 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented regiochemical control in the formation of aryl[1,2-a]imidazopyridines from alkynyliodonium salts: mechanistic insights†
Luke I.
Dixon
a,
Michael A.
Carroll
*a,
Thomas J.
Gregson‡
b,
George J.
Ellames‡
b,
Ross W.
Harrington
a and
William
Clegg
a
aSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: m.a.carroll@ncl.ac.uk; Fax: +44 (0)191 222 6929
bDepartment of Isotope Chemistry and Metabolite Synthesis, sanofi-aventis, Alnwick, Northumberland NE66 2JH, UK
First published on 15th July 2013
Abstract
Aryl(alkynyl)iodonium salts have been demonstrated to be valuable precursors to a diverse range of heteroaromatic ring systems including aryl[1,2-a]imidazopyridines. Successful application, using the recently described aryl(alkynyl)iodonium trifluoroacetate salts, is described, highlighting for the first time that the regioselectivity of this process is both counter-ion and concentration dependent. Studies with a carbon-13 labelled substrate established that the reactions of alkynyliodonium salts are highly complex and that multiple mechanistic processes appear to be underway simultaneously.
Introduction
Alkynyliodonium salts, first discovered in 1965 by Beringer and Galton,1 are a highly versatile class of compounds and have found widespread application in both organic and inorganic syntheses and as such have been the subject of numerous reviews.2–10As highly electron-deficient acetylenic species, alkynyliodonium salts are reactive partners in cycloaddition reactions3 including 1,3-dipolar cycloadditions11–14 and Diels–Alder chemistry.15–17 The hypernucleofuge nature of the iodoarene in alkynyliodonium salts also makes them excellent sources of carbenes,10 providing access to a wealth of cyclic species such as cyclopentenes18,19 and pyrroles;20 recently highly synthetically challenging cyanocarbenes have also been generated.21,22
In the same fashion, indoles, furopyridines, indenes, imidazopyridines, imidazopyrimidines, furotropones, furonaphthoquinones, thiazoles, selenazoles and benzofurans (Scheme 1) may also be formed.2,4 Cumulatively, these ring systems account for a wide range of known pharmacophores, yet the potential for alkynyliodonium salts in the preparation of heterocycles remains to be exploited.
 | ||
| Scheme 1 Heteroaromatics from alkynyliodonium salts.2,4 | ||
Commonly used anions in alkynyliodonium salts include mesylates,23 tosylates,24,25 triflates26,27 and tetrafluoroborates28,29 due to their low nucleophilicity, though in many of these cases addition of the anion to the β-acetylenic position was still observed.30 As such the development of alkynyliodonium salts with an intramolecular anion has been an area of continuing interest as a means of restraining the addition reaction.30–36 In contrast, alkynyliodonium trifluoroacetates (TFA) have received little attention,3,37 though it was recently shown by us that they are not only readily prepared38 but also available on a large scale (>0.5 mol).39
Despite the wealth of alkynyliodonium salts reported to date, very little is known about the effects of the counter-ion used or their solution state behaviour. A novel comparison is presented herein between the TFA salts and some of the more common alkynyliodonium salts, demonstrating for the first time that the anion used imparts a profound effect on the regioselectivity of arylimidazo[1,2-a]pyridine formation; it was also found that the outcome of the reaction could be manipulated through substrate concentration. This study highlights a dynamic solution state for alkynyliodonium salt derivatives and that, through control of experimental conditions, access to a plethora of substituted heteroaromatics may be achieved, with alternative regioisomers possible from the same starting material.
In 2004 Liu and co-workers reported the synthesis of a range of 2-arylimidazo[1,2-a]pyridines (cf. 3a) from the reaction of alkynyliodonium tosylates and 2-aminopyridine.40 Surprisingly, the analogous reaction of 1a afforded both 2- and 3-substituted imidazo[1,2-a]pyridines in roughly equal amounts (Scheme 2), as confirmed by X-ray crystallography (ESI† and Fig. 1).
![Synthesis of imidazo[1,2-a]pyridines from 1a.](/image/article/2013/OB/c3ob41112e/c3ob41112e-s2.gif) | ||
| Scheme 2 Synthesis of imidazo[1,2-a]pyridines from 1a. | ||
 | ||
| Fig. 1 Structures of the cations of 3a·HCl·H2O (left) and 4a·HCl·2H2O (right), with 40% probability displacement ellipsoids; N atoms are shown with shading. | ||
This intriguing production of two regioisomers led us to repeat the procedure reported by Liu and co-workers40 using the tosylate, 5, to confirm that just one regioisomer had indeed been produced. In our hands, the experiment showed that the major product of the reaction was in fact 4a and that, although this was dominant, trace amounts of 3a were also present (2%, 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44%, 4a); comparison of the 1H-NMR data with other reports supports this regiochemical assignment.41–44 Having optimized the reaction shown in Scheme 2 for a range of solvents and bases (see ESI†), fluorobenzene (PhF) (to minimise intermolecular insertion) and K2CO3 were chosen respectively, and at room temperature to minimize decomposition of the alkynyliodonium salts.
:44%, 4a); comparison of the 1H-NMR data with other reports supports this regiochemical assignment.41–44 Having optimized the reaction shown in Scheme 2 for a range of solvents and bases (see ESI†), fluorobenzene (PhF) (to minimise intermolecular insertion) and K2CO3 were chosen respectively, and at room temperature to minimize decomposition of the alkynyliodonium salts.
Heterogeneous bases and aprotic,45 non-coordinating solvents gave the best results. Despite slightly lower yields, PhF was chosen as it resulted in a ‘cleaner’ reaction facilitating isolation of the products. Comparison with other alkynyliodonium salts showed that 3a was undetectable using the triflate (6), whereas the cyclic iodane, 7 (see ESI†), produced a ratio of products between that observed for the trifluoroacetate and tosylate salts (Table 1). Only the phenyliodonium derivatives were investigated since variation of the second aromatic ring was previously found to have no effect in the preparation of 2-arylfuro[3,2-c]pyridines.38

Such a dependence on the anion used has never been reported previously in the formation of heteroaromatics from alkynyliodonium salts, though the literature suggests that the conversion of alkynyliodonium salts to alkenyliodonium salts appears to demonstrate stereoselectivity for the E- or Z-isomer depending on the counter-ion used.45–55 The influence of solvent on ion pair separation, and hence choice of counter-ion, has also been shown to affect the reactions of diaryliodonium salts.56
In addition to the counter-ion dependence of the reaction it was found that the concentration of the reactants also exerted an unexpected degree of control over the product distribution (Table 2 and Fig. 2).
 | ||
| Fig. 2 Concentration dependence for the reaction of 1a and 2. | ||

It should be noted that 1a was soluble in DCM and CHCl3 at all the concentrations tested, though not in PhF; however, in all three solvents, rapid dissolution of 1a was observed on addition of 2. Under the reaction conditions outlined in Scheme 1 the K2CO3 remained as a solid throughout. This suggests that the key reactive species is generated in situ as the same concentration dependence was observed for all three solvents (see ESI†).
Although some increase in yield was observed at higher dilutions, far more noticeable was the concentration-dependent regioselectivity favouring the 2-substituted regioisomer, 3a. To confirm this unexpected dependence, reactions were conducted using five different iodonium salt concentrations (Table 2 and Fig. 2). This ‘tuning’ of the reaction conditions has potential value, for example in drug discovery, where both regioisomers are accessible from a single set of reagents.
To establish whether the observed concentration dependence was restricted to the production of phenylimidazo[1,2-a]pyridines (3a and 4a) several alternative alkynyliodonium salts were also studied (1b–1d) and it was pleasing to note that a similar trend was found (Table 3).

The mechanism of imidazopyridine formation presented by Liu40 follows that previously reported by Wipf57 (and later Togo30) for the formation of thiazoles. An alternative route to the observed product has also been proposed by Ochiai.58 All of these options invoke a monomeric form of the aryl(alkynyl)iodonium salt as the starting species even though kinetic and spectroscopic evidence for other hypervalent iodine compounds has been reported that indicates the presence, in solution, not only of associated counter-ions, but also of higher-order structures (dimers, oligomers etc.).59–61 Such species have also been shown to be highly concentration-dependent60,61 and therefore contributions from these structures cannot be ruled out.
In addition these proposals rapidly result in loss of the counter-ion. However, to retain the influence of this component, and taking into account these prior mechanistic studies, we propose that intermediates such as 9 and 14 (Fig. 3) and 8 and 13 (Fig. 4) should also be considered in the mechanistic rationale since both the amino- and pyridinyl-nitrogen atoms of 2-aminopyridine are viable nucleophiles62 (Scheme 3: there may also be influence of the counter-ion in the subsequent steps due to the charged nature of the proposed intermediates).
![Proposed [10-I-4] intermediates, 14 and 18.](/image/article/2013/OB/c3ob41112e/c3ob41112e-f3.gif) | ||
| Fig. 3 Proposed [10-I-4] intermediates, 14 and 18. | ||
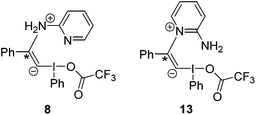 | ||
| Fig. 4 Potential intermediates of Michael addition. | ||
![Distribution of products from the reaction of [7′-1313C]-1.](/image/article/2013/OB/c3ob41112e/c3ob41112e-s3.gif) | ||
| Scheme 3 Distribution of products from the reaction of [7′-1313C]-1. | ||
Further complexity is introduced following alkylidene carbene formation (the carbene can either cyclize directly or undergo 1,2-migration prior to cyclization), resulting in the formation of 3a or 4avia several different pathways.
As the acetylenic products of 1,2-migration have been reported to be highly reactive,58,63 especially within a basic environment, they may prove difficult to observe and as such we prepared the isotopically labelled [7′-1313C]-1a to investigate the process.
This preliminary 13C-labelling study generated [2-1313C]-3a and [3-1313C]-4a as expected (Scheme 3); however, the isotopomer [3-1313C]-3a was also present, highlighting that a competitive 1,2-migration was occurring (Scheme 4) and suggesting that at least three reaction pathways are in operation.
![Mechanism of isotopomer formation; [3-1313C]-3a.](/image/article/2013/OB/c3ob41112e/c3ob41112e-s4.gif) | ||
| Scheme 4 Mechanism of isotopomer formation; [3-1313C]-3a. | ||
Conclusions
In summary, we have presented the first example of regiochemical control in the synthesis of heteroaromatics from alkynyliodonium salts. A protocol based on the counter-ion and concentration dependence of the process has been identified for the selective formation of 2-arylimidazo[1,2-a]pyridines and 3-arylimidazo[1,2-a]pyridines. In addition, initial studies using carbon-13 labelled substrates have demonstrated that, even though well studied, the reactions of alkynyliodonium salts are highly complex and that multiple mechanistic processes appear to be underway simultaneously.This new-found understanding is being applied to the preparation of a diverse range of heterocyclic ring systems which are of interest to our drug discovery programmes. Further work to resolve and differentiate the many mechanistic options is on-going.
Experimental
Reactions requiring anhydrous conditions were performed using oven- or flame-dried glassware and conducted under a positive pressure of nitrogen. Anhydrous solvents were prepared thus: DCM and MeCN were refluxed over CaH2; THF, ether and hexane were refluxed over sodium/benzophenone; toluene was refluxed over sodium; and dibromomethane, chloroform, 1,4-dioxane and fluorobenzene were stored over 3 Å molecular sieves. Infrared spectra were recorded on a Varian Scimitar Series 800 FT-IR with internal calibration. 1H and 13C-NMR spectra were recorded on a Bruker Advance 300 MHz spectrometer, a Jeol ECS 400 MHz spectrometer or a Jeol Lamda 500 MHz spectrometer with residual tetramethylsilane solvent as the reference for 1H and 13C. All coupling constants are given in Hz. Elemental analyses were carried out at London Metropolitan University. Mass spectrometry was recorded at the EPSRC Mass Spectrometry Service, Swansea or on a Waters LCT Premier (TOF-MS) operating in ‘W’ mode. Melting points were recorded on a Gallenkamp MF-370 melting point apparatus and are uncorrected. Automated flash chromatography was performed using a Varian IntelliFlash 971-FP discovery scale flash purification system. The terms ‘ether’ and ‘petrol’ refer to diethyl ether and the fractions boiling between 40 and 60 °C (unless otherwise specified) respectively. X-ray crystallographic data were measured on an Agilent Technologies Gemini A Ultra diffractometer at 150 K, using Mo or CuKα radiation; full details are in the ESI† and deposited with CCDC.CAUTION: Some hypervalent iodanes are potentially explosive and should be handled taking appropriate precautions.64–67
2-Phenylimidazo[1,2-a]pyridine (3a)40,68,69
K2CO3 (1.05 g, 7.62 mmol) and 2 (0.31 g, 3.27 mmol) were stirred together in dry PhF (250 mL) for 45 min before the addition of 1a (1.05 g, 2.51 mmol) by powder funnel. The solution was then stirred in darkness, at RT, under nitrogen overnight before being washed with water (300 mL) and extracted into DCM (2 × 75 mL). The organic fractions were dried (MgSO4) and concentrated in vacuo to give a brown oil. The crude product was purified by column chromatography (SiO2, Grace Resolve™ 80 g cartridge;§ sample loaded in DCM, 1:0 hexane–ether for 5 min then increasing to 3:7 over 120 min and holding at this solvent mixture until elution was complete) to give the product as a white crystalline solid (0.24 g, 1.22 mmol, 49%). Mp 132–133 °C (from DCM–petrol) (lit.,70 136–137 °C from cyclohexane); Rf 0.55 (4:1 ether–petrol); Found: C, 80.4; H, 5.3; N, 14.4. Calc. for C13H10N2: C, 80.4; H, 5.2; N, 14.4%; IR νmax/cm−1 (neat) 3130, 1632, 1502, 1475, 1447, 1369, 1353, 1304, 1273, 1246, 1203, 1145, 1077, 1027; δH (300 MHz, CDCl3; Me4Si) 8.10 (1H, d, H5 J 6.9), 7.97 (2H, d, H2′/H6′ J 7.2), 7.85 (1H, s, H3), 7.65 (1H, d, H8 J 9.0), 7.45 (2H, tapp., H3′/H5′ J 7.5), 7.34 (1H, t, H4′ J 7.5), 7.17 (1H, tapp., H7 J 6.9), 6.77 (1H, tapp., H6 J 6.0); δC (75 MHz, CDCl3; Me4Si) 146.41 (C2), 146.09 (C9), 134.27 (C1′), 128.99 (C3′/C5′), 128.27 (C4′), 126.55 C2′/C6′), 125.86 (C5), 124.79 (C7), 118.00 (C8), 112.66 (C6), 108.38 (C3); m/z (CI) 195 ([M + H]+, 100%), 95 (3), 80 (2), 52 (4). Found: [M + H]+, 195.0917. C13H11N2 requires 195.0917.
3-Phenylimidazo[1,2-a]pyridine (4a)43,71
Using K2CO3 (2.15 g, 15.55 mmol), 2-aminopyridine (0.61 g, 6.50 mmol), PhF (5 mL) and 1a (2.07 g, 4.95 mmol). White crystalline solid (0.36 g, 1.84 mmol, 37%). Mp 95–97 °C (from MeOH–H2O) (lit.,71 97–98 °C from petroleum ether); Rf 0.13 (4:1 ether–petrol); Found: C, 80.3; H, 5.1; N, 14.3. Calc. for C13H10N2: C, 80.4; H, 5.2; N, 14.4%; IR νmax/cm−1 (neat) 1634, 1603, 1540, 1499, 1480, 1450, 1442, 1352, 1296, 1272, 1262, 1175, 1148, 1134, 1074, 1009; δH (400 MHz, d6-DMSO; Me4Si) 8.45 (1H, d, H5 J 6.9), 7.72 (1H, s, H2), 7.61 (1H, d, H8 J 8.7), 7.57 (2H, d, H2′/H6′ J 7.3), 7.46 (2H, tapp., H3′/H5′ J 7.8), 7.34 (1H, t, H4′ J 7.4), 7.21 (1H, tapp., H7 J 7.8), 6.86 (1H, tapp., H6 J 6.6); δC (100 MHz, d6-DMSO; Me4Si) 146.06 (C9), 133.07 (C2), 129.74 (C3′/C5′), 129.42 (C1′), 128.37 (C4′), 127.97 (C2′/C6′), 125.58 (C3), 125.00 (C7), 124.45 (C5), 118.09 (C8), 113.30 (C6); m/z (ESI) 195 ([M + H]+, 100%). Found: [M + H]+, 195.0905. C13H11N2 requires 195.0922.
2-(4′-Methylphenyl)imidazo[1,2-a]pyridine (3b)69,72
Using K2CO3 (1.09 g, 7.89 mmol), 2 (0.32 g, 3.39 mmol), PhF (113 mL) and 1b (1.11 g, 2.56 mmol). White crystalline solid (0.26 g, 1.25 mmol, 49%) (as well as 4b (17%)). Mp 138–140 °C (from acetone) (lit.,72 145–146 °C); Rf 0.23 (4:1 ether–petrol); Found: C, 80.9; H, 5.7; N, 13.4. Calc. for C14H12N2: C, 80.7; H, 5.8; N, 13.5%.; IR νmax/cm−1 (neat) 3132, 1633, 1506, 1483, 1372, 1349, 1268, 1245, 1202, 1139; δH (500 MHz, CDCl3; Me4Si) 8.05 (1H, dtapp., H5 J 6.8, J 1.2), 7.84 (2H, d, H3′/H5′ J 8.1), 7.77 (1H, s, H3), 7.60 (1H, dd, H8 J 9.1, J 0.8), 7.23 (2H, d, H2′/H6′ J 8.1), 7.12 (1H, ddd, H7 J 9.1, J 6.8, J 1.3), 6.71 (1H, dtapp., H6 J 6.8, J 1.1), 2.38 (3H, s, Me); δC (125 MHz, CDCl3; Me4Si) 145.86, 145.55, 137.72, 130.89, 129.36, 125.90, 125.45, 124.41, 117.36, 112.21, 107.69, 21.22; m/z (ESI) 209 ([M + H]+, 100%). Found: [M + H]+, 209.1071. C14H13N2 requires 209.1073.
3-(4′-Methylphenyl)imidazo[1,2-a]pyridine (4b)
Using K2CO3 (1.06 g, 7.65 mmol), 2 (0.31 g, 3.29 mmol), PhF (24 mL) and 1b (1.07 g, 2.47 mmol). White crystalline solid (0.18 g, 0.88 mmol, 36%) (as well as 3b (27%)). Mp 84–86 °C (from DCM–ether); Rf 0.09 (4:1 ether–petrol); Found: C, 80.9; H, 5.7; N, 13.4. Calc. for C14H12N2: C, 80.7; H, 5.8; N, 13.5%; IR νmax/cm−1 (neat) 2981, 1634, 1545, 1490, 1353, 1295, 1255, 1166, 1148, 1013; δH (500 MHz, CDCl3; Me4Si) 8.30 (1H, dtapp., H5 J 6.9, J 1.2), 7.66 (1H, overlapped s, H2), 7.65 (1H, overlapped d, H8 J 8.0), 7.44 (2H, d, H3′/H5′ J 8.0), 7.32 (2H, dd, H2′/H6′ J 8.0, J 0.6), 7.17 (1H, ddd, H6 J 9.1, J 6.9, J 1.3), 6.78 (1H, tdapp., H7 J 6.9, J 1.2), 2.43 (3H, s, Me); δC (125 MHz, CDCl3; Me4Si) 145.99, 138.13, 132.28, 129.88, 128.00, 126.36, 125.73, 123.94, 123.34, 118.21, 112.34, 21.27. m/z (ESI) 209 ([M + H]+, 100%). Found: [M + H]+, 209.1072. C14H13N2 requires 209.1073.
2-(3′-Thienyl)imidazo[1,2-a]pyridine (3c)
Using K2CO3 (1.06 g, 7.64 mmol), 2 (0.32 g, 3.36 mmol), PhF (113 mL) and 1c (1.03 g, 2.43 mmol). White crystalline solid (0.25 g, 1.21 mmol, 50%) (as well as 4c (10%)). Mp 163–165 °C (from acetone); Rf 0.12 (4:1 ether–petrol); Found: C, 66.1; H, 3.9; N, 13.8. Calc. for C11H8N2S: C, 66.0; H, 4.0; N, 14.0%; IR νmax/cm−1 (neat) 3124, 1632, 1508, 1476, 1338, 1306, 1272, 1242, 1144, 1090; δH (500 MHz, d6-DMSO; Me4Si) 8.49 (1H, d, H5 J 6.7), 8.23 (1H, s, H3), 7.89 (1H, d, H2′ J 2.8), 7.61–7.55 (2H, m, H4′/H5′), 7.54 (1H, d, H8 J 9.0), 7.21 (1H, tapp., H7 J 6.6), 6.86 (1H, tapp., H6 J 6.7); δC (125 MHz, d6-DMSO; Me4Si) 144.94, 141.44, 136.38, 127.16, 127.07, 126.46, 125.15, 121.43, 116.76, 112.46, 109.31; m/z (ESI) 201 ([M + H]+, 100%). Found: [M + H]+, 201.0480. C11H9N2S requires 201.0481.
3-(3′-Thienyl)imidazo[1,2-a]pyridine (4c)
Using K2CO3 (1.07 g, 7.76 mmol), 2 (0.32 g, 3.36 mmol), PhF (24 mL) and 1c (1.05 g, 2.48 mmol). White crystalline solid (0.12 g, 0.62 mmol, 25%) (as well as 3c (33%)). Mp 54–57 °C (from DCM); Rf 0.06 (4:1 ether–petrol); IR νmax/cm−1 (neat) 3090, 1690, 1637, 1576, 1501, 1483, 1343, 1330, 1299, 1264, 1225, 1169, 1154, 1128, 1087, 1019; δH (500 MHz, d6-DMSO; Me4Si) 8.61 (1H, d, H5 J 7.0), 7.93 (1H, dd, H2′ J 1.7, J 1.3), 7.85 (1H, s, H2), 7.76 (1H, dd, H5′ J 5.0, J 2.1), 7.64 (1H, d, H8 J 8.5), 7.54 (1H, dd, H4′ J 5.0, J 1.3), 7.29 (1H, ddd, H7 J 8.5, J 6.7, J 1.7), 6.99 (1H, tdapp., H6 J 6.8, J 1.1); δC (125 MHz, d6-DMSO; Me4Si) 145.18, 132.58, 128.97, 127.20, 127.08, 124.57, 124.27, 121.18, 121.06, 117.40, 112.88; m/z (ESI) 201 ([M + H]+, 100%). Found: [M + H]+, 201.0479. C11H9N2S requires 201.0481.
2-(4′-Bromophenyl)imidazo[1,2-a]pyridine (3d)73
Using K2CO3 (0.96 g, 6.91 mmol), 2 (0.28 g, 2.95 mmol), PhF (102 mL) and 1d (1.13 g, 2.27 mmol). White crystalline solid (0.29 g, 1.47 mmol, 65%) (as well as 4d (13%)). Mp 196–198 °C (from acetone) (lit.,73 215–216 °C from heptane); Rf 0.34 (4:1 ether–petrol); Found: C, 57.3; H, 3.2; N, 10.1. Calc. for C13H9BrN2: C, 57.2; H, 3.3; N, 10.3%; IR νmax/cm−1 (neat) 2955, 2924, 1679, 1635, 1428, 1401, 1371, 1321, 1203, 1065, 1006; δH (500 MHz, d6-DMSO; Me4Si) 8.51 (1H, dtapp., H5 J 6.8, J 1.2), 8.42 (1H, s, H3), 7.91 (2H, d, H3′/H5′ J 8.7), 7.61 (2H, d, H2′/H6′ J 8.7), 7.56 (1H, dd, H8 J 9.1, J 1.0), 7.24 (1H, ddd, H7, J 9.1, J 6.8, J 1.3), 6.89 (1H, tdapp., H6 J 6.8, J 1.0); δC (125 MHz, d6-DMSO; Me4Si) 144.82, 143.13, 133.16, 131.55, 127.47, 126.87, 125.11, 120.59, 116.61, 112.34, 109.42; m/z (ESI) 275 ([81Br][M + H]+, 97%), 273 ([79Br][M + H]+, 100%). Found: [M + H]+, 273.0026. C13H10BrN2 requires 273.0022.
3-(4′-Bromophenyl)imidazo[1,2-a]pyridine (4d)69
Using K2CO3 (1.04 g, 7.55 mmol), 2 (0.31 g, 3.29 mmol), PhF (24 mL) and 1d (1.23 g, 2.48 mmol). White crystalline solid (0.18 g, 0.65 mmol, 26%) (as well as 3d (29%)). Mp 89–92 °C (from acetone); Rf 0.25 (4:1 ether–petrol); Found: C, 57.1; H, 3.2; N, 10.2. Calc. for C13H9BrN2: C, 57.2; H, 3.3; N, 10.3%; IR νmax/cm−1 (neat) 3023, 1537, 1499, 1478, 1398, 1351, 1303, 1291, 1264, 1174, 1151, 1101, 1074, 1007; δH (500 MHz, CDCl3; Me4Si) 8.25 (1H, dtapp., H5 J 7.0, J 1.2), 7.67 (1H, s, H2), 7.66 (1H, d, H8 J 9.1, J 1.1), 7.62 (2H, d, H2′/H6′ J 8.6), 7.40 (2H, d, H3′/H5′ J 8.6), 7.19 (1H, ddd, H7 J 9.1, J 6.7, J 1.3), 6.80 (1H, tdapp., H6 J 6.8, J 1.1); δC (125 MHz, CDCl3; Me4Si) 146.24, 132.68, 132.38, 129.35, 128.15, 124.49, 124.39, 123.06, 122.05, 118.29, 112.76; m/z (ESI) 275 ([81Br][M + H]+, 98%), 273 ([79Br][M + H]+, 100%). Found: [M + H]+, 273.0026. C13H10BrN2 requires 273.0022.
1′,1′-Dibromo-2′-[13C]-styrene ([131313131313C]-19)74
Triphenylphosphine (5.04 g, 19.22 mmol) and dry carbon tetrabromide (3.10 g, 9.34 mmol) were dissolved in dry DCM (30 mL) at 0 °C under an atmosphere of nitrogen. The solution was stirred for 30 minutes before the dropwise addition of benzaldehyde [13C]-carbonyl (0.50 g, 4.67 mmol) over 5 minutes. The solution was stirred at 0 °C for 1 hour before washing with an aqueous 5 M solution of CuSO4 (300 mL) followed by extraction into DCM (3 × 50 mL). The organic layers were combined, dried (MgSO4) and concentrated in vacuo. The resulting orange oily solid was dry loaded onto silica and purified by column chromatography (silica) to give the product as a pale orange clear oil which crystallized on standing (1.21 g, 4.60 mmol, 98%). Rf 0.74 (petrol 40/60); δH (300 MHz, CD2Cl2; Me4Si) 7.50–7.39 (2H, m), 7.44 (1H, d, J 159.07), 7.33–7.23 (3H, m); δC (75 MHz, CD2Cl2; Me4Si) 137.86 (C1′-label). m/z (EI) 265 ([81Br,81Br]M+, 8%), 263 ([81Br,79Br]M+, 18%), 261 ([79Br,79Br]M+, 8%), 184 (18), 182 (18), 103 (100). Found: M+, 260.8868. C713C1 H679Br2 requires 260.8864.Phenyl-α-[13C]acetylene ([1313131313C]-20)74
1′,1′-Dibromo-2′-[13C]-styrene ([1313131313C]-20) (1.21 g, 4.60 mmol) was dissolved in dry ether (30 mL) and cooled to −78 °C under an atmosphere of nitrogen. n-Butyllithium (2.17 M in hexanes, 5.41 mL, 11.75 mmol) was added dropwise over 10 minutes and the solution stirred for a further 30 minutes then for 1 hour at room temperature. The reaction was quenched with water (50 mL), washed with water (50 mL) and extracted into ether (3 × 50 mL). The organic layers were combined, dried (MgSO4) and concentrated in vacuo to give the product as a pale yellow oil (0.46 g, 4.43 mmol, 96%)¶ with sufficient purity to be used in subsequent reactions. δH (300 MHz, CDCl3; Me4Si) 7.54–7.49 (2H, m), 7.37–7.34 (3H, m), 3.09 (1H, d, J 49.52); δC (75 MHz, CDCl3; Me4Si) 84.05 (C1′-label). m/z (EI) 103 ([M]+, 100%). Found: M+, 103.0496. C713C1H6 requires 103.0498.Phenyl(phenyl-β-[13C]-ethynyl)iodonium trifluoroacetate ([13131313C]-1a)
Trifluoroacetic acid (1.01 g, 8.82 mmol) was added dropwise at −30 °C to a stirred solution of phenyliodonium bis(acetate) (1.35 g, 4.20 mmol) in dry DCM (25 mL) over a period of 10 minutes. After a further 30 minutes the solution was allowed to warm to room temperature and stirred for 1 hour before being re-cooled to −30 °C for the injection of a solution of phenyl[α-13C]acetylene, [1313131313C]-20, (0.46 g, 4.43 mmol) in dry DCM (5 mL) over 5 minutes. The resulting mixture was then allowed to reach room temperature over 3.5 hours in darkness before concentration in vacuo (to about 5 mL) followed by crystallization to give the product as a white, crystalline solid (0.63 g, 1.50 mmol, 36%). Mp 79–81 °C (dec.) (from DCM-ether-petrol) δH (400 MHz, CDCl3; Me4Si) 8.14 (2H, d, H2/H6, J 8.7), 7.58 (1H, dt, H4, J 7.8, J 0.9), 7.49–7.39 (5H, m), 7.34 (2H, t, H3/H5 J 7.3); δC (100 MHz, CDCl3; Me4Si) 162.55 (q, (CO) J 36.2), 133.55 (s, C2/C6), 132.90 (d, C3′/C5′, J 2.4), 132.14 (s, C3/C5), 131.96 (s, C4), 130.86 (d, C4′, J 1.4), 128.72 (d, C2′/C6′, J 5.5), 120.67 (s, C1), 120.40 (d, C1′, J 86.2), 104.10 (s, C7′-label), 45.14 (d, C8′, J 160.6); m/z (ESI) 306 ([M − TFA]+, 100%), 294 (14), 179 (19). Found: [M − TFA]+, 305.9861. C1313C1 H10I requires 305.9855.2-Phenyl-2/3-[13C]-imidazo[1,2-a]pyridine ([131313C]-3a) and 3-phenyl-3-[13C]-imidazo[1,2-a]pyridine ([1313C]-4a)
Potassium carbonate (0.30 g, 2.18 mmol) and 2-aminopyridine (0.09 g, 0.97 mmol) were stirred together in dry fluorobenzene (6.3 mL) for 45 minutes under an atmosphere of nitrogen before the addition of phenyl(phenyl[β-13C]ethynyl)iodonium trifluoroacetate (0.30 g, 0.70 mmol) by powder funnel. The solution was then stirred in darkness, at room temperature, overnight before being washed with water (150 mL) and extracted into DCM (4 × 30 mL). The organic fractions were combined, dried (NaSO4), filtered and concentrated in vacuo to a brown oil. The crude product was purified by column chromatography (Grace Resolve™ 80 g, 150 mL silica cartridge; 1:0 hexane–ether for 5 min then to 3:7 over 120 min and holding at this solvent mixture until elution was complete), loading the sample in DCM, to give the products as a white, crystalline solids; 2-Phenyl-2/3-[13C]-imidazo[1,2-a]pyridine ([131313C]-3a) (0.03 g, 0.14 mmol, 20%) and 3-Phenyl-3-[13C]-imidazo[1,2-a]pyridine ([1313C]-4a) (0.04 g, 0.21 mmol, 30%).
2-Phenyl-2/3-[13C]-imidazo[1,2-a]pyridine ([131313C]-3a)
R f 0.55 (4:1 ether–petrol); δH (400 MHz, CDCl3; Me4Si) 8.09 (1H, dt, H5 J 6.8, J 1.2), 7.97–7.93 (2H, m, H2′/H6′), 7.84 (0.82H, dd, H3 J 8.4, J 0.8), 7.84 (0.18H, d, H3 J 190.7) 7.63 (1H, d, H8 J 9.2), 7.43 (2H, tapp., H3′/H5′ J 8.0), 7.32 (1H, t, H4′ J 7.6), 7.15 (1H, ddd., H7 J 9.2, J 6.6, J 1.2), 6.75 (1H, dtapp., H6 J 6.6, J 0.8); δC (100 MHz, CDCl3; Me4Si) 145.82 (C2-label), 145.47 (d, C9 J 4.5), 133.76 (d, C1′ J 67.9), 133.76 (s, C1′), 128.82 (d, C3′/C5′ J 4.4), 128.82 (s, C3′/C5′), 128.08 (s, C4′), 126.14 (d, C2′/C6′ J 2.5), 125.66 (d, C5 J 7.9), 125.66 (s, C5), 124.79 (C7), 117.62 (d, C8 J 6.1), 117.62 (s, C8) 112.55 (C6), 108.21 (C3-label); m/z (CI) 196 ([M + H]+, 100%), 184 (12). Found: [M + H]+, 196.0947. C1213C1H11N2 requires 196.0950.
3-Phenyl-3-[13C]-imidazo[1,2-a]pyridine ([1313C]-4a)
R f 0.13 (4:1 ether–petrol); δH (400 MHz, CDCl3; Me4Si) 8.31 (1H, dd, H5 J 6.8, J 1.2), 7.68 (1H, od, H2 J 12.4), 7.65 (1H, od, H8 J 8.8), 7.55–7.52 (2H, m, H2′/H6′), 7.49 (2H, tapp., H3′/H5′ J 8.0), 7.39 (1H, tt, H4′ J 7.2, J 1.2), 7.17 (1H, ddd., H7 J 9.2, J 6.4, J 1.2), 6.86 (1H, tapp., H6 J 6.8); δC (100 MHz, CDCl3; Me4Si) 146.21 (d, C9 J 9.1), 132.57 (d, C2 J 69.0), 132.52 (s, C9), 129.40 (d, C1′ J 67.4), 129.33 (d, C3′/C5′ J 4.2), 129.33 (s, C3′/C5′), 128.27 (s, C4′), 128.11 (d, C2′/C6′ J 2.7), 125.84 (C3-label), 124.34 (s, C7), 123.43 (s, C5), 118.09 (C8), 113.30 (C6); m/z (CI) 196 ([M + H]+, 100%). Found: [M + H]+, 196.0945. C1213C1H11N2 requires 196.0950.
X-ray crystal structures are available for compounds 3a, 3a·HCl·H2O, 4a·HCl·2H2O and 7 (see ESI†). CCDC 907271–907274 contain the supplementary crystallographic data for this paper.
We thank sanofi-aventis, Newcastle University and the EPSRC (equipment grant EP/F03637X/1) for funding. We also thank Prof. W. McFarlane and Dr C. Wills for contributions in the field of NMR and the EPSRC National Mass Spectrometry Service Centre, Swansea, UK.
Notes and references
- S. A. Galton and F. M. Beringer, J. Org. Chem., 1965, 30, 1930–1934 CrossRef.
- T. Wirth, M. Ochiai, A. Varvoglis, V. V. Zhdankin, G. F. Koser, H. Tohma and Y. Kita, Hypervalent Iodine Chemistry, Springer, London, 2003 Search PubMed.
- P. J. Stang, Angew. Chem., Int. Ed. Engl., 1992, 31, 274–285 CrossRef.
- T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656–3665 CrossRef CAS.
- P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123–1178 CrossRef CAS.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523–2584 CrossRef CAS.
- P. J. Stang, J. Org. Chem., 2003, 68, 2997–3008 CrossRef CAS.
- T. Kitamura and Y. Fujiwara, Org. Prep. Proced. Int., 1997, 29, 409–458 CrossRef CAS.
- T. Wirth and U. H. Hirt, Synthesis, 1999, 1271–1287 CrossRef CAS.
- V. V. Zhdankin and P. J. Stang, Tetrahedron, 1998, 54, 10927–10966 CrossRef CAS.
- T. Kitamura, Y. Mansei and Y. Fujiwara, J. Organomet. Chem., 2002, 646, 196–199 CrossRef CAS.
- A. Varvoglis, E. Kotali and A. Bozopoulos, J. Chem. Soc., Perkin Trans. 1, 1989, 827–832 Search PubMed.
- P. J. Stang and P. Murch, Tetrahedron Lett., 1997, 38, 8793–8794 CrossRef CAS.
- G. Maas, M. Regitz, U. Moll, R. Rahm, F. Krebs, R. Hector, P. J. Stang, C. M. Crittell and B. L. Williamson, Tetrahedron, 1992, 48, 3527–3540 CrossRef CAS.
- P. J. Stang, P. Murch and A. M. Arif, J. Org. Chem., 1997, 62, 5959–5965 CrossRef.
- P. J. Stang, B. L. Williamson and A. M. Arif, J. Am. Chem. Soc., 1993, 115, 2590–2597 CrossRef.
- M. Shimizu, Y. Takeda and T. Hiyama, Chem. Lett., 2008, 1304–1305 CrossRef CAS.
- P. J. Stang, B. L. Williamson and R. R. Tykwinski, J. Am. Chem. Soc., 1994, 116, 93–98 CrossRef.
- M. Ochiai, M. Kunishima, S. Tani and Y. Nagao, J. Am. Chem. Soc., 1991, 113, 3135–3142 CrossRef CAS.
- K. S. Feldman, M. M. Bruendl and K. Schildknegt, J. Org. Chem., 1995, 60, 7722–7723 CrossRef CAS.
- I. F. D. Hyatt and M. P. Croatt, Angew. Chem., Int. Ed., 2012, 51, 7511–7514 CrossRef CAS.
- K. Banert, R. Arnold, M. Hagedorn, P. Thoss and A. A. Auer, Angew. Chem., Int. Ed., 2012, 51, 7515–7518 CrossRef CAS.
- P. J. Stang, B. W. Surber, Z. C. Chen, K. A. Roberts and A. G. Anderson, J. Am. Chem. Soc., 1987, 109, 228–235 CrossRef CAS.
- G. F. Koser, L. Rebrovic and R. H. Wettach, J. Org. Chem., 1981, 46, 4324–4326 CrossRef CAS.
- T. Kitamura and P. J. Stang, J. Org. Chem., 1988, 53, 4105–4106 CrossRef CAS.
- P. J. Stang and V. V. Zhdankin, J. Am. Chem. Soc., 1990, 112, 6437–6438 CrossRef CAS.
- M. D. Bachi, N. Barner, C. M. Crittell, P. J. Stang and B. L. Williamson, J. Org. Chem., 1991, 56, 3912–3915 CrossRef CAS.
- V. V. Zhdankin, R. Tykwinski, R. Caple, B. Berglund, A. S. Kozmin and N. S. Zefirov, Tetrahedron Lett., 1988, 29, 3717–3720 CrossRef CAS.
- V. V. Zhdankin, R. Tykwinski, B. Berglund, M. Mullikin, R. Caple, N. S. Zefirov and A. S. Kozmin, J. Org. Chem., 1989, 54, 2609–2612 CrossRef CAS.
- Y. Ishiwata and H. Togo, Synlett, 2008, 2637–2641 CAS.
- V. V. Zhdankin, P. J. Persichini, R. Cui and Y. Jin, Synlett, 2000, 719–721 CAS.
- V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547–6551 CrossRef CAS.
- G. F. Koser, G. P. Sun, C. W. Porter and W. J. Youngs, J. Org. Chem., 1993, 58, 7310–7312 CrossRef CAS.
- M. Ochiai, Y. Masaki and M. Shiro, J. Org. Chem., 1991, 56, 5511–5513 CrossRef CAS.
- J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346–9349 CrossRef CAS.
- S. Nicolai, S. Erard, D. F. Gonzalez and J. Waser, Org. Lett., 2010, 12, 384–387 CrossRef CAS.
- E. B. Merkushev, L. G. Karpitskaya and G. I. Novosel'tseva, Dokl. Akad. Nauk SSSR, 1979, 245, 607–610 CAS.
- L. I. Dixon, M. A. Carroll, T. J. Gregson, G. J. Ellames, R. W. Harrington and W. Clegg, Eur. J. Org. Chem., 2013, 2334–2345 CrossRef CAS.
- L. I. Dixon, M. A. Carroll, T. J. Gregson, G. J. Ellames, R. W. Harrington and W. Clegg, Org. Synth., 2013, in press Search PubMed.
- Z. Liu, Z. C. Chen and Q. G. Zheng, Synth. Commun., 2004, 34, 361–367 CrossRef CAS.
- M. Ueno, T. Nabana and H. Togo, J. Org. Chem., 2003, 68, 6424–6426 CrossRef CAS.
- Y. Y. Xie, Z. C. Chen and Q. G. Zheng, J. Chem. Res., Synop., 2003, 614–615 CrossRef CAS.
- B. B. Toure, B. S. Lane and D. Sames, Org. Lett., 2006, 8, 1979–1982 CrossRef CAS.
- C. Enguehard, J.-L. Renou, V. Collot, M. Hervet, S. Rault and A. Gueiffier, J. Org. Chem., 2000, 65, 6572–6575 CrossRef CAS.
- T. Kitamura and P. J. Stang, Tetrahedron Lett., 1988, 29, 1887–1890 CrossRef CAS.
- P. J. Stang, H. Wingert and A. M. Arif, J. Am. Chem. Soc., 1987, 109, 7235–7236 CrossRef CAS.
- M. Ochiai, M. Kunishima, K. Fuji and Y. Nagao, J. Org. Chem., 1988, 53, 6144–6145 CrossRef CAS.
- M. Yoshida and S. Hara, Org. Lett., 2003, 5, 573–574 CrossRef CAS.
- M. Yoshida, A. Komata and S. Hara, J. Fluorine Chem., 2004, 125, 527–529 CrossRef CAS.
- M. Ochiai, K. Uemura, K. Oshima, Y. Masaki, M. Kunishima and S. Tani, Tetrahedron Lett., 1991, 32, 4753–4756 CrossRef CAS.
- M. Ochiai, Y. Kitagawa, M. Toyonari, K. Uemura, K. Oshima and M. Shiro, J. Org. Chem., 1997, 62, 8001–8008 CrossRef CAS.
- S. Hara, M. Yoshida, T. Fukuhara and N. Yoneda, Chem. Commun., 1998, 965–966 RSC.
- M. Yoshida, A. Komata and S. Hara, Tetrahedron, 2006, 62, 8636–8645 CrossRef CAS.
- T. Guan, M. Yoshida, D. Ota, T. Fukuhara and S. Hara, J. Fluorine Chem., 2005, 126, 1185–1190 CrossRef CAS.
- M. Ochiai, Y. Nishi and M. Hirobe, Tetrahedron Lett., 2005, 46, 1863–1866 CrossRef CAS.
- T. L. Ross, J. Ermert, C. Hocke and H. H. Coenen, J. Am. Chem. Soc., 2007, 129, 8018–8025 CrossRef CAS.
- P. Wipf and S. Venkataraman, J. Org. Chem., 1996, 61, 8004–8005 CrossRef CAS.
- K. Miyamoto, Y. Nishi and M. Ochiai, Angew. Chem., Int. Ed., 2005, 44, 6896–6899 CrossRef CAS.
- M. A. Carroll, S. Martin-Santamaria, V. W. Pike, H. S. Rzepa and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 2, 1999, 2707–2714 RSC.
- T. Okuyama, S. Imamura and M. Fujita, J. Org. Chem., 2006, 71, 1609–1613 CrossRef CAS.
- M. Ochiai, M. Kida, K. Sato, T. Takino, S. Goto, N. Donkai and T. Okuyama, Tetrahedron Lett., 1999, 40, 1559–1562 CrossRef CAS.
- P. Kowalski and J. Korchowiec, J. Mol. Struct. (THEOCHEM), 1993, 288, 119–131 CrossRef.
- S. Nikas, N. Rodios and A. Varvoglis, Molecules, 2000, 5, 1182–1186 CrossRef CAS.
- P. J. Stang, Chem. Eng. News, 1989, 67, 4 CAS.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- H. Tohma, S. Takizawa, T. Maegawa and Y. Kita, Angew. Chem., 2000, 112, 1362–1364 CrossRef.
- H. J. Lucas and E. R. Kennedy, Org. Synth. Coll. Vol., 1955, 3, 485–487 Search PubMed.
- J. S. Yadav, B. V. Subba Reddy, Y. Gopal Rao, M. Srinivas and A. V. Narsaiah, Tetrahedron Lett., 2007, 48, 7717–7720 CrossRef CAS.
- S. Kumar and D. P. Sahu, ARKIVOC, 2008, 88–98 CrossRef CAS.
- R. Adams and I. J. Patcher, J. Am. Chem. Soc., 1954, 76, 1845–1847 CrossRef CAS.
- Gol'dfarb and Kondakowa, Zh. Prikl. Khim., 1942, 15, 151–158 CAS.
- E. S. Hand and W. W. Paudler, Tetrahedron, 1982, 38, 49–56 CrossRef CAS.
- V. A. Artyomov, A. M. Shestopalov and V. P. Litvinov, Synthesis, 1996, 927–929 CrossRef CAS.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 3769–3772 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and spectra, tables of X-ray crystallographic data and results. CCDC 907271–907274. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41112e |
| ‡ Current address: Department of Isotope Chemistry, Covance Laboratories, Alnwick, Northumberland, NE66 2JH, UK. |
| § Although several column packings were evaluated, e.g. reverse phase silica, alumina (neutral, basic and acidic), with a range of solvents and additives, the Grace cartridges were found to provide satisfactory purification. |
| ¶ Due to volatility, solvent could not be fully removed; yield calculated from 1H-NMR. |
| This journal is © The Royal Society of Chemistry 2013 |