 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of a non-selective adenosine receptor antagonist into A3-selective high affinity fluorescent probes using peptide-based linkers†
Andrea J.
Vernall‡
a,
Leigh A.
Stoddart‡
b,
Stephen J.
Briddon
b,
Hui Wen
Ng
c,
Charles A.
Laughton
a,
Stephen W.
Doughty
c,
Stephen J.
Hill§
*b and
Barrie
Kellam§
*a
aSchool of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: barrie.kellam@nottingham.ac.uk; Tel: +44(0)-115-9513026
bSchool of Pharmacy, University of Nottingham Malaysia Campus, Jalan Broga, 43500 Semenyih, Selangor Darul Ehsan, Malaysia. E-mail: stephen.hill@nottingham.ac.uk; Tel: +44-115-8230082
cInstitute of Cell Signalling, School of Biomedical Science, Queen's Medical Centre, University of Nottingham, Nottingham NG1 1GF, UK
First published on 4th July 2013
Abstract
Advances in fluorescence-based imaging technologies have helped propel the study of real-time biological readouts and analysis across many different areas. In particular the use of fluorescent ligands as chemical tools to study proteins such as G protein-coupled receptors (GPCRs) has received ongoing interest. Methods to improve the efficient chemical synthesis of fluorescent ligands remain of paramount importance to ensure this area of bioanalysis continues to advance. Here we report conversion of the non-selective GPCR adenosine receptor antagonist Xanthine Amine Congener into higher affinity and more receptor subtype-selective fluorescent antagonists. This was achieved through insertion and optimisation of a dipeptide linker between the adenosine receptor pharmacophore and the fluorophore. Fluorescent probe 27 containing BODIPY 630/650 (pKD = 9.12 ± 0.05 [hA3AR]), and BODIPY FL-containing 28 (pKD = 7.96 ± 0.09 [hA3AR]) demonstrated clear, displaceable membrane binding using fluorescent confocal microscopy. From in silico analysis of the docked ligand-receptor complexes of 27, we suggest regions of molecular interaction that could account for the observed selectivity of these peptide-linker based fluorescent conjugates. This general approach of converting a non-selective ligand to a selective biological tool could be applied to other ligands of interest.
Introduction
Discrete molecular probes are one mechanism by which a receptor's role and function in biological processes can be interrogated, and as such, rational design-based approaches to develop these chemical tools are vital. In particular, developing molecular probes with a high affinity and selectivity for a particular cell surface receptor target is paramount for unravelling processes of interest in physiologically relevant systems that contain mixed receptor subtype populations. G protein-coupled receptors (GPCRs) are the largest family of transmembrane signalling proteins in the human genome and are the target of 30–40% of currently marketed drugs. Probes that target these receptors are therefore regarded as extremely valuable tools. Unsurprisingly therefore, fluorescent ligands have been increasingly used in studying GPCRs; for example to determine receptor expression levels in diseased tissues,1 real-time receptor–receptor interactions and signaling,2,3 and as a tracer ligand in a competition binding assay.4 A common approach to the design and synthesis of receptor probes involves tethering a known orthosteric binding moiety to a second ligand or fluorophore via a linker to form a conjugate.5,6 It is desirable to develop general methods that can increase conjugate affinity and selectivity for a target receptor, especially if a receptor subtype-selective pharmacophore is not available. Indeed, recent advances in X-ray crystallography of GPCRs7–11 have further reignited interest in structure-based design approaches for more selective synthetic ligands.12From the earlier observations of Jacobson et al.,13 we were encouraged to investigate whether minor chemical changes to a peptide-based linker component of a fluorescent ligand conjugate could potentially be used to fine tune affinity and/or selectivity of the final fluorescent probe for a given receptor; since the linker passes through regions of the receptor capable of engaging in productive molecular recognition events by the very nature of the functional group chemistries present there. In this study, we chose to explore our hypothesis by using the human A1 adenosine receptor (AR) and hA3AR subtypes as our model system. The ARs are Class A GPCRs, and there are four characterised receptor subtypes – the A1, A2A, A2B, and A3.14 There is growing interest in the A1AR15 and A3AR16 as AR drug targets,17 therefore it remains pertinent to further refine chemical methods to produce high affinity and selective tools to probe these receptors. In addition, these receptors are closely related in terms of amino acid sequence and therefore pose a significant challenge in terms of designing selective probes.18
Following the now well established chemical biology practice of exploiting amino acids in biological conjugates, for example in drug delivery systems19 and hydrogels,20 we herein report that the affinity and selectivity of fluorescent conjugates for the human A1AR versus A3AR can be modulated by single amino acid changes in a dipeptide linker region connecting the orthosteric binding moiety and fluorophore. This approach for increasing affinity and tuning selectivity is likely to have a broad applicability for developing fluorescent probes for other biological targets, in particular, ligands for other Class A GPCRs.
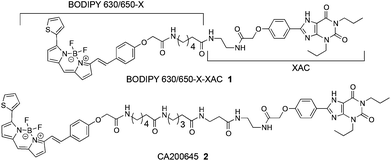
We commenced this study by considering the previously reported AR fluorescent antagonist 121 alongside the recently reported 24 (CA200645) (Fig. 1), which are based on the non-subtype-selective xanthine amine congener (XAC) (3) and are themselves non-selective for the A3AR/A1AR. Previously, Baker et al. indicated that when 3 was tethered to different fluorophores using the same linker, significant differences in conjugate affinity for the human A1AR were detected.22 Here we sought to expand upon this observation by also addressing receptor affinity and selectivity imparted by subtle changes in linker composition for conjugates containing the boron-dipyrromethene moiety. The BODIPY 630/650-X (6-(((4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)amino hexanoic acid) fluorophore was selected based on previous successes with a number of A3AR and A1AR agonists and antagonists.4,21–24 However, multicolour imaging applications are made easier if a range of ligands with varying emission wavelengths are available, and we were conscious that the only previously reported example of pharmacophore 3 tethered to the BODIPY-FL (4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid) fluorophore22 exhibited low affinity for the A1AR and made a poor imaging tool due to substantial membrane penetration. Therefore in this study we also sought to optimise the linker component in order to generate a usable green fluorescent imaging tool with improved receptor affinity.
Results and discussion
Synthesis
We had become increasingly mindful that synthesis of any fluorescent compound library containing different linkers could become very costly, as large amounts of fluorophore (for example BODIPY 630/650-X) are often required. We therefore chose to investigate whether the inexpensive and commonly available amino-protecting group 9-fluorenylmethyloxycarbonyl (Fmoc) could be employed as an inexpensive alternative for the hydrophobic BODIPY 630/650 moiety, enabling a ‘pre-screening’ of the optimal peptide-linker congener prior to introduction of the expensive fluorophore. As BODIPY 630/650-X is only commercially available with an internal 6-aminohexanoyl linker already present, N-Fmoc-aminohexanoic acid (Fmoc-Ahx) was proposed as an alternative to BODIPY 630/650-X in order to mirror the hexanoyl linker present in any final conjugates. To assess the suitability of this fluorophore substitute approach, commercially available 3 was coupled to N-Fmoc-aminohexanoic acid using O-(7-azabenzotriazol-1-yl)-N,N,N′N′-tetramethyluronium hexafluorophosphate (HATU) to furnish 4 (Scheme 1). The Fmoc-protected congener 5 was synthesised by coupling 3 to N-protected aminopentanoic acid, followed by N-deprotection and coupling to N-Fmoc-aminohexanoic acid. Congener 5 represents the hydrocarbon chain-length equivalent of the dipeptide moiety we planned to introduce.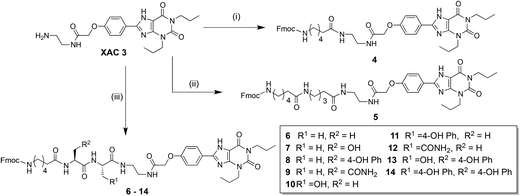 | ||
| Scheme 1 Synthesis of Fmoc-Ahx-XAC scaffold containing varying linkers. (i) N-Fmoc-Ahx-OH, HATU, N,N-diisopropylethylamine, DMF, 83%; (ii) Three steps from 3, 35% over three steps; (iii) (a) N-Fmoc-amino acid-OH (side-chain protected), HATU, N,N-diisopropylethylamine, DMF; (b). Diethylamine, CH2Cl2; (c) Repeat steps (a) and (b); (d) Fmoc-Ahx-OH, HATU, DIPEA, DMF; (e) Trifluoroacetic acid, CH2Cl2. | ||
The encouraging pharmacology observed for Fmoc-Ahx-congeners 4 and 5 (see later discussion) encouraged us to investigate if replacement of the aminopentanoyl moiety of 5 with a dipeptide could modulate receptor affinity and/or selectivity. Our rationale being that introduction of functional side chains (via the chosen amino acids) would provide additional regions of molecular interactivity between the conjugate and the region of receptor space it navigates through. A library of N-Fmoc-aminohexanoic-dipeptidyl-XAC conjugates was therefore synthesised (Scheme 1).
Initially, alanine–alanine (Ala–Ala) linked 6 was synthesised to ascertain if introduction of a dipeptide maintained affinity for the AR. As the pharmacology of this ligand confirmed this was the case, the N-terminal alanine residue from 6 was substituted with serine (Ser) (7), tyrosine (Tyr) (8), or asparagine (Asn) (9). The same three substitutions were also performed at the C-terminal amino acid position, to generate 10, 11, and 12. The prerequisite for amino acid selection was that the side-chain functional group should be polar but not requiring a protecting group for a coupling reaction with a fluorophore-succinimidyl ester (SE) (Scheme 2). Ser, Tyr, and Asn were selected as their side-chain functional groups are capable of hydrogen bonding interactions. Additionally, with Tyr we could explore potential cation–pi or pi–pi interactions between the aromatic side-chain and receptor. Based on the observed structure–activity relationships (Table 1), two other Fmoc-Ahx-dipeptide-XAC compounds were synthesised; containing a Tyr–Ser (13) or Tyr–Tyr (14) dipeptide linker.
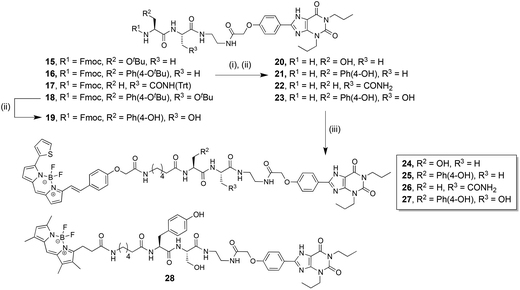 | ||
| Scheme 2 Synthesis of fluorescent dipeptide-XAC conjugates. (i) Diethylamine, CH2Cl2, quantitative. (ii) Trifluoroacetic acid, CH2Cl2, quantitative. (iii) BODIPY 630/650-X-SE, N,N-diisopropylethylamine, DMF; 20 gave 24 (40% after RP-HPLC purification), 21 gave 25 (28% after RP-HPLC purification), 22 gave 26 (52% after RP-HPLC purification), 23 gave 27 (25% after RP-HPLC purification). Or BODIPY-FL-X-SE, N,N-diisopropylethylamine, DMF; 23 gave 28 (25% after RP-HPLC purification). | ||
| Compound | hA3ARa,d | n | hA1ARb,c,e | n | Fold Selectivity A3AR/A1ARg | |
|---|---|---|---|---|---|---|
| a pKi values were calculated from inhibition of the binding of 2 (A3AR Ki = 3.11 nM) to CHO-A3 CRE-SPAP cells. b pKi values were calculated from inhibition of the binding of 2 (A1AR Ki = 17.0 nM) to CHO-A1 cells. c % inhibition of binding of 2 by 10 μM 14. d pKD values were obtained from global Schild analysis of NECA-mediated inhibition of FSK-stimulated CRE-SPAP responses in CHO-A3 CRE-SPAP cells. e pKi values calculated from inhibition of [3H]DPCPX (Ki = 2.0 nM) binding in CHO-A1 cells. All values represent mean ± SEM for n separate experiments performed in duplicate (a–c) or triplicate (d and e). f Due to apparent non-competitive antagonism at higher concentrations of 1, the pKD value was estimated from a shift in the NECA concentration response curves to a single concentration of 100 nM of 1 as described in the ESI. g Fold selectivity was calculated from the Ki (nM) value at A1AR divided by the Ki at A3AR. | ||||||
| 3 | XAC | 7.80 ± 0.07a | 6 | 7.30 ± 0.10b | 4 | 3.1 |
| 4 | Fmoc-Ahx-XAC | 6.94 ± 0.06a | 5 | 6.85 ± 0.07b | 4 | 1.2 |
| 5 | Fmoc-Ahx-pentyl -XAC | 7.45 ± 0.12a | 4 | 6.96 ± 0.17b | 4 | 3.1 |
| 6 | Fmoc-Ahx-Ala-Ala-XAC | 7.41 ± 0.15a | 4 | 7.00 ± 0.19b | 5 | 2.6 |
| 7 | Fmoc-Ahx Ser-Ala-XAC | 7.88 ± 0.17a | 4 | 7.02 ± 0.10b | 4 | 7.2 |
| 8 | Fmoc-Ahx-Tyr-Ala-XAC | 8.95 ± 0.06a | 4 | 7.49 ± 0.15b | 4 | 28.8 |
| 9 | Fmoc-Ahx-Asn-Ala-XAC | 7.60 ± 0.17a | 4 | 6.84 ± 0.03b | 4 | 5.7 |
| 10 | Fmoc-Ahx-Ala-Ser-XAC | 7.61 ± 0.15a | 5 | 7.30 ± 0.09b | 4 | 2.0 |
| 11 | Fmoc-Ahx-Ala-Tyr-XAC | 7.69 ± 0.03a | 5 | 6.95 ± 0.10b | 4 | 5.5 |
| 12 | Fmoc-Ahx-Ala-Asn-XAC | 7.35 ± 0.03a | 4 | 7.08 ± 0.07b | 4 | 1.8 |
| 13 | Fmoc-Ahx-Tyr-Ser-XAC | 8.49 ± 0.21a | 4 | 7.66 ± 0.12b | 4 | 6.8 |
| 14 | Fmoc-Ahx-Tyr-Tyr-XAC | 6.80 ± 0.09a | 5 | 61.6 ± 5.2%c | 5 | — |
| 19 | Fmoc-Tyr-Ser-XAC | 7.26 ± 0.12a | 4 | 7.04 ± 0.12b | 4 | 1.7 |
| 23 | Tyr-Ser-XAC | 7.27 ± 0.19a | 4 | 6.86 ± 0.13b | 4 | 2.6 |
| 1 | BODIPY 630/650-X-XAC | 7.51 ± 0.21f | 7 | 8.03 ± 0.14e | 7 | 0.3 |
| 2 | CA200645 | 8.38 ± 0.15d | 4 | 7.79 ± 0.07e | 4 | 3.9 |
| 24 | BODIPY 630/650-X-Ser-Ala-XAC | 9.29 ± 0.17d | 5 | 8.39 ± 0.09e | 7 | 8.0 |
| 25 | BODIPY 630/650-X-Tyr-Ala-XAC | 8.41 ± 0.09d | 4 | 7.62 ± 0.11e | 4 | 6.2 |
| 26 | BODIPY 630/650-X-Ala-Asn-XAC | 8.58 ± 0.11d | 4 | 7.82 ± 0.07e | 4 | 5.8 |
| 27 | BODIPY 630/650-X-Tyr-Ser-XAC | 9.12 ± 0.05d | 4 | 7.62 ± 0.13e | 4 | 31.6 |
| 28 | BODIPY FL-X-Tyr-Ser-XAC | 7.96 ± 0.09d | 4 | 6.50 ± 0.04e | 4 | 28.7 |
The Fmoc-Ahx-dipeptide-XAC compounds were not direct precursors for conjugation to our chosen fluorophore, since BODIPY 630/650 is commercially available with the aminohexanoyl linker pre-installed. Preliminary pharmacological evaluation of the N-Fmoc-aminohexanoyl-dipeptidyl-XAC library conjugates showed a general trend of higher affinity for the A3AR rather than the A1AR. Therefore the three dipeptide linkers that imparted the highest affinity and selectivity for the A3AR were selected for incorporation into an equivalent BODIPY 630/650 fluorescent conjugate; Ser–Ala from 7, Tyr–Ala from 8, and Tyr–Ser from 13. The Ala–Asn dipeptide from 12 was also chosen, as it was one of the least selective Fmoc-dipeptide-congeners, and thereby allowed us to investigate if this translated into a comparably non-selective fluorescent conjugate.
Compounds 15–17 and 18 (intermediates in the synthesis of 6–14, Scheme 1) were therefore globally deprotected to generate 20–23 respectively, and coupled to commercially available BODIPY 630/650-X-SE to afford 24–27 (Scheme 2). For completeness, and to analyse how effectively the affinity and selectivity profile of the Fmoc-Ahx compounds matched the BODIPY-630/650-X-compounds, Fmoc-containing 19 (the analogous compound to 13 lacking the aminohexanoyl linker) was also synthesised. Preliminary pharmacological evaluation of these four BODIPY 630/650 conjugates indicated that 27 possessed superior selectivity for the A3AR over the A1AR, therefore Tyr–Ser-based 23 was additionally coupled to commercially available BODIPY-FL-X-SE to afford the green fluorescent conjugate 28.
Pharmacology
Pharmacological characterisation of the compounds was carried out in Chinese Hamster Ovary (CHO) cells expressing either the human A1AR (CHO-A1 cells25), or the human A3AR and a reporter gene consisting of 6 × cyclic adenosine monophosphate response elements (6 × CRE) promoting the expression of a human-secreted placental alkaline phosphatase (SPAP; CHO-A3 SPAP cells23). The affinity of each of the non-fluorescent compounds (3–14, 19, 23) for the human A1AR and A3AR was determined in an established fluorescence-based live cell competition binding assay4 using the fluorescent adenosine receptor antagonist 2 as the labelling ligand (as detailed in the ESI†).It was not possible to obtain affinity values of the new BODIPY 630/650-containing compounds using tracer 2 in this particular assay as 2 also contained the BODIPY 630/650 fluorophore. Affinity values of all the BODIPY-containing compounds at the A1AR were therefore determined using a [3H]1,3-dipropyl-9-cyclopentylxanthine ([3H]DPCPX) whole cell competition binding assay in CHO-A1 cells. Affinity estimates for the new BODIPY-containing compounds at the A3AR were initially obtained by measuring their ability to antagonise agonist-stimulated functional responses in CHO-A3 SPAP cells. SPAP levels are driven through the CRE promoter in these cells and can be used as an indirect measure of cyclic 3′,5′-adenosine monophosphate (cAMP) levels. The agonist adenosine-5-N-ethylcarboxamide (NECA) effected a dose-dependent inhibition of forskolin (FSK) stimulated SPAP production in these cells (ESI Fig. S1†), which was antagonised by all of the fluorescent XAC conjugates (Table 1). It has already been demonstrated that pKi values obtained with this fluorescence-based competition binding assay show an excellent correlation to affinity values measured using different assay platforms,4 and this was confirmed for non-fluorescent compounds 3, 8, 13, and 19 (ESI Table S2†). In addition, we later show in this report that, after confirming 28 possessed suitable fluorescent confocal imaging properties (refer to Confocal Microscopy section), it could be used as the competing ligand to measure and cross-validate the affinity of the new BODIPY 630/650-containing ligands for the AR (refer to later discussion, and Fig. 3). All test compounds were confirmed as stable under the fluorescent binding assay conditions as no significant degradation was seen when analysed using reversed-phase high-performance liquid chromatography (RP-HPLC) (ESI Fig. S2†).
All of the Fmoc-compounds (4–19), with the exception of 14 at the A1AR, were able to displace the binding of 2 to non-specific levels at both A1AR and A3AR. The affinity of the Fmoc-containing conjugate 4 was similar at both receptor subtypes (pKi ≈ 6.9) and addition of the BODIPY 630/650 moiety to afford conjugate 1 increased affinity at both receptors and displayed only marginal A1-receptor selectivity. Likewise, Fmoc-containing 5 was 3-fold A3-AR selective and displayed a slightly increased affinity for each receptor subtype on conversion to the BODIPY 630/650 conjugate 2, but maintained a similar A3AR/A1AR selectivity. These initial examples suggested that Fmoc-Ahx might potentially be suitable as an alternative to the BODIPY 630/650-X fluorophore in permitting a cost-effective pre-screening for optimal linker composition. We therefore set out to address two questions – (i) can introduction of a dipeptide moiety in the linker tune the affinity and/or selectivity of the conjugate, and (ii) does the use of Fmoc in place of BODIPY 630/650 give a translatable affinity/selectivity profile for dipeptide-containing conjugate screening?
Ala–Ala-linked 6 represented an equivalent congener to 5, but with the 5-aminopentanoyl moiety of the latter replaced with a dipeptide. The selectivity profile and affinity of 6 at the A1AR and A3AR was comparable to 5, demonstrating that this linker substitution was tolerated but offered no benefit in relation to either conjugate affinity or selectivity. This result was encouraging however, since it provided scope to change the amino acid side-chains in an iterative fashion, and therefore an Fmoc-based mini-library was synthesised (Scheme 1). Changing the N-terminal Ala of 6 to Ser (7), Tyr (8), or Asn (9) resulted in an interesting spectrum of pharmacological effects. For 7, the additional side chain hydroxyl resulted in an increased affinity at the A3AR whilst exerting no significant change at the A1AR. Taken together, this effected a 4-fold enhancement in selectivity for the former receptor. With 8, insertion of the phenol moiety produced an even more pronounced enhancement of A3AR affinity (pKi = 8.95 ± 0.06) coupled with a modest affinity increase at the A1AR; when taken together however this ultimately resulted in a 26-fold increase in A3AR selectivity. For conjugate 9, the observed improvement in A3AR selectivity is born out of a modest increase in A3AR affinity coupled with the only example in this ligand series of a drop in A1AR affinity.
Alternatively, swapping the C-terminal Ala of 6 to Ser (10), Tyr (11), or Asn (12) resulted in a less pronounced effect on receptor affinity than the N-terminal amino acid substitution described above. Of these three compounds, 10 and 11 showed a small increase in affinity for the A3AR compared to 6, with 12 possessing the lowest A3AR affinity (pKi = 7.35 ± 0.03) of the Fmoc-Ahx-dipeptide compounds tested. Conjugates 10 and 12 therefore displayed decreased A3AR/ A1AR selectivity profiles; the former as a result of a marginally greater enhancement of A1AR affinity compared to A3AR affinity, whereas for 12 it was a consequence of a modest decrease in A3AR affinity being coupled with a correspondingly small increase in A1AR affinity. From the results of these N- and C-terminal iterations, we elected two additional dipeptide-linked compounds for synthesis; Tyr–Ser containing 13 (N-terminal Tyr-conjugate 8 displayed the highest affinity A3AR, whilst the C-terminal Ser-conjugate 10 displayed the second highest affinity at the A3AR for their respective 3-ligand series) and Tyr–Tyr containing 14 (N-terminal Tyr-conjugate 8 displayed the highest affinity A3AR, whilst the C-terminal Tyr-conjugate 11 exhibited the highest affinity at the A3AR for their respective 3-ligand series). The Tyr–Ser-linked 13 revealed a modest decrease in A3AR affinity alongside a slight increase in A1AR affinity when compared to Tyr–Ala-linked 8. Compared to the Ala–Ser-containing 10 however, 13 showed a higher affinity at both receptors and a 5-fold increase in selectivity towards the A3AR. The remaining dipeptide-conjugate containing Tyr–Tyr (14) showed a substantial loss of affinity for both the A1AR and A3AR compared to 8 and 11. Taken together these results identify a complex interplay between each of the two amino acid side-chain contributions to the ultimate observed receptor affinity.
The pharmacology of the Fmoc-Ahx-dipeptide series demonstrated that inclusion and subsequent amino acid iteration of a dipeptide linker does indeed influence the affinity and selectivity of the conjugate towards the A3AR and A1AR. Before investigating if an Fmoc-dipeptide conjugate library is a good predictor of the pharmacological properties of the corresponding BODIPY 630/650 conjugates, we first considered the influence of the Ahx linker and Fmoc group in a Fmoc-Ahx-dipeptide conjugate. A Tyr–Ser-congener without the Ahx linker (19) and another with a free N-terminus (23) were synthesised. Congeners 19 and 23 both showed very similar receptor subtype affinities and hence selectivities, yet a substantial reduction in affinity for the A3AR and a minor drop in affinity for the A1AR when compared to Fmoc-Ahx-linked 13. This suggests that the potentially protonated N-terminus of 23 may not be a significant factor in the loss of AR affinity compared to 13, but rather the lack of an “optimally positioned” Ahx and/or Fmoc moiety.
The Fmoc-dipeptide based mini-compound library members (6–14) showed a general trend of A3AR selectivity over the A1AR. The three Fmoc-compounds that possessed the highest A3AR affinity and selectivity (Ser–Ala 7, Tyr–Ala 8 and Tyr–Ser 13) were subsequently selected for conversion of the Fmoc moiety to BODIPY 630/650 (giving 24, 25, and 27 respectively). The Fmoc moiety of the compound with the lowest A3AR affinity and minimal selectivity (Ala–Asn-12) was also exchanged with BODIPY 630/650 to give 26, to ascertain if the non-selective and lower affinity profile of 12 was faithfully translated into the fluorescent conjugate. Parallel shifts in the agonist concentration-response curves at the A3AR were observed for fluorescent XAC-conjugates 2 and 24–27 (Table 1, ESI Fig. S2†), with a Schild slope value not differing from unity; indicating that they act as competitive antagonists. However non-parallel shifts in the NECA concentration-response curves were observed in the presence of the previously reported 1, suggesting non-competitive antagonism at the A3AR. In a [3H]DPCPX whole cell binding assay all compounds (2, 24–27) including 1 could displace radio-ligand binding to non-specific levels, indicating that they were binding to the same site on the A1AR.
BODIPY 630/650 conjugate 24, containing the Ser–Ala linkage, displayed the highest affinity for both the A3AR (pKD = 9.29 ± 0.17) and A1AR (pKi = 8.39 ± 0.09) of all the compounds in this study. There was a substantial increase in the affinity of 24 (1.4 log unit increase in pKi at each receptor) compared to Fmoc-analogue 7. The selectivity profile was reproduced, with both 24 and 7 showing 7- and 8-fold A3AR/A1AR selectivity respectively. An enhanced A3AR and A1AR affinity of the BODIPY 630/650 conjugate compared to the Fmoc analogue was also observed for the 4/1 and 5/2 compound pairs. It is interesting to note this gain in affinity by the presence of BODIPY 630/650 has also been observed for other fluorescent AR ligands, for example quinoxaline-based antagonists23 and even for agonist-based conjugates.24 Tyr–Ala-linked 25 was the only BODIPY 630/650-conjugate that had a reduced affinity (0.5 log unit decrease) for the A3AR compared to the corresponding Fmoc-compound (8). Therefore since the A1AR affinity of 25 was unchanged, there was a resultant decrease in A3AR selectivity from 29-fold to 6-fold. The Ala–Asn dipeptide was chosen based on 12 being the least selective of all the Fmoc-Ahx-dipeptide compounds. When Ala–Asn was incorporated into BODIPY 630/650 conjugate 26, again its affinity increased and its A3AR selectivity shifted from 2-fold to 6-fold. The BODIPY 630/650-conjugate with the greatest A3AR selectivity (32-fold) was Tyr–Ser-linked 27 (A3AR, pKD = 9.12 ± 0.05; A1AR, pKi = 7.62 ± 0.13) (Fig. 2). Compared to Fmoc-containing 13, 27 retained a similar affinity for the A1AR but an increased affinity for the A3AR, thereby improving its A3AR selectivity. A comparison of the BODIPY 630/650-peptide conjugates (24–27), highlighted the significant effect of the dipeptide linker composition on their pharmacological profile, as previously observed for the Fmoc-dipeptide compound series (6–14).
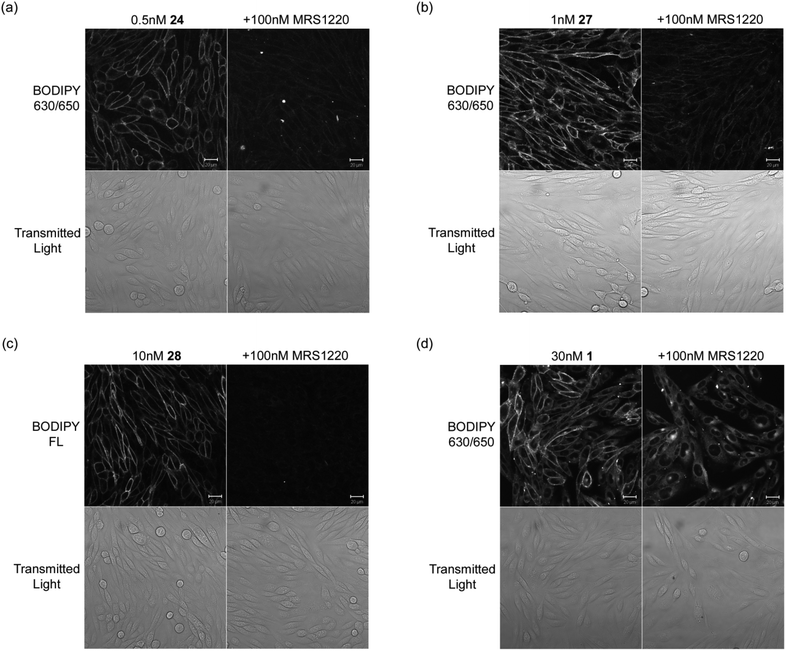 | ||
| Fig. 2 Live cell confocal imaging of the human A3AR expressed in CHO cells using (a) 24 (b) 27 (c) 28 and compared to (d) the non-peptide linked 1. CHO-A3 SPAP cells were incubated with fluorescent ligand for 30 min at 22 °C in the absence (left columns) or presence (right columns) of MRS1220. Single equatorial confocal images (BODIPY 630/650 or BODIPY FL) and their corresponding transmitted light images (Transmitted Light, lower rows) were obtained in the continued presence of the fluorescent ligand and/or unlabelled antagonist. For each compound, images in the presence and absence of MRS1220 were obtained using identical settings for laser power, detector offset and gain. Images shown are from a single experiment representative of 3–5 performed. Scale bars = 20 μm. | ||
Returning to our original hypothesis, iterative chemical changes in the linker component of a conjugate can therefore be used to fine tune affinity and/or selectivity for a given receptor. However results obtained in this study clearly suggest that the Fmoc-conjugate pharmacology does not reliably predict the corresponding BODIPY 630/650-conjugate pharmacology. It is interesting to note though that in the majority of cases, addition of the BODIPY fluorophore increased the affinity by more than 3-fold; adding further substance to the hypothesis that the fluorophore is implicated in some form of exosite binding, with a significant influence on overall ligand affinity for the receptor. It is highly likely that the BODIPY fluorophore is sampling different AR residues to the Fmoc moiety. Indeed, when one considers the molecular overlay of the two groups it is conceivable that the Fmoc portion could be interacting with similar AR residues to the styryl group that is contained within the BODIPY 630/650 entity.
Since conjugate 27 was our leading fluorescent compound in terms of the maximum A3AR selectivity, we incorporated the same Tyr–Ser linker into a BODIPY-FL conjugate to furnish 28 (Scheme 2, Table 1). Previous attempts in our laboratory to construct a BODIPY-FL-containing AR fluorescent tool have been unsuccessful. For example Baker et al.22 reported that a conjugate of 3 linked to BODIPY-FL via an 8-(2-aminoethylamino)-8-oxooctanoyl spacer showed no displacement of [3H]DPCPX specific binding at concentrations up to 10 μM in a CHO-A1 whole-cell binding assay. Utilising our new peptidic-linker approach, BODIPY-FL conjugate 28 showed an affinity for the A3AR (A3AR, pKD = 7.96 ± 0.09) that was acceptable and promising in terms of an exploitable fluorescent probe for imaging, albeit with a reduced affinity for the A3AR and A1AR compared to both 13 and 27. The 29-fold A3AR/A1AR selectivity of 28 was similar to the 32-fold selectivity observed for the equivalent BODIPY 630/650 conjugate (27).
Live cell confocal microscopy
The measured A3AR affinity of 24, 27, and 28 does not necessarily imply that these conjugates will be useful as fluorescent probes for the A3AR, as the physiochemical and photochemical properties must also be appropriate. A good fluorescent ligand must show low levels of nonspecific membrane binding, should not significantly diffuse into the cell cytosol and should have a sufficiently high quantum yield when bound to the receptor to provide a good signal to noise ratio when imaged.We therefore used confocal microscopy to examine the ability of 24, 27 and 28 to detect the human A3AR in CHO-A3 SPAP cells, and in particular to compare their imaging properties to those of our original non-peptide conjugated XAC-derivative 121 (Fig. 2). In each case, CHO-A3 SPAP cells were incubated with fluorescent ligand at a concentration equivalent to its KD for the receptor (Table 1) to ensure equivalent receptor occupancies. Following incubation of CHO-A3 SPAP cells with 0.5 nM 24 and 1 nM 27 for 30 min, strong defined membrane fluorescence was seen, whilst levels of intracellular fluorescence remained low (Fig. 2a,b). The bulk of the observed membrane binding was to the A3AR, since when cells were pre-treated with the non-fluorescent A3AR antagonist MRS1220, the binding was significantly reduced. The conjugate 28 containing the BODIPIY-FL fluorophore also produced clear distinct and displaceable membrane binding. In comparison to the dipeptide-linked conjugates (24, 27, 28), whilst a 30 min incubation with 1 also produced significant and bright membrane-localised fluorescence, there was also substantially more cytoplasmic fluorescence. This was particularly evident in cells pre-treated with MRS1220, where the membrane-localised fluorescence was prevented, but there was a substantial increase in cytoplasmic signal (Fig. 2d).
Fluorescent binding assay using compound 28
The success of 24, 27 and 28 as fluorescent chemical tools for the A3AR demonstrates that this design-based approach of using side-chain functionalised peptidic linkers has significant benefits over non-peptidic linkers. In addition to tuning the affinity and selectivity of the conjugate, the increased levels of specific membrane binding at the low concentrations and the propensity of the compounds to remain localised to cell membrane even after extended incubation is a significant advantage over earlier non-peptidic fluorescent derivatives of 3 (for example 1). Because of the refined imaging properties, now for the first time we were able to use a green ligand, 28, as the tracer ligand in place of the previously employed 2 (Table 1) in an analogues competition binding assay (Fig. 3). This enabled measurement of the affinity values for the new BODIPY 630/650-containing compounds (24, 25, 27) for the A3AR using 28 as the tracer, as there is a large separation in the excitation/emission wavelengths of the BODIPY FL and BODIPY 630/650 fluorophores.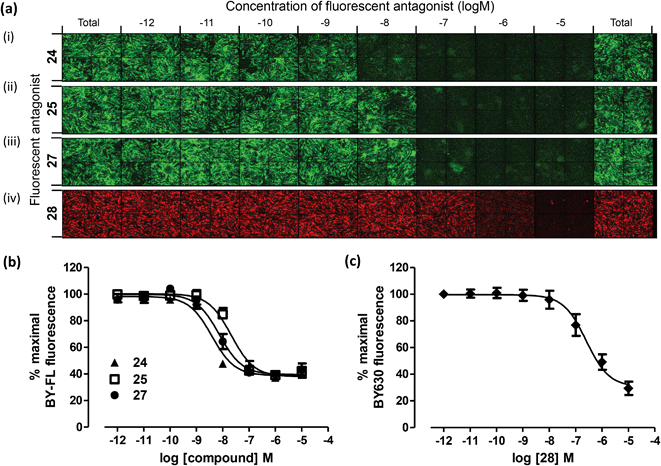 | ||
| Fig. 3 Measurement of the affinity of 24, 25, 27 and 28 at the A3AR using a fluorescence based competition binding assay. (a) Representative montages of images of CHO-A3 SPAP cells with increasing concentrations of 24 (i), 25 (ii) or 27 (iii) using tracer 28 and BODIPY-FL fluorescence measured; or with increasing concentrations of 28 (iv) using tracer 2 and BODIPY 630/650 fluorescence measured. Competition curves generated from the total BODIPY FL (b) or BODIPY 630/650 (c) image intensity. Data are normalised to maximal 28 (b) or 2 (c) fluorescence in the absence of any competing ligand. Each data point represents the mean ± SEM from four (24), five (25, 27 and 28) or six (d) experiments performed in duplicate. | ||
Clear concentration-dependent displacement of 28 by increasing concentrations of 24, 25 and 27 was observed, which enabled generation of competition binding curves and estimation of pKi values (pKi = 8.96 ± 0.03, 8.20 ± 0.06, 8.70 ± 0.10 for 24, 25 and 27 respectively (Fig. 3a i–iii, 4b). The pKi values measured for 24, 25 and 27 using 28 as the competing tracer are of the same order of magnitude as the values obtained for 24, 25, and 27 at the A3AR in the CRE-SPAP gene transcription assay (Table 1). Using the same competition assay that was originally used to analyse the Fmoc-based compounds with tracer 2 (Table 1) the affinity of the BODIPY FL-labelled 28 for the A3AR was also determined (pKi = 7.55 ± 0.19) (Fig. 3a iv, 3c), and again showed good correlation with the A3AR CRE-SPAP measurements (Table 1).
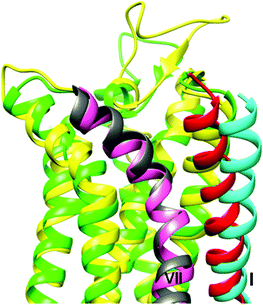 | ||
| Fig. 4 Comparison of the homology models of the A1AR (yellow) and A3AR (green), highlighting the different positions of the extracellular ends of helix I (red for A1AR, cyan for A3AR). For the latter protein this results in the formation of a cleft and groove at the interface with helix VII (grey for A1AR, pink for A3AR) that is absent in the A1AR model. | ||
Molecular modelling
Molecular modelling of the A1AR and A3AR as well as docking of the BODIPY 630/650-containing compounds 27 and 26 (greatest and least A3AR/A1AR selectivity respectively) was carried out in an attempt to rationalise the pharmacological data obtained in this study in terms of the selectivity imbued by the peptide linker. The homology models for the two proteins (see ESI Fig. S3–S6, Tables S3–S4†) were generally quite similar, however in the A3AR model the extracellular end of helix I was predicted to be further away from the neighbouring helix VII, generating a cleft and groove that was absent in the A1AR model (Fig. 4) and as is discussed below, this difference proved to be significant.Docking of 27 to the A1AR failed to reveal a single high-scoring pose. While the XAC-component of the molecule remained in its crystallographic position, the dipeptide linker and terminal BODIPY sampled a variety of alternative conformations, none of which featured particularly significant ligand–protein interactions, and all of which placed the BODIPY moiety in a rather solvent-exposed environment (Fig. 5a). However for the A3AR, docking of 27 produced a clear single cluster of poses (Fig. 5b) which revealed similar interacting regions within the pocket and featured the dipeptide/BODIPY portion exiting through the transmembrane (TM) I/ TM VII cleft and groove to the space otherwise occupied by the aliphatic chains of the membrane lipids (ESI Fig. S7†).
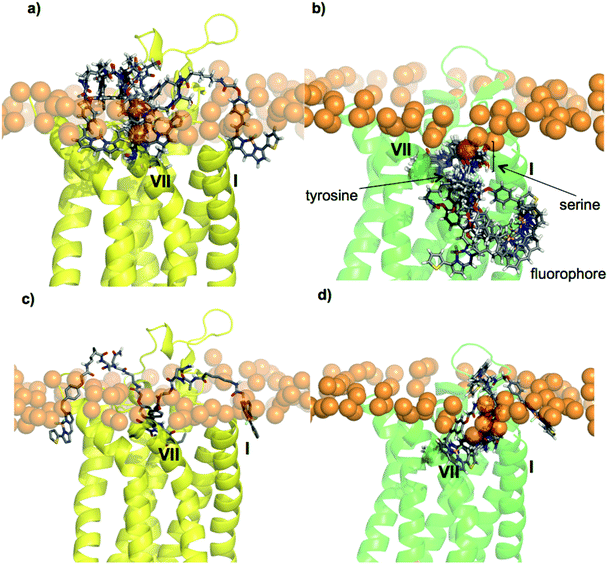 | ||
| Fig. 5 Docking of fluorescent conjugates using Glide into the AR homology models. (a) Five poses generated for 27 in A1AR, (b) six poses generated for 27 in A3AR, (c) two poses generated for 26 in A1AR, (d) three poses generated for 26 in A3AR. The phosphorus atoms of the POPC upper leaflet headgroups are shown in orange, to mark the water–lipid interface. | ||
Docking of 26 to both the A1AR and the A3AR provided some rationalisation for why this compound possesses the lowest A3AR/A1AR selectivity. Only two poses could be generated for 26 bound to A1AR (Fig. 5c) and three for it bound to A3AR (Fig. 5d). In neither case was a single, well-defined binding mode identified, nor was the BODIPY portion buried deep in the lipid bilayer. In the absence of single, well-defined predictions for the binding poses it is not appropriate to make detailed analyses of how the various structural features in these two proteins and ligands contribute to the patterns of affinity and selectivity, however some general insights are possible. Primarily, the cleft and groove between helices I and VII of the A3AR may provide an opportunity for a suitably designed fluorescent ligand to bury its fluorophore in the lipid environment in a way much harder to achieve for the A1AR. For effective binding to the A3AR the nature of the dipeptide linker is important for two separate reasons. Firstly, the amino acids present in the ligand should have suitable functionality to interact with the protein in the region of the cleft and groove. Secondly, the docking studies suggest that the dipeptide moiety of the conjugate is quite exposed to the surrounding environment as it exits the protein and this environment includes areas within the lipid bilayer, and in close proximity to the phospholipid headgroups. Interestingly, it has been established that certain amino acids, such as serine and in particular tyrosine, have a greater propensity to feature in this environment26 while in contrast amino acids such as asparagine are not as favourable.
Conclusions
A key feature of the current state of research into GPCRs is an increasing awareness of the need to scrutinise their cellular location and involvement in complex signalling interactions. As such, fluorescence-based approaches have developed apace to meet these needs.27 The necessity to supply high quality fluorescent ligands as chemical tools to study this important area of bioanalysis therefore remains a primary research goal.28 The methods applied to fluorescent ligand chemical synthesis have matured significantly and this study has further highlighted the important contribution of the linker to the overall pharmacology of fluorescently labelled GPCR ligands. Specifically we have shown that the non-selective A1AR and A3AR antagonist 3 can be used as the parent ligand to ultimately generate higher affinity and subtype-selective fluorescent probes (for example 27 and 28) using advantageous amino acid selection within a dipeptide linker.We have also demonstrated that fluorescent ligand measurements can be undertaken using live cells with ligands conjugated to spectrally separated fluorophores. This allowed affinity data to be obtained for each ligand using the alternative as the competing probe. In silico analysis of BODPIY-630/650-X dipeptide-conjugates docked into homology models of the A1AR and A3AR has identified potential sites of molecular interaction between the peptidic linker moiety and the receptor, which may help rationalise their observed selectivity. For GPCRs where there are no subtype discriminating orthosteric ligands to select as a starting point for conjugate synthesis, the methods established here could prove even more valuable for the design of sub-type selective fluorescent probes. One could postulate that further SAR refinement of the dipeptide sequence, or extending the peptide length may impart even greater subtype selectivity and this is the on-going focus of work within our laboratories.
Acknowledgements
This work was support by the Medical research Council, UK (Grant Number G0800006).Notes and references
- V. O. Nikolaev, A. Moshkov, A. R. Lyon, M. Miragoli, P. Novak, H. Paur, M. J. Lohse, Y. E. Korchev, S. E. Harding and J. Gorelik, Science, 2010, 327, 1653–1657 CrossRef CAS.
- L. T. May, T. J. Self, S. J. Briddon and S. J. Hill, Mol. Pharmacol., 2010, 78, 511–523 CrossRef CAS.
- L. T. May, L. J. Bridge, L. A. Stoddart, S. J. Briddon and S. J. Hill, FASEB J., 2011, 25, 3465–3476 CrossRef CAS.
- L. A. Stoddart, A. J. Vernall, J. L. Denman, S. J. Briddon, B. Kellam and S. J. Hill, Chem. Biol., 2012, 19, 1105–1115 CrossRef CAS.
- K. A. Jacobson, Bioconjugate Chem., 2009, 20, 1816–1835 CrossRef CAS.
- R. J. Middleton and B. Kellam, Curr. Opin. Chem. Biol., 2005, 9, 517–525 CrossRef CAS.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS.
- V.-P. Jaakola, M. T. Griffith, M. A. Hanson, V. Cherezov, E. Y. T. Chien, J. R. Lane, A. P. Ijzerman and R. C. Stevens, Science, 2008, 322, 1211–1217 CrossRef CAS.
- E. Y. T. Chien, W. Liu, Q. Zhao, V. Katritch, G. W. Han, M. A. Hanson, L. Shi, A. H. Newman, J. A. Javitch, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1091–1095 CrossRef CAS.
- S. Granier, A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, W. I. Weis and B. K. Kobilka, Nature, 2012, 485, 400–404 CrossRef CAS.
- A. C. Kruse, J. Hu, A. C. Pan, D. H. Arlow, D. M. Rosenbaum, E. Rosemond, H. F. Green, T. Liu, P. S. Chae, R. O. Dror, D. E. Shaw, W. I. Weis, J. Wess and B. K. Kobilka, Nature, 2012, 482, 552–556 CrossRef CAS.
- J. S. Mason, A. Bortolato, M. Congreve and F. H. Marshall, Trends Pharmacol. Sci., 2012, 33, 249–260 CrossRef CAS.
- (a) K. A. Jacobson, K. L. Kirk, W. L. Padgett and J. W. Daly, Mol. Pharmacol., 1986, 29, 126–133 CAS.
- B. B. Fredholm, A. P. Ijzerman, K. A. Jacobson, J. Linden and C. E. Müller, Pharmacol. Rev., 2011, 63, 1–34 CrossRef CAS.
- S. Schenone, C. Brullo, F. Musumeci, O. Bruno and M. A. Botta, Curr. Top. Med. Chem., 2010, 10, 878–901 CrossRef CAS.
- S. Cohen, S. M. Stemmer, G. Zozulya, A. Ochaion, R. Patoka, F. Barer, S. Bar-Yehuda, L. Rath-Wolfson, K. A. Jacobson and P. Fishman, J. Cell. Physiol., 2011, 226, 2438–2447 CrossRef CAS.
- J-F. Chen, H. K. Eltzschig and B. B. Fredholm, Nat. Rev. Drug Discovery, 2013, 12, 265–286 CrossRef CAS.
- B. B. Fredholm, A. P. Ijzerman, K. A. Jacobson, K. N. Klotz and J. Linden, Pharmacol. Rev., 2001, 53, 527–552 CAS.
- D. Xin, Y. Wang and J. Xiang, Pharm. Res., 2010, 27, 380–389 CrossRef CAS.
- S.-H. Lee, J. J. Moon, J. S. Miller and J. L. West, Biomaterials, 2007, 28, 3163–3170 CrossRef CAS.
- S. J. Briddon, R. J. Middleton, Y. Cordeaux, F. M. Flavin, J. A. Weinstein, M. W. George, B. Kellam and S. J. Hill, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4673–4678 CrossRef CAS.
- J. G. Baker, R. J. Middleton, L. Adams, M. T. May, S. J. Briddon, B. Kellam and S. J. Hill, Br. J. Pharmacol., 2010, 159, 772–786 CrossRef CAS.
- A. J. Vernall, L. A. Stoddart, S. J. Briddon, S. J. Hill and B. Kellam, J. Med. Chem., 2012, 55, 1771–1782 CrossRef CAS.
- C. L. Dale, S. J. Hill and B. Kellam, MedChemCommun, 2012, 3, 333 RSC.
- Y. Cordeaux, S. J. Briddon, A. E. Megson, J. McDonnell, J. M. Dickenson and S. J. Hill, Mol. Pharmacol., 2000, 58, 1075–1084 CAS.
- S. Mitaku, T. Hirokawa and T. Tsuji, Bioinformatics, 2002, 18, 608–616 CrossRef CAS.
- S. J. Briddon and S. J. Hill, Trends Pharmacol. Sci., 2007, 28, 637–645 CrossRef CAS.
- M. Leopoldo, E. Lacivita, F. Berardi and R. Perrone, Drug Discovery Today, 2009, 14, 706–712 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation of synthetic compounds, further pharmacological analysis, and details of the homology modelling and docking. See DOI: 10.1039/c3ob41221k |
| ‡ These authors contributed equally to this work. |
| § B.K. and S.J.H. are founding directors of the University of Nottingham spinout company CellAura Technologies Ltd. |
| This journal is © The Royal Society of Chemistry 2013 |