 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient syntheses of halo-dibenz[b,f]azepines and carbamazepine analogues via N-arylindoles†‡
Emma-Claire
Elliott§
a,
James L.
Maggs
b,
B. Kevin
Park
b,
Paul M.
O'Neill
a and
Andrew V.
Stachulski
*a
aDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: stachuls@liv.ac.uk; Fax: +44 (0)151 794 3588; Tel: +44 (0)151 794 3482
bMRC Centre for Drug Safety Science, Institute of Translational Medicine, University of Liverpool, Liverpool L69 3GE, UK
First published on 28th October 2013
Abstract
The dibenz[b,f]azepine heterocyclic system and related molecules with a single 10,11-bond are important templates for well-prescribed drug molecules, notably carbamazepine (anticonvulsant), clomipramine and imipramine (antidepressants). We synthesised a range of halogenated carbamazepine analogues, in connection with metabolic and immunological studies, as probes for structure-metabolism and hypersensitive effects and have published on their metabolic behaviour. While a number of synthetic routes to such analogues are possible, we naturally sought short and efficient methods for our target compounds. In the following report we present an effective two-step synthesis of a range of dibenz[b,f]azepines from appropriate indoles via N-arylation, then acid-catalysed rearrangement, with a critical analysis of other approaches. We showed earlier that this route was effective for fluoro analogues and here present a broader review of its scope. The 5-(carboxamido) side chain of carbamazepine may be added in various ways, affording overall a convenient access to drug molecules.
Introduction
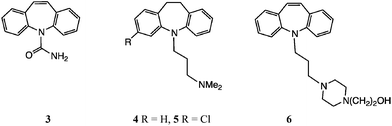
The tricyclic heterocycles dibenzo[b,f]azepine 1 and its 10,11-dihydro version 21 are major structural elements for a number of important CNS-active drugs, such as carbamazepine (CBZ) 3,2 imipramine 4,3 clomipramine 54 and opipramol 6.5
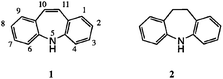
![Synthetic routes to dibenz[b,f] azepines: (1) via isatins; (2) from IDB; (3) via styrenes; (4) via stilbenes.](/image/article/2013/OB/c3ob41252k/c3ob41252k-s1.gif) | ||
| Scheme 1 Synthetic routes to dibenz[b,f] azepines: (1) via isatins; (2) from IDB; (3) via styrenes; (4) via stilbenes. | ||
Tsvelikhovsky and Buchwald recently devised a synthesis from an o-bromostyrene and an o-chloroaniline, route (3), published while our studies were in progress.12a,b The key to this was ligand 7, which diverted the bracketed intermediate toward the desired [6,7,6] ring system and away from other possible ring closures that would generate acridines or indoles. Although a most elegant route, this does require in general the availability of trisubstituted aromatics for both fragments, which could be difficult of access, depending on X1 and X2. Furthermore, the authors later published a correction12b in which they stated that the yield of dibenzazepine from 2-chloroaniline and 2-bromostyrene, whether by the one or two-step procedure, was significantly lower than reported earlier (70% rather than 99%), and that selectivity was incomplete. This leaves a clear concern that more highly substituted examples may also give lower yields and selectivity. Similarly, route (4) of Liang et al. requires the synthesis of mono/disubstituted o/o′-dibromo-Z-stilbenes and applies only to N-alkyl or N-aryl amines, though the ring closure is impressively efficient.13
Regarding the general availability of mono- and disubstituted IDBs [we applied route (2) for Br/Cl analogues only], Jorgensen et al. prepared a number of examples, including fluoro and methoxy analogues, in seven linear steps via appropriate o-nitrostilbenes; for our purpose, the 10,11-double bond would still need to be reintroduced.14 This and other older routes are summarised by Kricka and Ledwith.1
In our studies, we employed both routes (1) and (2)8,9 but also extensively studied a much shorter and attractive route, shown in Scheme 2, namely the acid-catalysed (polyphosphoric acid, PPA) rearrangement of N-aryl indoles. A number of efficient syntheses of N-aryl indoles are now available (v.i.), and the rearrangement step was discovered and evaluated for a number of analogues by Tokmakov and Grandberg.15 Although there are restrictions on the substitution patterns compatible with these conditions, the attractions of a two-step synthesis are obvious. We now report in detail on our findings, including especially a discussion of the scope of the rearrangement step and substituent effects, with finally a survey of methods for addition of the carboxamide group present in 3.
![Synthesis of dibenz[b,f]azepines via N-aryl indoles.](/image/article/2013/OB/c3ob41252k/c3ob41252k-s2.gif) | ||
| Scheme 2 Synthesis of dibenz[b,f]azepines via N-aryl indoles. | ||
Results and discussion
N-Arylindole synthesis
We studied various conditions for the N-arylation reaction, first optimising conditions for N-phenyl indole itself; initially employing the palladium-catalysed route of Watanabe et al.16 and Buchwald et al.17Bulky, electron-rich ligands, e.g. PBut3, are favoured for this chemistry16 as they allow reaction at lower temperatures and greatly reduce dehalogenation as a side reaction. Buchwald et al. preferred the ligand 7 noted above, and related o,o′-disubstituted biphenyls, with aryl bromides, chlorides or triflates as the donors.17
|
|
||||||
|---|---|---|---|---|---|---|
| Indole | Aryl iodide | Temp (°C)/time (h) | Reference if known compound | Product no. | Yield (%) | Yield (%) (other methods)a |
| a Other conditions: A1, Pd(OAc)2 (1 mol%), PBut3, o-xylene, 110 °C; A2, as A1, using the corresponding aryl bromide; B. CuI (5 mol%), (CH2NH2)2, K2CO3, PEG-400, 160 °C. b Yield on a 1 g scale; this dropped to 22% on a 5 g scale. c A 53% yield was obtained at 85 °C. d A 22% yield was obtained on a 5 g scale. e No useful product isolated at 80 °C. f A 45% yield was obtained using 4-bromofluorobenzene. g A 12% yield was obtained on a 5 g scale. h No product isolated. | ||||||
| 8 | 16 | 90/24 | 19b | 23 | 97 | 96 (A1),b,c 94 (A2) |
| Fluorinated examples | ||||||
| 8 | 17 | 100/24 | 8 | 24 | 81 | 88 (A1),d 59 (B)e,f |
| 8 | 18 | 100/28 | 8 | 25 | 78 | |
| 9 | 16 | 100/24 | 8 | 26 | 70 | |
| 10 | 16 | 100/24 | 8 | 27 | 76 | 73 (A1)g |
| 11 | 16 | 100/24 | 8 | 28 | 83 | |
| 10 | 17 | 100/24 | 8 | 29 | 75 | |
| 11 | 18 | 100/24 | 8 | 30 | 85 | |
| Chloro, bromo and alkoxy examples | ||||||
| 8 | 19 | 90/36 | 19a | 31 | 68 | |
| 12 | 16 | 90/36 | 20 | 32 | 71 | |
| 13 | 16 | 90/36 | 21 | 33 | 62 | |
| 8 | 20 | 90/36 | — | 34 | —h | |
| 8 | 21 | 80/24 | 15 | 35 | 87 | |
| 14 | 16 | 80/24 | — | 36 | 92 | |
| 14 | 21 | 80/24 | — | 37 | 85 | |
| 8 | 22 | 90/36 | 22 | 38 | 79 | |
| 15 | 16 | 90/36 | 20 | 39 | 83 | |
| 15 | 22 | 90/36 | — | 40 | 51 | |
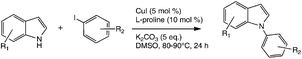
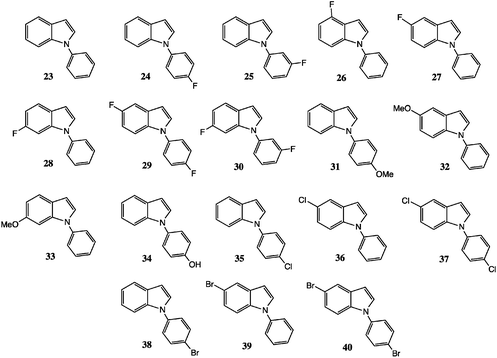
Cyclisation reaction
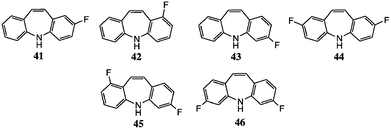
We first prepared a sample of 1 from 23 using the conditions of Tokmakov and Grandberg,15 then applied this method for the halo analogues. A number of experimental details were significant. Commercial PPA is not anhydrous, but we deoxygenated the reagent by passage of dry argon before use and performed the reactions under argon as the products are oxygen-sensitive. In earlier experiments23 we had conducted the rearrangement at 150 °C, but this was found to give significant amounts of 9-methylacridine by-products. This side-reaction was not commented on by Tokmakov and Grandberg,15 but they did generally work at significantly lower temperatures, viz. 75–120 °C, and generally at 90–100 °C. In the present studies we worked at 100 °C as closely as possible: this generally minimized the formation of 9-methylacridines, although traces (<5%) were still seen by NMR, especially the N-methyl group, e.g. δH (CDCl3) 2.98 for 2,7-difluoro-9-methylacridine. Scheme 3 presents feasible mechanisms for the cyclisation step (a) and the by-product formation (b), and Table 2 summarises our results for products 1 and 41–51.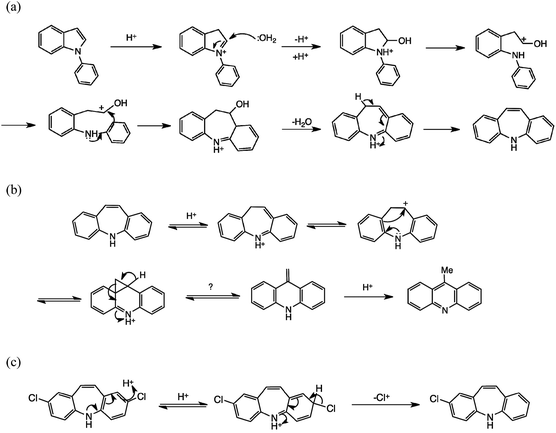 | ||
| Scheme 3 Mechanistic schemes. (a) N-Aryl indole to DBA rearrangement, after ref. 15. (b) Suggested mechanism for 9-methylacridine by-product formation, based on ref. 1. (c) Proposed dehalogenation mechanism. | ||
|
|
|||
|---|---|---|---|
| N-Aryl indole | Product(s)b | Yield (%) | Reference if known compound |
| a Reaction carried out at 100 °C and for 36–72 h, until judged complete by TLC, unless stated. b In cases where X2 is a meta-substituent, two regioisomers are produced: see text. c An 8% yield was reported. d A 10% yield was reported. e Also 49 (15%) was produced. f Reaction carried out at 65 °C. | |||
| 23 | 1 | 67 | 15 |
| Fluorinated examples | |||
| 24 | 41 | 40 | 8 |
| 25 | 42, 43 | 18, 22 | 8 |
| 26 | 42 | 24 | 8 |
| 27 | 41 | 47 | 8 |
| 28 | 43 | 48 | 8 |
| 29 | 44 | 66 | 8 |
| 30 | 45, 46 | 16, 35 | 8 |
| Chloro, bromo and alkoxy examples | |||
| 31 | 47 | 25 | 15 |
| 32 | 47 | 37 | 15 |
| 33 | 48 | 22 | 24 |
| 35 | 49 | 41 | 9 |
| 36 | 49 | 60 | 9 |
| 37 | 50 | 32e | 9 |
| 39 | 51 | 5f | 9 |
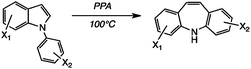
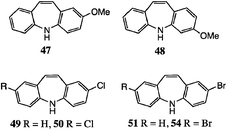
As stated above, we were particularly interested in halo analogues, and in addition to the fluoro compounds8 we considered the possibility of obtaining bromo- and chloro-DBAs in this way, with interesting results. In the earlier work,15 2-chloro-DBA 49 was obtained from 35 in 25% yield; we isolated a somewhat better yield of 41%. Here, however, because of the pseudo-symmetry of the product, 36 should be an equally good precursor and indeed we obtained a very satisfactory yield of 49 (61%) from 36. Thus the undoubted electron-withdrawing nature of Cl is not a barrier to successful reaction, especially if the substituent resides in the indole ring of the substrate. The main limitation is actually dehalogenation: in fact only trace amounts of 1 were observed from 36, but the 5,4′-dichloro substrate 37 afforded a separable mixture of 2,8-dichloro-DBA 50 (32%) and 2-chloro-DBA 49 (15%).
We suggest that protonation of the DBA products in the strongly acidic medium is the likely cause of loss of Cl. Where possible, protonation will occur on the non-chlorinated ring; when both rings are chlorinated [as Scheme 3c], protonation allows subsequent dechlorination as a side reaction. Nevertheless, the yields of both 49 and 50 are now at least competitive with those obtained via chlorination of 2,9 and only two steps as opposed to five are needed.
However, bromine was not compatible with the reaction conditions. From the reaction of 5-bromo-(N-phenyl)indole 39, only a 5% yield of 51 was isolable and milder conditions (65 °C) were necessary. Nevertheless, even this modest yield was useful for a reference sample, as it proved very difficult to obtain pure 2-bromo-DBA 51via the halogenation route from 2, as noted earlier.9 This is probably owing to radical-induced rearrangement during the introduction of the 10,11-double bond.
Carboxamidation
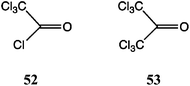
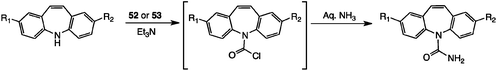
A number of methods for adding the carboxamide unit to dibenz[b,f]azepines, generating carbamazepine analogues, have been investigated. Many of these have appeared in the patent literature, particularly after carbamazepine 3 itself came off patent. We showed earlier9 that by varying the traditional procedure using an alkali metal isocyanate,25 in particular employing TFA as a stronger acid catalyst,26 a range of halogenated analogues of 3 was readily accessible. Nevertheless, we screened a number of other reagents, and this led to some interesting chemistry. In particular, we found good alternative methods for the synthesis of dibromo and dichloro analogues, involving the generation of an activated N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O·X intermediate and its reaction, possibly following isolation, with an appropriate nucleophile.
O·X intermediate and its reaction, possibly following isolation, with an appropriate nucleophile.
|
|
|||||
|---|---|---|---|---|---|
| Dibenzazepine | Solvent | Temperature (°C) | Time (h) prior to NH3 addition | Product | Yield (%) |
| a Using 0.5 eq. 52 or 0.33 eq. 53 with an equivalent amount of Et3N unless stated. b No Et3N added. | |||||
| Diphosgene | |||||
| 1 | PhMe | 10–15 | 8 | 3 | 79 |
| Triphosgene | |||||
| 1 | THF | 10–15 | 6 | 3 | 27 |
| 1 | PhMe | 10–15 | 6 | 3 | 92 |
| 1 | PhMe | 10–110b | 6 | 3 | — |
| Triphosgene with halogenated DBAs | |||||
| 50 | PhMe | 60 | 12 | 55 | 12 |
| 54 | PhMe | 10–15 | 6 | 56 | 24 |
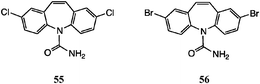
Substituted organic isocyanates were more promising, but here it was important to be able to isolate the N-acyl (or sulfonyl) urea intermediate. Thus on reaction of 2,8-dibromo-DBA 54 with N-(chlorosulfonyl)isocyanate30 in CH2Cl2, the presumed N-chlorosulfonyl intermediate was filtered off and reacted directly with dilute aq. NaHCO3. We obtained a low yield (ca. 10%) of the CBZ analogue, but preparative TLC was necessary to purify the product.23
The best alternative to the acid-catalysed alkali metal isocyanate reaction was the use of (trichloroacetyl)isocyanate (Table 4).31 For the symmetrical substrates (viz. 1, 50 and 54) the initial reaction was fast and the N-acyl ureas were obtained in high yield by simple filtration. The difficulty, however, was the lack of selectivity in the hydrolysis step, Table 4. Thus, in the unsubstituted case, treatment of the N-acylurea derived from 1 with either dilute acid or base lead to mixtures of 1 and 3. Relatively best was 5% aq. Na2CO3, leading to 3 as the main product. Similarly, for the dibromo and dichloro examples, the N-acylureas could be isolated in good yields from 50 and 54 and afforded 50–60% of 55 and 56 on mild basic hydrolysis. The method was not suitable for mono-halo DBAs.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| DBA | Temp. (°C) | Time (h) | Yield of N-acylurea (%) | Hydrolysis conditions | Product ratio A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B :B |
Desired product | Yield (%) |
| a Using toluene as solvent in all cases. DBA = dibenz[b,f]azepine. | |||||||
| 1 | 20–0 | 6 | 89 | 0.1 M HCl | 10:90 |
||
| 0.1 M NaOH | 12:88 |
||||||
| 5% aq. Na2CO3 | 57:33 |
3 | 56 | ||||
| 5% aq. K2CO3 | 34:56 |
||||||
| 50 | 20–0 | 24 | 75 | 5% aq. Na2CO3 | 60:40 |
55 | 45 |
| 54 | 20–0 | 8 | 78 | 5% aq. Na2CO3 | 60:40 |
56 | 47 |
Although the N-acylureas are moderately stable for short periods at room temperature, they are readily hydrolysed, even during NMR acquisition to some extent. Nevertheless, it was possible to obtain satisfactory 1H NMR spectra and HRMS for the dichloro example, and this data was adjudged sufficient since both final products 55 and 56 gave identical spectroscopic data to material obtained via the NaNCO/TFA method.9
Conclusions
There are a number of possible syntheses of dibenz[b,f]azepines from available starting materials. We developed very successfully a simple two-step synthesis, starting from readily available indole derivatives. N-Arylation of the indoles was effected in good to excellent yield using the procedure of Ma et al.,19a,b employing CuI with proline as ligand. The conditions were fully compatible with other halogens. Rearrangement of the N-aryl indoles to dibenz[b,f]azepines was effected using strong acid catalysis (PPA, 100 °C), in yields from 22–66%. We developed the original conditions of Tokmakov and Grandberg:15 a significant by-product was a 9-methyl acridine but this could be minimized. Both fluoro and chloro substituents were compatible with the conditions, though with the latter the site of Cl substitution (viz. on the indole ring or in the N-aryl substituent) was an important factor. With an N-(3-fluoro)phenyl substituent, both 1- and 3-fluoro products resulted. The simplicity of this two-step sequence compensates for moderate yields in the rearrangement step. Finally, attachment of the carboxamide substituent in carbamazepine 1 and its 2,8-dibromo and dichloro analogues can be effected using various reagents, including triphosgene and (trichloroacetyl)isocyanate, with a following ammonolysis or hydrolysis step. However, the combination of KNCO or NaNCO and TFA is most versatile and gives access to monohalo analogues also.9Experimental
Organic extracts were finally washed with saturated brine and dried over anhydrous Na2SO4 prior to rotary evaporation at <30 °C. Moisture sensitive reactions were carried out in anhydrous organic solvents (purchased from Sigma-Aldrich) under a N2 or Ar atmosphere. Reactions were monitored by analytical thin-layer chromatography using Merck Kieselgel 60 F254 silica plates, and were viewed under UV or by staining with anisaldehyde, vanillin, KMnO4, iodine, or bromocresol green. Preparative flash column chromatography was performed on either VWR Prolabo silica gel or Sigma-Aldrich silica gel (particle size 40–63 Å). Melting points were recorded using a Bibby-Sterlin Stuart SMP3 melting point apparatus and are uncorrected. High resolution mass spectrometry for the N-aryl indoles was performed by the EPSRC National Mass Spectrometry Service, Swansea University. Other mass spectra were obtained in either electrospray mode (ES) with a Micromass LCT or chemical ionization (CI) mode with a Micromass Trio 1000 using ammonia; two final high-resolution mass spectra (compounds 47, 48) were obtained in CI mode using an Agilent QTOF 7200 instrument in this Department. Elemental analyses were performed by Mr Steve Apter, University of Liverpool. 1H and 13C NMR spectra were obtained using a Bruker Avance or a Bruker DPX 400 instrument operating at 400 and 101 MHz, respectively; chemical shifts are reported in ppm (δ) relative to Me4Si. Coupling constants (J) are reported in Hz.General method for N-arylindole synthesis
A mixture of the appropriate indole (2.2 mmol), K2CO3 (5.0 mmol), CuI (0.1 mmol) and L-proline (0.2 mmol) was stirred in anhydrous DMSO (4 mL) and heated to 100 °C. After 10 min the appropriate iodobenzene (2.0 mmol) was added dropwise over 20 min, then the mixture was stirred at this temperature for 24 h. The mixture was cooled and partitioned between EtOAc (3 × 50 mL) and water; the combined organic extracts were evaporated to dryness and the product isolated by chromatography, eluting with EtOAc–hexane mixtures.Spectroscopic and analytical data, including photocopy NMR spectra, for compounds 24–30 have been given earlier.8 Data for compounds 23, 31–33, 35, 36 and 38–40 are placed in the ESI.‡ All yields are in Table 1.
5-Chloro-N-(4-chloro)phenylindole 37
M.p. 66–67 °C. Found: C, 64.1; H, 3.5; N, 5.2; m/z 262.0183. C14H9Cl2N requires C, 64.2; H, 3.5; N, 5.3%; C14H10NCl2 (MH+) requires m/z, 262.0185; δH 6.62 (1 H, dd, J = 3.3 and 0.8 Hz, 3-H), 7.17 (1 H, dd, J = 8.8 and 2.0 Hz, 6-H), 7.30 (1 H, d, J = 3.3 Hz, 2-H), 7.37–7.42 and 7.46–7.51 (4 H, approx. dd, 2′-, 3′-, 5′- and 6′-H), 7.40 (1 H, d, J = 8.7 Hz, 7-H) and 7.64 (1 H, d, J = 1.8 Hz, 4-H); δC 104.0, 111.7, 121.0, 123.2, 125.9, 126.6, 129.4, 130.3, 130.8, 132.8, 134.6 and 138.3.General method for dibenz[b,f]azepine synthesis
Polyphosphoric acid (1 mL per 100 mg N-arylindole) was purged with Ar and heated to 100 °C for 30 min. The appropriate N-arylindole was then added via a syringe and the reaction was stirred at 100 °C, generally for 36–72 h (see Table 2). Once it was judged complete by TLC, the reaction was cooled to ambient temperature, poured cautiously onto ice-cold NaHCO3 aq. and stirred vigorously for 1 h, then extracted with CH2Cl2 (2 × 100 mL). The combined organic extracts were washed with water and evaporated; the resulting crude product was purified by chromatography, eluting with 10% EtOAc–hexane.Spectroscopic data for compounds 41–46,849,9509 and 519 have been given earlier; since we now report the synthesis of 49, 50 and 51 by the N-aryl indole rearrangement route, NMR data and photocopy spectra for these compounds and for compound 48 are included in the ESI.‡
2-Methoxy-5H-dibenz[b,f]azepine 4715
This was prepared from either N-aryl indole 31 (25% yield) or 32 (37% yield), Table 2. Found: m/z, 224.1075. C15H14NO requires m/z, 224.1070 (MH+); δH [(CD3)2SO] 3.63 (3 H, s, CH3O), 6.13, 6.18 (2 H, ABq, 10-H + 11-H), 6.41 (1 H, d, J = 2.1 Hz, 6-H), 6.59 (1 H, d, J = 2.6 Hz, 9-H), 6.62 (1 H, d, J = 7.6 Hz, 4-H), 6.69 (1 H, t, J = 7.2 Hz, 8-H), 6.74–6.79 (2 H, m, 1-H + 3-H) and 6.97 (1 H, t, J = 7.2 Hz, 7-H); δC [(CD3)2SO] 55.1, 114.4, 115.3, 118.9, 120.0, 121.6, 128.9, 129.5, 130.4, 131.6, 132.7, 134.1, 142.4, 150.3, and 154.7; m/z (CI) 224 (MH+, 100%).General procedure for carboxamidation reactions
The N-(trichloroacetyl)intermediates derived from 1 and 54 were sufficiently stable on isolation to allow characterization: N-(2,2,2-trichloroacetyl)-5H-dibenz[b,f]azepine: white powder. Found: m/z, 402.9784. C17H1135Cl3N2O2Na requires m/z, 402.9784; δH [(CD3)2SO] 6.05 (2H, s, 10-H + 11-H), 6.52 (2 H, d, J = 8.4 Hz), 6.88–6.98 (2 H, m), 7.06–7.21 (4 H, m) and 10.85 (1 H, br s, NH); δC [(CD3)2SO] 93.1, 113.6, 121.0, 131.1, 131.9, 132.2, 132.7, 148.4 and 162.9; m/z (ES +ve mode) 403 (MNa+, 100%). N-(2,2,2-Trichloroacetyl)-2,8-dibromo-5H-dibenz[b,f]azepine: white powder. Found: m/z, 558.8021, C17H979Br235Cl3N2O2Na requires m/z, 558.7994; δH [(CD3)2SO] 7.08(2 H, s, 10-H + 11-H), 7.35–7.60 (6 H, m) and 10.46 (I H, br s, NH); δC [(CD3)2SO] 91.8, 119.1, 121.9, 127.1, 129.3, 129.3, 129.5, 130.4, 132.1, 133.8, 135.1, 138.2, 140.5, 149.5, 150.3, 158.4, and 162.9; m/z (ES +ve mode) 559(MNa+, 23%) and 561 (100%).
We give NMR data for 55 and 56, as prepared by the trichloroacetyl isocyanate route, together with photocopy NMR spectra in the ESI‡ to show identity with material obtained earlier9a using the alkali metal isocyanate route.
Acknowledgements
We are grateful to the EPSRC for funding (DTA maintenance grant to EE).Notes and references
- L. J. Kricka and A. Ledwith, Chem. Rev., 1974, 74, 101–123 CrossRef CAS.
- (a) F. Albani, R. Riva and A. Baruzzi, Pharmacopsychiatry, 1995, 28, 235–244 CrossRef CAS PubMed; (b) A. Sidebottom and S. Maxwell, J. Clin. Pharm. Ther., 1995, 20, 31–35 CrossRef CAS.
- (a) M. R. Mavissakalian and J. M. Perel, Am. J. Psychiatry, 1995, 152, 673–682 CAS; (b) D. H. Barlow, J. M. Gorman, M. K. Shear and S. W. Woods, J. Am. Med. Assoc., 2000, 283, 2529–2536 CrossRef CAS.
- (a) P. Thoren, M. Asberg, B. Cronholm, L. Joenestedt and L. Traskman, Arch. Gen. Psychiatry, 1980, 37, 1281–1285 CrossRef CAS; (b) D. McTavish and P. Benfield, Drugs, 1990, 39, 136–153 CrossRef CAS PubMed.
- T. S. Rao, J. A. Cler, S. J. Mick, D. M. Ragan, T. H. Lanthorn, P. C. Contreras, S. Iyengar and P. L. Wood, Neuropharmacology, 1990, 29, 1199–1204 CrossRef CAS.
- (a) K. Lertratanangkoon and M. G. Horning, Drug Metab. Dispos., 1982, 10, 1–10 CAS; (b) J. L. Maggs, M. Pirmohamed, N. R. Kitteringham and B. K. Park, Drug Metab. Dispos., 1997, 25, 275–280 CAS.
- (a) S. Madden, J. L. Maggs and B. K. Park, Drug Metab. Dispos., 1996, 24, 469–479 CAS; (b) R. E. Pearce, J. P. Uetrecht and J. S. Leeder, Drug Metab. Dispos., 2005, 33, 1819–1826 CAS; (c) H. Z. Bu, P. Kang, A. J. Deese, P. Zhao and W. F. Pool, Drug Metab. Dispos., 2005, 33, 1920–1924 CrossRef CAS PubMed.
- E. Elliott, E. R. Bowkett, J. L. Maggs, J. Bacsa, B. K. Park, S. L. Regan, P. M. O'Neill and A. V. Stachulski, Org. Lett., 2011, 13, 5592–5595 CrossRef CAS PubMed.
- (a) E. Elliott, S. L. Regan, J. L. Maggs, E. R. Bowkett, L. Parry, D. P. Williams, B. K. Park and A. V. Stachulski, J. Med. Chem., 2012, 55, 9773–9784 CrossRef CAS PubMed; (b) J. Farrell, M. Lichtenfels, A. Sullivan, E. Elliott, A. Alfirevic, A. V. Stachulski, M. Pirmohamed, D. J. Naisbitt and B. K. Park, J. Allergy Clin. Immunol., 2013, 132, 493–495 CrossRef CAS PubMed.
- E.g., (a) P. N. Craig, B. M. Lester, A. J. Saggiomo, C. Kaiser and C. L. Zirkle, J. Org. Chem., 1961, 26, 135–138 CrossRef CAS; (b) W. Schindler and H. Blattner, Helv. Chim. Acta, 1961, 44, 753–763 CrossRef CAS.
- Cf. K. Smith, D. M. James, A. G. Mistra, M. R. Bye and D. J. Faulkner, Tetrahedron, 1992, 48, 7479–7488 CrossRef CAS ; up to four Br atoms may be introduced into 2 by acid-catalysed bromination.
- (a) D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14048–14051 CrossRef CAS PubMed; (b) D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 16917–16917 CrossRef CAS PubMed.
- X. Zhang, Y. Yang and Y. Liang, Tetrahedron Lett., 2012, 53, 6406–6408 CrossRef CAS PubMed.
- T. K. Jorgensen, K. E. Andersen, J. Lau, P. Madsen and P. O. Huusfeldt, J. Heterocycl. Chem., 1999, 36, 57–64 CrossRef CAS.
- G. P. Tokmakov and I. I. Grandberg, Tetrahedron, 1995, 51, 2091–2098 CrossRef CAS.
- (a) M. Watanabe, M. Nishiyama, T. Yamamoto and Y. Koie, Tetrahedron Lett., 2000, 41, 481–483 CrossRef CAS; (b) M. Nishiyama, T. Yamamoto and Y. Koie, Tetrahedron Lett., 1998, 39, 617–620 CrossRef CAS.
- D. W. Old, M. C. Harris and S. L. Buchwald, Org. Lett., 2000, 2, 1403–1406 CrossRef CAS PubMed.
- S. Chandrasekhar, S. S. Sultana, S. R. Yaragorla and N. R. Reddy, Synthesis, 2006, 839–842 CrossRef CAS PubMed.
- (a) D. Ma and Q. Cai, Synlett, 2004, 128–130 CrossRef PubMed; (b) H. Zhang, Q. Cai and D. Ma, J. Org. Chem., 2005, 70, 5164–5173 CrossRef CAS PubMed; (c) A related procedure uses CuI without a ligand, in DMF at 120 °C: L. Zhu, P. Guo, G. Li, J. Lan, R. Xie and J. You, J. Org. Chem., 2007, 62, 8535–8538 CrossRef PubMed.
- R. K. Rao, A. B. Naidu, E. A. Jaseer and G. Sekar, Tetrahedron, 2009, 65, 4619–4624 CrossRef CAS PubMed.
- G. Tarzia, A. Duranti, G. Gatti, G. Piersanti, A. Tontini, S. Rivara, A. Lodola, P. V. Plazzi, M. Mor, S. Kathuria and D. Piomelli, ChemMedChem, 2006, 1, 130–139 CrossRef CAS PubMed.
- W. Deng, Y. F. Wang, C. Zhang, L. Liu and Q. X. Guo, Chin. Chem. Lett., 2006, 17, 313–316 CAS.
- E. R. Bowkett, unpublished observations; cf. ref. 8.
- V. Lucini, M. Pannacci, F. Scaglione, F. Fraschini, S. Rivara, M. Mor, F. Bordi, P. V. Plazzi, G. Spadoni, A. Bedini, G. Piersanti and G. Tarzia, J. Med. Chem., 2004, 47, 4202–4212 CrossRef CAS PubMed.
- R. Eckardt and H.-J. Jaensch, Process for the preparation of 5H-dibenz[b,f]azepine-5-carboxamide from iminostilbene and alkali cyanates, DE 4307181, 10th Nov. 1994.
- G. J. Durant, Improved synthesis of N,N-diarylureas, Chem. Ind., 1965, 1428–1429 CAS . These conditions were originally employed for N,N-diarylamines.
- W. Raml and G. Eichberger, Preparation of carbamazepine by the phosgenation and amidation of 5H-dibenz[b,f]azepine, AT01174B, 1996.
- (a) L. Cotarca and H. Eckert, Phosgenations – A Handbook, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) G. Skorna and I. Ugi, Angew. Chem., Int. Ed. Engl., 1977, 16, 259–260 CrossRef.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26, 894–895 CrossRef.
- (a) Cf. N. Ghiasi, A. A. Oskouie and H. Saeidian, Carbohydr. Res., 2012, 348, 47–54 CrossRef PubMed; (b) D. Che, N. Corelli-Rennie, B. R. Guntoori and J. Faught, Process for the preparation of oxcarbazepine and related intermediates, US 20050282797, 2005.
- This reagent has generally been used for carbamoylation of alcohols, e.g. T. M. Tagmose and M. Bols, Chem.–Eur. J., 1997, 3, 453–462 CrossRef CAS.
Footnotes |
| † This paper is dedicated with affection to Drs. Tom Gilchrist and Richard Storr, two great colleagues and leading exponents of heterocyclic chemistry. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob41252k |
| § Current address: Unilever Research and Development Laboratories, Port Sunlight, Wirral, UK. |
| This journal is © The Royal Society of Chemistry 2013 |