20 A similar reaction product was obtained when the cycloaddition reaction between pyridinium ylide 2l with 3-alkylideneoxindole 1a was performed in ethanol at room temperature, resulting in clean conversion to the elimination product 5 in 60% yield (Scheme 3). Presumably, the reaction proceeds via 1,4-conjugate addition and not cycloaddition, with subsequent elimination of pyridine.21,22
 | ||
| Scheme 3 Reagents and conditions: a, Et3N (2 equiv.), ethanol, rt, 60%; b, Et3N (2 equiv.), CH2Cl2, rt. | ||
Significantly, crude cycloadduct 4 obtained from cycloaddition in dichloromethane at room temperature did not undergo elimination when stirred in ethanol with triethylamine. This result, when considered alongside the observation for cycloadduct 3g′ above, suggests that the reversible mechanism is not occurring in these cases. Overall, these studies underline that, for these reagents, cycloaddition and 1,4-conjugate addition reaction outcomes are under the subtle influence of solvent.
Conclusions
In summary, 1,3-dipolar cycloadditions between pyridinium ylides and 3-alkenyloxindoles that give spiroindolizidine oxindole products in high yield and with excellent regioselectivity and diastereoselectivity are reported for the first time. These cycloadducts are highly reminiscent of biologically active alkaloids, especially isorhynchophylline and strychnofoline. Specific modification of this general cycloaddition route to facilitate highly convergent access to such alkaloids and related targets is underway.Experimental
General information
Experimental describing precursor preparation is included within the ESI.†![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 6.10 (1H, d, J = 7.0 Hz, CH–CHCBr), 5.06 (1H, d, J = 7.0 Hz, NCH(CO2Et), 5.00 (1H, s, NCHC–Br), 4.45 (1H, app t, J = 7.0 Hz, NCHCH), 4.21 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.65 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 3.45 (1H, d, J = 7.0 Hz, CCH(CO2Et), 1.23 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.62 (3H, t, J = 7.1 Hz, CCO2CH2CH3); NMR δC (100 MHz, d6-DMSO) 176.2 (C), 171.4 (C), 168.4 (C), 144.0 (C), 134.9 (CH), 129.5 (CH), 128.9 (CH), 126.5 (C), 124.7 (CH), 121.5 (CH), 109.9 (CH), 106.9 (C), 93.2 (CH), 72.6 (CH), 64.8 (CH), 64.2 (C), 62.0 (CH2), 61.4 (CH2), 52.8 (CH), 14.5 (CH3), 13.5 (CH3); m/z (HRMS-ESI+) 461.0697 (M + H C21H2279BrN2O5 requires 461.0712), 463.0700 (M + H C21H2281BrN2O5 requires 463.0692).CH), 6.14 (1H, app dt, J = 6.4, 1.0 Hz, CH–CHCBr), 5.20 (1H, d, J = 1.0 Hz, NCHCBr), 5.05 (1H, d, J = 6.6 Hz, NCH(CO2Et), 4.58 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.76 (2H, q, J = 7.1 Hz, CCHCO2CH2CH3), 3.69 (1H, d, J = 6.6 Hz, CCHCO2Et), 1.29 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.78 (3H, t, J = 7.1 Hz, CCHCO2CH2CH3); NMR δC (100 MHz, d6-acetone) 175.3 (C), 170.6 (C), 168.0 (C), 143.0 (C), 134.0 (CH), 131.9 (CH), 129.0 (C), 128.8 (CH), 127.5 (CH), 113.3 (C), 111.3 (CH), 106.8 (C), 93.3 (CH), 72.8 (CH), 64.8 (CH), 64.4 (C), 61.6 (CH2), 61.1 (CH2), 52.8 (CH), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 538.9808 (M + H C21H2179Br2N2O5 requires 538.9812), 540.9806 (M + H C21H2181Br2N2O5 requires 540.9792).CH), 6.15 (1H, d, J = 6.4 Hz, CH–CHCBr), 5.27 (1H, s, NCHC–Br), 5.15 (1H, d, J = 6.5 Hz, NCH(CO2Et)), 4.62 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.28 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.77 (1H, d, J = 6.5 Hz, CCH(CO2Et)), 3.76 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 1.30 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.77 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 176.0 (C), 170.4 (C), 167.9 (C), 149.9 (C), 142.4 (C), 134.0 (CH), 129.1 (CH), 127.5 (C), 126.3 (CH), 120.2 (CH), 109.7 (CH), 106.4 (C), 93.6 (CH), 73.2 (CH), 65.0 (CH), 64.2 (C), 61.7 (CH2), 61.4 (CH2), 52.8 (CH), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 506.0569 (M + H C21H2179BrN3O7 requires 506.0558), 508.0552 (M + H C21H2181BrN3O7 requires 508.0537).CH), 6.08 (1H, dt, J = 6.4, 0.9 Hz, CH–CHCBr), 5.21 (1H, d, J = 0.9 Hz, NCHC–Br), 5.06 (1H, d, J = 6.9 Hz, NCH(CO2Et)), 4.63 (1H, d, J = 17.8, 2.6 Hz, NCHaHb), 4.57 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.50 (1H, d, J = 17.8, 2.5 Hz, NCHaHb), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.74 (3H, s, OCH3), 3.73 (1H, d, J = 6.9 Hz, CCH(CO2Et), 3.72 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 2.80 (1H, dd, J = 2.6, 2.5 Hz, C
CH), 6.10 (1H, d, J = 7.0 Hz, CH–CHCBr), 5.06 (1H, d, J = 7.0 Hz, NCH(CO2Et), 5.00 (1H, s, NCHC–Br), 4.45 (1H, app t, J = 7.0 Hz, NCHCH), 4.21 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.65 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 3.45 (1H, d, J = 7.0 Hz, CCH(CO2Et), 1.23 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.62 (3H, t, J = 7.1 Hz, CCO2CH2CH3); NMR δC (100 MHz, d6-DMSO) 176.2 (C), 171.4 (C), 168.4 (C), 144.0 (C), 134.9 (CH), 129.5 (CH), 128.9 (CH), 126.5 (C), 124.7 (CH), 121.5 (CH), 109.9 (CH), 106.9 (C), 93.2 (CH), 72.6 (CH), 64.8 (CH), 64.2 (C), 62.0 (CH2), 61.4 (CH2), 52.8 (CH), 14.5 (CH3), 13.5 (CH3); m/z (HRMS-ESI+) 461.0697 (M + H C21H2279BrN2O5 requires 461.0712), 463.0700 (M + H C21H2281BrN2O5 requires 463.0692).CH), 6.14 (1H, app dt, J = 6.4, 1.0 Hz, CH–CHCBr), 5.20 (1H, d, J = 1.0 Hz, NCHCBr), 5.05 (1H, d, J = 6.6 Hz, NCH(CO2Et), 4.58 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.76 (2H, q, J = 7.1 Hz, CCHCO2CH2CH3), 3.69 (1H, d, J = 6.6 Hz, CCHCO2Et), 1.29 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.78 (3H, t, J = 7.1 Hz, CCHCO2CH2CH3); NMR δC (100 MHz, d6-acetone) 175.3 (C), 170.6 (C), 168.0 (C), 143.0 (C), 134.0 (CH), 131.9 (CH), 129.0 (C), 128.8 (CH), 127.5 (CH), 113.3 (C), 111.3 (CH), 106.8 (C), 93.3 (CH), 72.8 (CH), 64.8 (CH), 64.4 (C), 61.6 (CH2), 61.1 (CH2), 52.8 (CH), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 538.9808 (M + H C21H2179Br2N2O5 requires 538.9812), 540.9806 (M + H C21H2181Br2N2O5 requires 540.9792).CH), 6.15 (1H, d, J = 6.4 Hz, CH–CHCBr), 5.27 (1H, s, NCHC–Br), 5.15 (1H, d, J = 6.5 Hz, NCH(CO2Et)), 4.62 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.28 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.77 (1H, d, J = 6.5 Hz, CCH(CO2Et)), 3.76 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 1.30 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.77 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 176.0 (C), 170.4 (C), 167.9 (C), 149.9 (C), 142.4 (C), 134.0 (CH), 129.1 (CH), 127.5 (C), 126.3 (CH), 120.2 (CH), 109.7 (CH), 106.4 (C), 93.6 (CH), 73.2 (CH), 65.0 (CH), 64.2 (C), 61.7 (CH2), 61.4 (CH2), 52.8 (CH), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 506.0569 (M + H C21H2179BrN3O7 requires 506.0558), 508.0552 (M + H C21H2181BrN3O7 requires 508.0537).CH), 6.08 (1H, dt, J = 6.4, 0.9 Hz, CH–CHCBr), 5.21 (1H, d, J = 0.9 Hz, NCHC–Br), 5.06 (1H, d, J = 6.9 Hz, NCH(CO2Et)), 4.63 (1H, d, J = 17.8, 2.6 Hz, NCHaHb), 4.57 (1H, dd, J = 7.1, 6.4 Hz, NCHCH), 4.50 (1H, d, J = 17.8, 2.5 Hz, NCHaHb), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.74 (3H, s, OCH3), 3.73 (1H, d, J = 6.9 Hz, CCH(CO2Et), 3.72 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 2.80 (1H, dd, J = 2.6, 2.5 Hz, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 1.30 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.73 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 172.8 (C), 170.7 (C), 167.9 (C), 155.6 (C), 137.0 (C), 133.9 (CH), 128.5 (CH), 126.9 (C), 113.3 (CH), 111.8 (CH), 109.3 (CH), 106.4 (C), 93.3 (CH), 76.9 (C), 72.8 (CH), 72.7 (C), 64.8 (CH), 61.5 (CH2), 61.0 (CH2), 52.3 (CH), 55.1 (CH), 52.3 (CH3), 29.2 (CH2), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 529.0957 (M + H C25H2679BrN2O6 requires 529.0969), 531.0951 (M + H C25H2681BrN2O6 requires 531.0949).CH), 6.11 (1H, d, J = 6.5 Hz, CH–CHCBr), 5.37 (1H, s, NCHC–Br), 5.05 (1H, d, J = 7.3 Hz, NCH(CO2Et)), 4.58 (1H, dd, J = 7.1, 6.5 Hz, NCHCH), 4.29 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.86 (1H, d, J = 7.3 Hz, CCH(CO2Et), 3.69 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 2.66 (3H, s, NCOCH3), 1.31 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.69 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 175.3 (C), 170.4 (C), 170.3 (C), 167.4 (C), 141.5 (C), 133.8 (CH), 129.4 (CH). 129.1 (CH), 125.4 (C), 124.7 (CH), 123.9 (CH), 115.9 (CH), 106.3 (C), 93.5 (CH), 74.3 (CH), 64.5 (CH), 63.8 (C), 61.7 (CH2), 61.2 (CH2), 53.6 (CH), 25.8 (CH3), 13.5 (CH3), 12.7 (CH3); m/z (HRMS-ESI+) 503.0800 (M + H C23H2479BrN2O6 requires 503.0813), 505.0788 (M + H C23H2481BrN2O6 requires 505.0792).CH), 6.07 (1H, d, J = 7.2 Hz, CHCBr), 5.38 (1H, s, CH), 4.62 (1H, app.t, J = 7.2 Hz, NCHCH), 4.44 (1H, dd, J = 8.4, 7.9 Hz, CHCO2Et), 4.29 (2H, q, J = 7.1 Hz, CH2CH3), 2.52 (1H, dd, J = 13.4, 7.9 Hz, CHaHb), 2.22 (1H, dd, J = 13.4, 8.4 Hz, CHaHb), 1.35 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, CDCl3) 178.0 (C), 171.7 (C), 141.1 (C), 132.8 (CH), 130.6 (C), 128.6 (CH), 127.4 (CH), 123.6 (CH), 122.5 (CH), 110.0 (CH), 109.3 (C), 94.9 (CH), 71.5 (CH), 64.1 (CH), 61.8 (CH2), 60.9 (C), 37.7 (CH2), 14.2 (CH3); m/z (HRMS-ESI+) 389.0494 (M + H C18H1879BrN2O3 requires 389.0501), 391.0476 (M + H C18H1881BrN2O3 requires 391.0480).:1 mixture of diastereoisomers, *indicates minor where different from major) 9.65 (1H, br s, NH), 9.50 (1H, br s, NH*), 7.35–7.20 (3H, m, ArH, NCCCH), 7.07–6.95 (2H, m, ArH), 5.80 (1H, app dt, J = 10.0, 1.5 Hz, CNC–CH*CH), 5.67 (1H, app dt, J = 10.1, 1.5 Hz, CNC–CHCH), 5.06 (1H, t, J = 2.1 Hz, NCH*), 4.98–4.93 (2H, m, CHCO2Et, NCH), 4.86 (1H, app t, J = 7.7 Hz, CH*CO2Et), 4.60 (1H, ddd, J = 10.0, 2.1, 1.0 Hz, CHCH*–CH), 4.52 (1H, ddd, J = 10.1, 2.1, 0.8 Hz, CHCH–CH), 4.35–4.24 (2H, m, CH2CH3), 2.57 (1H, dd, J = 13.4, 8.6 Hz, CHa*), 2.48–2.43 (2H, m, CH2CHCO2Et), 2.37 (1H, dd, J = 13.4, 7.7 Hz, CHb*), 1.30 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, d6-acetone) 179.3 (C), 176.5 (C), 171.8 (C), 171.6 (C), 147.8 (CH), 146.6 (CH), 143.5 (C), 142.3 (C), 132.1 (C), 129.8 (CH), 129.4 (CH), 127.9 (C), 126.4 (C), 124.7 (CH), 124.0 (CH), 123.7 (CH), 123.2 (CH), 123.0 (CH), 120.9 (C), 120.7 (C) 111.9 (CH), 111.5 (CH), 110.7 (CH), 110.2 (CH), 78.6 (C), 78.4 (C), 67.8 (CH), 67.4 (CH), 63.9 (CH), 63.2 (CH), 62.4 (CH2), 62.3 (CH2), 37.2 (CH2), 34.9 (CH2), 14.5 (CH3); m/z (HRMS-ESI+) 358.1162 (M + Na C19H17N3NaO3 requires 358.1162).:3 mixture of diastereoisomers, *indicates minor where different from major) 10.63 (1H, s, NH), 10.40 (1H, s, NH*), 7.38 (2H, s, CHCCONH2), 7.27–7.16 (4H, m, ArH), 7.02–6.84 (4H, m, ArH), 6.54 (4H, br s, NH2), 6.28 (1H, dt, J = 10.2, 2.1 Hz, NH2COC–CH*CH), 6.14 (1H, dt, J = 10.2, 2.1 Hz, NH2COC–CHCH), 4.95 (1H, dd, J = 8.8, 7.4 Hz, CHCO2Et), 4.92 (1H, dd, J = 8.8, 7.4 Hz, CH*CO2Et), 4.78 (2H, app dd, J = 2.1, 0.6 Hz, NCH), 4.38 (1H, ddd, J = 10.2, 2.1, 0.6 Hz, CHCH*–CH), 4.35 (1H, ddd, J = 10.2, 2.1, 0.6 Hz, CHCH–CH), 4.24 (4H, m, CH2CH3), 2.47 (1H, dd, J = 13.3, 8.8 Hz, CHa*Hb), 2.35–2.17 (2H, m, CHb*, CHaHb), 1.29 (3H, t, J = 7.1 Hz, CH2CH3*), 1.27 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, d6-DMSO) 179.3 (C), 176.6 (C), 172.2 (C), 172.1 (C), 167.4 (C), 167.3 (C), 143.0 (C), 142.6 (CH), 141.9 (C), 141.5 (CH), 132.0 (C) 129.0 (CH), 128.7 (CH), 128.0 (C) 124.8 (CH), 124.2 (CH), 124.1 (CH), 123.2 (CH), 122.1 (CH), 122.0 (CH) 110.0 (CH), 109.9 (CH), 109.1 (CH), 107.5 (CH), 101.1 (C), 101.0 (C), 68.1 (CH), 67.5 (CH), 63.5 (CH), 62.6 (CH), 61.8 (CH2), 61.7 (CH2), 55.6 (C), 55.4 (C), 36.5 (CH2), 34.3 (CH2), 14.6 (CH3), 14.5 (CH3); m/z (HRMS-ESI+) 354.1442 (M + H C19H20N3O4 requires 354.1454).:1.3 mixture of diastereoisomers, *indicates minor where different from major) 8.35 (1H, s, NH*), 8.21 (1 H, s, NH), 7.47 (1H, s, NCH*CCOCH3), 7.42 (1H, s, NCHCCOCH3), 7.27–6.94 (8H, m, ArH), 6.53 (1H, d, J = 10.3 Hz, (COCH3)CHCH–CH), 6.36 (1H, d, J = 10.3 Hz, (COCH3)CH*CH–CH), 5.18 (1H, t, J = 1.8 Hz, (COCH3)CHCH–CH*), 5.05 (1H, t, J = 1.8 Hz, (COCH3)CHCH–CH), 4.81 (1H, t, J = 7.8 Hz, CHCO2Et), 4.67 (1H, dd, J = 10.3, 1.8 Hz, (COCH3)CHCH–CH) 4.61 (1H, t, J = 7.8 Hz, CH*CO2Et) 4.53 (1H, dd, J = 10.3, 1.8 Hz, (COCH3)CHCH*–CH), 4.38–4.30 (4H, m, CH2CH3), 2.70–2.58 (2H, m, CH–CHaCHb), 2.45–2.33 (2H, m, CH–CHaCHb), 2.18 (3H, s, COCH3*), 2.13 (3H, s, COCH3), 1.41–1.34 (3H, m, CH2CH3); NMR δC (100 MHz, CDCl3) 191.8 (C), 179.6 (C), 176.8 (C), 171.2 (C), 170.6 (C), 146.2 (CH), 144.3 (CH), 141.6 (C), 140.3 (C), 130.7 (C), 129.2 (CH), 128.9 (CH), 126.8 (C), 123.9 (CH), 123.7 (CH), 123.5 (CH), 123.2 (CH), 122.9 (CH), 122.8 (CH), 122.6 (CH), 110.4 (CH), 110.3 (CH), 108.7 (CH), 68.1 (CH), 67.1 (CH), 63.3 (CH), 62.8 (CH), 62.4 (CH2), 62.2 (CH2), 58.8 (C), 58.4 (C), 36.9 (CH2), 34.4 (CH2), 14.2 (CH3); m/z (HRMS-ESI+) 353.1498 (M + H C20H21N2O4 requires 353.1501).CBr), 6.09 (1H, d, J = 7.0 Hz, N–CHCH), 4.88 (1H, s, NCH), 4.70 (1H, t, J = 7.0 Hz, NCHCH), 4.49 (1H, t, J = 6.8 Hz, CHCOCH3), 2.19 (3H, s, COCH3), 2.11–2.06 (2H, m, CHaHb); NMR δC (100 MHz, d6-DMSO) 207.3 (C), 177.7 (C), 142.8 (C), 135.2 (CH), 131.3 (C), 128.7 (CH), 128.2 (CH), 123.8 (CH), 121.8 (CH), 109.9 (CH), 108.0 (C), 93.3 (CH), 71.7 (CH), 70.5 (CH), 61.3 (C), 36.4 (CH2), 26.7 (CH3); m/z (HRMS-ESI+) 359.0370 (M + H C17H1679BrN2O2 requires 359.0395), 361.0359 (M + H C17H1681BrN2O2 requires 361.0375).CH), 5.39–5.34 (1H, m, CH–CHCF), 5.17 (1H, s, NCHC–F), 5.03 (1H, dd, J = 7.4, 1.2 Hz, NCH(CO2Et)), 4.54–4.49 (1H, m, NCHCH), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.81–3.66 (3H, m, NCH(CO2Et)CH(CO2Et), NCH(CO2Et)CH(CO2CH2CH3)), 1.29 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.70 (3H, t, J = 7.1 Hz, NCH(CO2Et)CH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 174.9 (C), 170.9 (C), 168.1 (C), 149.3 (d, JC–F = 252 Hz, C), 143.0 (C), 130.5 (d, JC–F = 4 Hz, CH), 129.0 (CH), 126.6 (C), 124.6 (CH), 121.3 (CH), 109.6 (CH), 103.1 (d, JC–F = 14 Hz, CH), 91.1 (d, JC–F = 5 Hz, CH), 68.0 (d, JC–F = 33 Hz, CH), 65.3 (d, JC–F = 1 Hz, CH), 62.6 (d, JC–F = 3 Hz, C), 61.5 (CH2), 60.9 (CH2), 52.9 (CH), 13.6 (CH3), 12.8 (CH3); m/z (HRMS-ESI+) 401.1507 (M + H C21H22FN2O5 requires 401.1507).
CH), 1.30 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.73 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 172.8 (C), 170.7 (C), 167.9 (C), 155.6 (C), 137.0 (C), 133.9 (CH), 128.5 (CH), 126.9 (C), 113.3 (CH), 111.8 (CH), 109.3 (CH), 106.4 (C), 93.3 (CH), 76.9 (C), 72.8 (CH), 72.7 (C), 64.8 (CH), 61.5 (CH2), 61.0 (CH2), 52.3 (CH), 55.1 (CH), 52.3 (CH3), 29.2 (CH2), 13.6 (CH3), 12.9 (CH3); m/z (HRMS-ESI+) 529.0957 (M + H C25H2679BrN2O6 requires 529.0969), 531.0951 (M + H C25H2681BrN2O6 requires 531.0949).CH), 6.11 (1H, d, J = 6.5 Hz, CH–CHCBr), 5.37 (1H, s, NCHC–Br), 5.05 (1H, d, J = 7.3 Hz, NCH(CO2Et)), 4.58 (1H, dd, J = 7.1, 6.5 Hz, NCHCH), 4.29 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.86 (1H, d, J = 7.3 Hz, CCH(CO2Et), 3.69 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 2.66 (3H, s, NCOCH3), 1.31 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.69 (3H, t, J = 7.1 Hz, CCH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 175.3 (C), 170.4 (C), 170.3 (C), 167.4 (C), 141.5 (C), 133.8 (CH), 129.4 (CH). 129.1 (CH), 125.4 (C), 124.7 (CH), 123.9 (CH), 115.9 (CH), 106.3 (C), 93.5 (CH), 74.3 (CH), 64.5 (CH), 63.8 (C), 61.7 (CH2), 61.2 (CH2), 53.6 (CH), 25.8 (CH3), 13.5 (CH3), 12.7 (CH3); m/z (HRMS-ESI+) 503.0800 (M + H C23H2479BrN2O6 requires 503.0813), 505.0788 (M + H C23H2481BrN2O6 requires 505.0792).CH), 6.07 (1H, d, J = 7.2 Hz, CHCBr), 5.38 (1H, s, CH), 4.62 (1H, app.t, J = 7.2 Hz, NCHCH), 4.44 (1H, dd, J = 8.4, 7.9 Hz, CHCO2Et), 4.29 (2H, q, J = 7.1 Hz, CH2CH3), 2.52 (1H, dd, J = 13.4, 7.9 Hz, CHaHb), 2.22 (1H, dd, J = 13.4, 8.4 Hz, CHaHb), 1.35 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, CDCl3) 178.0 (C), 171.7 (C), 141.1 (C), 132.8 (CH), 130.6 (C), 128.6 (CH), 127.4 (CH), 123.6 (CH), 122.5 (CH), 110.0 (CH), 109.3 (C), 94.9 (CH), 71.5 (CH), 64.1 (CH), 61.8 (CH2), 60.9 (C), 37.7 (CH2), 14.2 (CH3); m/z (HRMS-ESI+) 389.0494 (M + H C18H1879BrN2O3 requires 389.0501), 391.0476 (M + H C18H1881BrN2O3 requires 391.0480).:1 mixture of diastereoisomers, *indicates minor where different from major) 9.65 (1H, br s, NH), 9.50 (1H, br s, NH*), 7.35–7.20 (3H, m, ArH, NCCCH), 7.07–6.95 (2H, m, ArH), 5.80 (1H, app dt, J = 10.0, 1.5 Hz, CNC–CH*CH), 5.67 (1H, app dt, J = 10.1, 1.5 Hz, CNC–CHCH), 5.06 (1H, t, J = 2.1 Hz, NCH*), 4.98–4.93 (2H, m, CHCO2Et, NCH), 4.86 (1H, app t, J = 7.7 Hz, CH*CO2Et), 4.60 (1H, ddd, J = 10.0, 2.1, 1.0 Hz, CHCH*–CH), 4.52 (1H, ddd, J = 10.1, 2.1, 0.8 Hz, CHCH–CH), 4.35–4.24 (2H, m, CH2CH3), 2.57 (1H, dd, J = 13.4, 8.6 Hz, CHa*), 2.48–2.43 (2H, m, CH2CHCO2Et), 2.37 (1H, dd, J = 13.4, 7.7 Hz, CHb*), 1.30 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, d6-acetone) 179.3 (C), 176.5 (C), 171.8 (C), 171.6 (C), 147.8 (CH), 146.6 (CH), 143.5 (C), 142.3 (C), 132.1 (C), 129.8 (CH), 129.4 (CH), 127.9 (C), 126.4 (C), 124.7 (CH), 124.0 (CH), 123.7 (CH), 123.2 (CH), 123.0 (CH), 120.9 (C), 120.7 (C) 111.9 (CH), 111.5 (CH), 110.7 (CH), 110.2 (CH), 78.6 (C), 78.4 (C), 67.8 (CH), 67.4 (CH), 63.9 (CH), 63.2 (CH), 62.4 (CH2), 62.3 (CH2), 37.2 (CH2), 34.9 (CH2), 14.5 (CH3); m/z (HRMS-ESI+) 358.1162 (M + Na C19H17N3NaO3 requires 358.1162).:3 mixture of diastereoisomers, *indicates minor where different from major) 10.63 (1H, s, NH), 10.40 (1H, s, NH*), 7.38 (2H, s, CHCCONH2), 7.27–7.16 (4H, m, ArH), 7.02–6.84 (4H, m, ArH), 6.54 (4H, br s, NH2), 6.28 (1H, dt, J = 10.2, 2.1 Hz, NH2COC–CH*CH), 6.14 (1H, dt, J = 10.2, 2.1 Hz, NH2COC–CHCH), 4.95 (1H, dd, J = 8.8, 7.4 Hz, CHCO2Et), 4.92 (1H, dd, J = 8.8, 7.4 Hz, CH*CO2Et), 4.78 (2H, app dd, J = 2.1, 0.6 Hz, NCH), 4.38 (1H, ddd, J = 10.2, 2.1, 0.6 Hz, CHCH*–CH), 4.35 (1H, ddd, J = 10.2, 2.1, 0.6 Hz, CHCH–CH), 4.24 (4H, m, CH2CH3), 2.47 (1H, dd, J = 13.3, 8.8 Hz, CHa*Hb), 2.35–2.17 (2H, m, CHb*, CHaHb), 1.29 (3H, t, J = 7.1 Hz, CH2CH3*), 1.27 (3H, t, J = 7.1 Hz, CH2CH3); NMR δC (100 MHz, d6-DMSO) 179.3 (C), 176.6 (C), 172.2 (C), 172.1 (C), 167.4 (C), 167.3 (C), 143.0 (C), 142.6 (CH), 141.9 (C), 141.5 (CH), 132.0 (C) 129.0 (CH), 128.7 (CH), 128.0 (C) 124.8 (CH), 124.2 (CH), 124.1 (CH), 123.2 (CH), 122.1 (CH), 122.0 (CH) 110.0 (CH), 109.9 (CH), 109.1 (CH), 107.5 (CH), 101.1 (C), 101.0 (C), 68.1 (CH), 67.5 (CH), 63.5 (CH), 62.6 (CH), 61.8 (CH2), 61.7 (CH2), 55.6 (C), 55.4 (C), 36.5 (CH2), 34.3 (CH2), 14.6 (CH3), 14.5 (CH3); m/z (HRMS-ESI+) 354.1442 (M + H C19H20N3O4 requires 354.1454).:1.3 mixture of diastereoisomers, *indicates minor where different from major) 8.35 (1H, s, NH*), 8.21 (1 H, s, NH), 7.47 (1H, s, NCH*CCOCH3), 7.42 (1H, s, NCHCCOCH3), 7.27–6.94 (8H, m, ArH), 6.53 (1H, d, J = 10.3 Hz, (COCH3)CHCH–CH), 6.36 (1H, d, J = 10.3 Hz, (COCH3)CH*CH–CH), 5.18 (1H, t, J = 1.8 Hz, (COCH3)CHCH–CH*), 5.05 (1H, t, J = 1.8 Hz, (COCH3)CHCH–CH), 4.81 (1H, t, J = 7.8 Hz, CHCO2Et), 4.67 (1H, dd, J = 10.3, 1.8 Hz, (COCH3)CHCH–CH) 4.61 (1H, t, J = 7.8 Hz, CH*CO2Et) 4.53 (1H, dd, J = 10.3, 1.8 Hz, (COCH3)CHCH*–CH), 4.38–4.30 (4H, m, CH2CH3), 2.70–2.58 (2H, m, CH–CHaCHb), 2.45–2.33 (2H, m, CH–CHaCHb), 2.18 (3H, s, COCH3*), 2.13 (3H, s, COCH3), 1.41–1.34 (3H, m, CH2CH3); NMR δC (100 MHz, CDCl3) 191.8 (C), 179.6 (C), 176.8 (C), 171.2 (C), 170.6 (C), 146.2 (CH), 144.3 (CH), 141.6 (C), 140.3 (C), 130.7 (C), 129.2 (CH), 128.9 (CH), 126.8 (C), 123.9 (CH), 123.7 (CH), 123.5 (CH), 123.2 (CH), 122.9 (CH), 122.8 (CH), 122.6 (CH), 110.4 (CH), 110.3 (CH), 108.7 (CH), 68.1 (CH), 67.1 (CH), 63.3 (CH), 62.8 (CH), 62.4 (CH2), 62.2 (CH2), 58.8 (C), 58.4 (C), 36.9 (CH2), 34.4 (CH2), 14.2 (CH3); m/z (HRMS-ESI+) 353.1498 (M + H C20H21N2O4 requires 353.1501).CBr), 6.09 (1H, d, J = 7.0 Hz, N–CHCH), 4.88 (1H, s, NCH), 4.70 (1H, t, J = 7.0 Hz, NCHCH), 4.49 (1H, t, J = 6.8 Hz, CHCOCH3), 2.19 (3H, s, COCH3), 2.11–2.06 (2H, m, CHaHb); NMR δC (100 MHz, d6-DMSO) 207.3 (C), 177.7 (C), 142.8 (C), 135.2 (CH), 131.3 (C), 128.7 (CH), 128.2 (CH), 123.8 (CH), 121.8 (CH), 109.9 (CH), 108.0 (C), 93.3 (CH), 71.7 (CH), 70.5 (CH), 61.3 (C), 36.4 (CH2), 26.7 (CH3); m/z (HRMS-ESI+) 359.0370 (M + H C17H1679BrN2O2 requires 359.0395), 361.0359 (M + H C17H1681BrN2O2 requires 361.0375).CH), 5.39–5.34 (1H, m, CH–CHCF), 5.17 (1H, s, NCHC–F), 5.03 (1H, dd, J = 7.4, 1.2 Hz, NCH(CO2Et)), 4.54–4.49 (1H, m, NCHCH), 4.27 (2H, q, J = 7.1 Hz, NCHCO2CH2CH3), 3.81–3.66 (3H, m, NCH(CO2Et)CH(CO2Et), NCH(CO2Et)CH(CO2CH2CH3)), 1.29 (3H, t, J = 7.1 Hz, NCHCOCH2CH3), 0.70 (3H, t, J = 7.1 Hz, NCH(CO2Et)CH(CO2CH2CH3)); NMR δC (100 MHz, d6-acetone) 174.9 (C), 170.9 (C), 168.1 (C), 149.3 (d, JC–F = 252 Hz, C), 143.0 (C), 130.5 (d, JC–F = 4 Hz, CH), 129.0 (CH), 126.6 (C), 124.6 (CH), 121.3 (CH), 109.6 (CH), 103.1 (d, JC–F = 14 Hz, CH), 91.1 (d, JC–F = 5 Hz, CH), 68.0 (d, JC–F = 33 Hz, CH), 65.3 (d, JC–F = 1 Hz, CH), 62.6 (d, JC–F = 3 Hz, C), 61.5 (CH2), 60.9 (CH2), 52.9 (CH), 13.6 (CH3), 12.8 (CH3); m/z (HRMS-ESI+) 401.1507 (M + H C21H22FN2O5 requires 401.1507).Crude cycloadduct 4 (75 mg) and RANEY® nickel (10 mol%) were dissolved in anhydrous ethyl acetate (4 mL), under argon. The atmosphere was replaced with H2 and the mixture stirred until reduction was complete by TLC analysis and then filtered through a pad of Celite. The filtrate was concentrated in vacuo and purified via column chromatography (EtOAc–petrol; 1/4) to give spiroindolizidine 3l as a yellow oil; 48 mg (63%); IR (νmax/cm−1, CHCl3) 3440, 3198, 2942, 1732, 1621, 1471, 1373, 1339, 1319, 1184, 1097; NMR δH (400 MHz, CDCl3) 8.66 (1H, br s, NH), 7.31 (1H, d, J = 7.6 Hz, ArH), 7.22 (1H, td, J = 7.6, 1.0 Hz, ArH), 7.00 (1H, t, J = 7.6 Hz, ArH), 6.92 (1H, d, J = 7.6 Hz, ArH), 4.59 (1H, d, J = 5.6 Hz, NCHCO2Et), 4.35–4.20 (2H, m, NCH(COCH2CH3)), 4.00 (1H, d, J = 5.6 Hz, NCH(CO2Et)CH), 3.81–3.61 (2H, m, CH(COCH2CH3)), 3.41 (1H, dd, J = 10.9, 2.4 Hz, NCHC)), 3.09 (1H, d, J = 9.5 Hz, CHaHb), 2.45 (1H, t, J = 9.5 Hz, CHaHb), 1.64–1.53 (2H, m, CHaHb, CHaHb), 1.42–1.12 (6H, m, CHaHb, CHaHb, CHaCHb, CH2CH3), 0.74–0.64 (4H, m, CH2CH3, CHaCHb); NMR δC (100 MHz, CDCl3) 178.4 (C), 172.4 (C), 143.1 (C), 131.7 (C), 128.2 (CH), 126.5 (C), 125.8 (CH), 122.2 (CH), 109.4 (CH), 67.7 (CH), 64.1 (CH), 60.9 (CH2), 60.6 (CH2), 60.0 (C), 54.2 (CH), 47.5 (CH2), 25.9 (CH2), 25.3 (CH2), 22.6 (CH2), 14.4 (CH3), 13.3 (CH3); m/z (HRMS-ESI+) 387.1908 (M + H C21H27N2O5 requires 387.1920).
CH), 6.12 (1H, d, J = 6.4 Hz, CH–CHCBr), 5.48 (1H, d, J = 6.4 Hz, NCHCN, 5.25 (1H, s, NCHC–Br), 4.72 (1H, app t, J = 6.4 Hz, NCHCH), 3.77 (1H, d, J = 6.4 Hz, CHCO2CH2CH3), 3.72 (2H, q, J = 7.1 Hz, CCH(CO2CH2CH3)), 0.74 (3H, t, J = 7.1 Hz, NCHCOCH2CH3); NMR δC (100 MHz, d6-DMSO) 176.1 (C), 167.9 (C), 144.6 (C), 132.8 (CH), 130.3 (CH), 129.1 (CH), 126.7 (C), 125.5 (CH), 122.3 (CH), 119.1 (C), 110.6 (CH), 109.1 (C), 96.7 (CH), 72.9 (CH), 63.8 (C), 62.3 (CH2), 55.6 (CH), 54.9 (CH), 13.7 (CH3); m/z (HRMS-ESI+) 436.0273 (M + Na C19H1679BrN3NaO3 requires 436.0273), 438.0256 (M + Na C19H1681BrN3NaO3 requires 438.0256).CH), 6.07 (1H, d, J = 6.7 Hz, CH–CHCBr), 5.43 (1H, d, J = 8.5 Hz, NCHPh, 4.51 (1H, app t, J = 6.7 Hz, CHCHN), 3.78 (1H, dq, J = 10.8, 7.1 Hz, CHaHb), 3.67 (1H, dq, J = 10.8, 7.1 Hz, CHaHb), 3.4 (1H, d, J = 8.5 Hz, CHCO2Et), 0.77 (3H, t, J = 7.1 Hz, CH3); NMR δC (100 MHz, d6-acetone) 176.6 (C), 169.3 (C), 144.5 (C), 143.2 (C), 134.6 (CH), 130.0 (CH), 129.9 (CH), 129.4 (CH), 128.7 (CH), 127.5 (C), 127.2 (CH), 125.6 (CH), 122.2 (CH), 110.4 (CH), 108.3 (C), 94.1 (CH), 73.7 (CH), 68.1 (CH), 64.8 (C), 61.6 (CH2), 59.4 (CH), 13.8 (CH3); m/z (HRMS-ESI+) 465.0800 (M + H C24H2279BrN2O3 requires 465.0808), 467.0786 (M + H C24H2281BrN2O3 requires 465.0793).:1 mixture of diastereoisomers, only major diastereoisomer peaks are quoted) (νmax/cm−1, CHCl3) 3446, 3011, 2986, 1726, 1617, 1470, 1370, 1334, 1295, 1192; NMR δH (400 MHz, CDCl3) 8.38 (1H, s, NH), 7.66 (1H, d, J = 8.0 Hz, ArH), 7.29 (1H, td, J = 8.0, 1.2 Hz, ArH), 6.98 (1H, td, J = 8.0, 1.2 Hz, ArH), 6.85 (1H, d, J = 8.0 Hz, ArH), 4.43 (2H, q, J = 7.2 Hz, CH2CH3), 4.41 (2H, s, CH2CO2Et), 4.22 (2H, q, J = 7.2 Hz, CH2CH3), 1.40 (3H, t, J = 7.2 Hz, CH2CH3), 1.29 (3H, t, J = 7.2 Hz, CH2CH3); NMR δC (100 MHz, CDCl3) 169.7 (C), 169.2 (C), 167.4 (C), 141.0 (C), 135.0 (C), 130.9 (CH), 129.4 (C), 125.1 (CH), 122.3 (CH), 120.8 (C), 109.9 (CH), 61.9 (CH2), 61.2 (CH2), 34.7 (CH2), 14.2 (CH3), 14.0 (CH3); m/z (HRMS-ESI+) 326.0982 ([M + Na] 100%) requires C16H17NO5Na+ 326.0999.Acknowledgements
This work was supported by the EPSRC (MU, EP/J021008/1), AstraZeneca (JD) and the University of Nottingham (RAC).Notes and references
- B. Over, S. Wetzel, C. Grutter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Nat. Chem., 2013, 5, 21–28 CrossRef CAS PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- For recent examples and references: (a) A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schürmann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735–740 CrossRef CAS PubMed; (b) T.-L. Liu, Z.-Y. Xue, H.-Y. Tao and C.-J. Wang, Org. Biomol. Chem., 2011, 9, 1980–1986 RSC.
- A. B. Serov, V. G. Kartsev, Y. A. Aleksandrov and F. M. Dolgushin, Russ. Chem. Bull., 2005, 54, 2432–2436 CrossRef CAS PubMed.
- L. Wu, J. Sun and C.-G. Yan, Org. Biomol. Chem., 2012, 10, 9452–9463 CAS.
- B. X. Wang, X. C. Zhang, J. Li, X. Jiang, Y. F. Hu and H. W. Hu, J. Chem. Soc., Perkin Trans. 1, 1999, 1571–1575 RSC . See ref. 22 also.
- S. Kanemasa, S. Takenaka, H. Watanabe and O. Tsuge, J. Org. Chem., 1989, 54, 420–424 CrossRef CAS.
- S. J. Taylor, A. M. Taylor and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 1681–1685 CrossRef CAS PubMed.
- For recent developments: M. A. Ischay, M. K. Takase, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 2478–2481 CrossRef CAS PubMed and references therein.
- (a) For Wittig olefination for 1a–e: S. W. Duan, Y. Li, Y. Y. Liu, Y. Q. Zou, D. Q. Shi and W. J. Xiao, Chem. Commun., 2012, 48, 5160–5162 RSC; (b) For Peterson route to 1f: T. Watanabe, M. Arisawa, K. Narusuye, M. S. Alam, K. Yamamoto, M. Mitomi, Y. Ozoe and A. Nishida, Bioorg. Med. Chem., 2009, 17, 94–110 CrossRef CAS PubMed.
- A. R. Katritzky, N. E. Grzeskowiak and J. Alvarezbuilla, J. Chem. Soc., Perkin Trans. 1, 1981, 1180–1185 RSC.
- W. G. Phillips and K. W. Ratts, J. Org. Chem., 1970, 35, 3144–3147 CrossRef CAS.
- X.-M. Zhang, F. G. Bordwell, M. Van Der Puy and H. E. Fried, J. Org. Chem., 1993, 58, 3060–3066 CrossRef CAS.
- R. L. Hinman and C. P. Bauman, J. Org. Chem., 1964, 29, 2431–2437 CrossRef CAS.
- For a related synthetic process: B. Viswambharan, K. Selvakumar, S. Madhavan and P. Shanmugam, Org. Lett., 2010, 12, 2108–2111 CrossRef CAS PubMed.
- G. Laus, J. Chem. Soc., Perkin Trans. 2, 1998, 315–317 RSC.
- Diastereoisomers, such as 3g:3g′ could not be separated by column chromatography on silica gel. No change was observed to the diastereomeric ratio in the crude product obtained from cycloaddition reactions performed in various solvents at a range of temperatures.
- Q. Fu and C.-G. Yan, Tetrahedron, 2013, 69, 5841–5849 CrossRef CAS PubMed.
- For related solvent effects: N. E. Dontsova, V. N. Nesterov and A. M. Shestopalov, Tetrahedron, 2013, 69, 5016–5021 CrossRef CAS PubMed.
- A very recent paper describing stepwise reaction of pyridinium ylides with Michael acceptors: D. S. Allgäuer and H. Mayr, Eur. J. Org. Chem., 2013 DOI:10.1002/ejoc.201300784.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental and spectroscopic information. CCDC 927102–927103. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41415a |
‡ Crystal data for 3a. C21H21BrN2O5, M = 507.38, triclinic, a = 8.5537(12), b = 9.0560(14), c = 16.2097(17) Å, U = 1147.4(3) Å3. T = 120(2)K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 8522 reflections measured, 4490 unique with R = 0.0804, wR2 = 0.2241. CCDC 927103. Crystal data for 3g′. C19H17N3O3, M = 335.35, triclinic, a = 8.8865(5), b = 9.3125(3), c = 10.9460(5) Å, U = 826.26(7) Å3. T = 120(2) K, space group P, Z = 2, 15441 reflections measured, 3480 unique with R = 0.0346, wR2 = 0.0903. CCDC 927102. , Z = 2, 8522 reflections measured, 4490 unique with R = 0.0804, wR2 = 0.2241. CCDC 927103. Crystal data for 3g′. C19H17N3O3, M = 335.35, triclinic, a = 8.8865(5), b = 9.3125(3), c = 10.9460(5) Å, U = 826.26(7) Å3. T = 120(2) K, space group P, Z = 2, 15441 reflections measured, 3480 unique with R = 0.0346, wR2 = 0.0903. CCDC 927102. |
| This journal is © The Royal Society of Chemistry 2013 |