 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting folded RNA: a branched peptideboronic acid that binds to a large surface area of HIV-1 RRE RNA†
Wenyu
Zhang‡
,
David I.
Bryson‡
,
Jason B.
Crumpton‡
,
Jessica
Wynn
and
Webster L.
Santos
*
Department of Chemistry and Virginia Tech Center for Drug Discovery, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: santosw@vt.edu; Fax: +1 540 231 3255; Tel: +1 540 231 5041
First published on 2nd August 2013
Abstract
On-bead high-throughput screening of a medium-sized (1000–2000 Da) branched peptideboronic acid (BPBA) library consisting of 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 656 unique sequences against HIV-1 RRE RNA generated peptides with binding affinities in the low micromolar range. In particular, BPBA1 had a Kd of 1.4 μM with RRE IIB, preference for RNA over DNA (27 fold), and selectivity of up to >75 fold against a panel of RRE IIB variants. Structure–activity studies suggest that the boronic acid moiety and “branching” in peptides are key structural features for efficient binding and selectivity for the folded RNA target. BPBA1 was efficiently taken up by HeLa and A2780 cells. RNA-footprinting studies revealed that the BPBA1 binding site encompasses a large surface area that spans both the upper stem as well as the internal loop regions of RRE IIB.
656 unique sequences against HIV-1 RRE RNA generated peptides with binding affinities in the low micromolar range. In particular, BPBA1 had a Kd of 1.4 μM with RRE IIB, preference for RNA over DNA (27 fold), and selectivity of up to >75 fold against a panel of RRE IIB variants. Structure–activity studies suggest that the boronic acid moiety and “branching” in peptides are key structural features for efficient binding and selectivity for the folded RNA target. BPBA1 was efficiently taken up by HeLa and A2780 cells. RNA-footprinting studies revealed that the BPBA1 binding site encompasses a large surface area that spans both the upper stem as well as the internal loop regions of RRE IIB.
Introduction
Ribonucleic acid (RNA) is a unique macromolecular entity that plays key roles in a living cell. Serving in essential functions such as a carrier of genetic information, RNA also catalyzes protein synthesis and regulates gene expression. Along with protein–protein interactions, RNA–protein interactions are the gateway to the diversity of function that mediate a variety of biological effects. One attractive approach to perturbing the system is to inhibit RNA–protein interaction by disassembling the construct either through binding to the protein or RNA portion. Targeting RNA as a macromolecular entity with small molecules is a herculean task that is complicated by RNA structural dynamics—specific binding to a single conformation is difficult. Despite considerable campaigns, clinically effective small molecule inhibitors of RNA are rare outside of antibiotics binding to the ribosome.1,2In silico studies have emerged as a powerful tool in the rational design of RNA ligands. Such studies rely on virtual screening methods that examine binding preferences of ligands toward certain RNA motifs,3,4 or from docking experiments where ligand–RNA dynamic ensembles are generated by nuclear magnetic resonance and molecular dynamics studies.5,6 While these investigations are a significant leap forward, these approaches are still in their infancy. A complementary approach is high-throughput screening of chemical libraries against an RNA target.7–9 Chemical libraries that exploit chemical space outside the region used for protein-targeting small molecules are ideal since structural features present in RNA are vastly different than proteins.7
Although chemically similar, the presence of 2′-hydroxyl groups and other nucleotide modifications in RNA generate far more complex tertiary structures than those found in DNA.10 For example, DNA forms a double stranded helical structure while a single stranded RNA folds into a variety of secondary structures. Hairpins, bulges, loops, pseudoknots, and turns give rise to three-dimensional architecture akin to targetable regions of proteins; theoretically, these can create unique binding pockets suitable for intermolecular binding with small molecules. While attractive, discovery of small molecules that selectively bind to a well-folded RNA has proven difficult.1,2 New molecular scaffolds that can recognize three dimensional structures of RNA are needed. Recently, Disney and co-workers used a modular assembly approach to target r(CCUG) repeats that cause myotonic dystrophy type 2.4 Three copies of kanamycin A, tethered by a linker, bound to the internal loop, and resulted in the multivalent inhibition of the protein–RNA complex with an IC50 of 25 nM.
In contrast to molecules that target RNA via Watson–Crick base pairing, we surmise that an alternative mode of binding that recognizes the native three dimensional fold of RNA could be advantageous. Firstly, this will afford a complementary approach to targeting RNA molecules with inaccessible primary sequences as a consequence of RNA folding. Secondly, the tertiary structure of RNA could present multiple crevices or pockets suitable for medium sized molecules to penetrate and bind favorably—a collection of small binding interactions could accumulate to significant affinity that can also aid in selectivity. We previously developed a first generation branched peptide library (BP) that selectively bound with an HIV-1 related RNA tertiary structure, the transactivation response element (TAR) and demonstrated that medium-sized BPs (MW ∼ 1000–2000 Da) were cell permeable and displayed minimal to no toxicity.11,12 Moreover, our studies revealed that branching in peptides plays a significant role in increasing binding affinity to the target RNA. More recently, we reported the screening of a second generation BP library that was diversified with unnatural amino acids decorated with boronic acid moieties against HIV-1 RRE IIB RNA.13 These medium-sized branched peptideboronic acids (BPBAs) were capable of binding to the tertiary structure of HIV-1 RRE IIB in the low micromolar regime.
The Rev/RRE export pathway is essential for HIV-1 viral replication and has become a potential drug target.14 The Rev–RRE interaction is also completely viral in nature, which provides a high value therapeutic target completely independent from the natural cellular processes of the host. This is a huge advantage that could allow the interaction to be targeted selectively with minimal risk of side effects. Owing to the therapeutic potential of the Rev/RRE export pathway, many ligands have been designed to interrupt the Rev–RRE interaction with limited clinical success. Small molecules such as neomycin B, as well as other aminoglycosides, are demonstrated submicromolar binding ligands of RRE; however, their lack of binding specificity, poor cell permeability, and toxicity make them therapeutically undesirable.2,15–17 Other inhibitors such as aromatic heterocycles, antisense oligonucleotides, transdominant negative Rev mutant proteins, RRE-based decoys, cyclic peptides, α-helical peptidomimetics, and others have also been identified yet none of these have found clinical success.18–32
Studies directed toward understanding the fundamental interactions between RNA and its ligand at the molecular level is critical. These investigations will reveal concepts that will inform the design of next generation RNA ligands with the desired selectivity, potency, and permeability properties suitable for eventual clinical use in the treatment of various diseases. From an academic standpoint, RNA ligands that minimize nonspecific electrostatic interactions are highly desirable. We hypothesized that the empty p-orbital of boron would be a surrogate for a positive charge and act as an acceptor for the 2′-hydroxyl group or other Lewis bases of the RNA (Fig. 1A). Further, we expect that the boronic acid moiety will bias against DNA and prefer binding to RNA. In this report, we detail the biophysical characterization of a branched peptideboronic acid (BPBA1) against HIV-1 RRE IIB to understand the molecular interactions between our peptide toward RNA and to determine its selectivity against a panel of closely related RNA and DNA sequences. Our investigations suggest that BPBA1 forms complex interactions that span a large surface area of the structured RNA and that its binding affinity is highly dependent on the BP sequence.
 | ||
| Fig. 1 (A) RNA-branched peptideboronic acid interaction: formation of a possible reversible covalent bond between the 2′-hydroxyl group of ribose and empty p-orbital of boronic acids. (B) Structures of boronic acid monomers used in this study. | ||
Results and discussion
Library design and on-bead high-throughput screening
We synthesized a 46656-membered BPBAs library by split-and-pool synthesis and subjected it to an on-bead high-throughput screening assay.13 Each library member featured two identical N-termini branched by a Lys residue and a single C-terminus for facile deconvolution using MALDI/MS.33 The peptides were linked to beads through the C-termini via a photocleavable linker (3-amino-3-(2-nitrophenyl)propionic acid, ANP). There were six variable amino acid positions at N- and C-termini (A1–A3 and A4–A6, respectively) and six possible side chains in each position (Fig. 2). The identity of each side chain was chosen for its potential to interact with the RRE IIB target RNA through hydrophobic interaction, electrostatic attraction, hydrogen bonding, or pi-stacking. In addition, two unnatural amino acids containing boron side chains were incorporated at each variable position to provide reversible covalent bond formation between boron and a Lewis base presented by the RNA target, such as the 2′ hydroxyl group of the ribose sugars. By varying the side chain length and boron Lewis acidity of these residues (FBPA and KBBA, Fig. 1B), we were able to probe their effects on binding to our RNA target. Finally, a Tyr was included at position A7 to quantify peptide concentrations. Presentation of diverse moieties on the bead surface would allow interaction of DY547-labeled RRE IIB with all possible modes of binding. We first minimized non-specific binding by blocking the library-on-bead with excess bovine serum albumin and competitor tRNA. DY547-labeled RRE-IIB RNA was added during the second incubation.
 | ||
| Fig. 2 High-throughput screening of the 3.3.4-branched peptideboronic acid library reveal hit compounds with varying binding affinities. | ||
Increased fluorescence of beads, which was monitored by fluorescence microscopy, indicated specific binding to the target RNA. Eleven beads were selected, photocleaved and sequenced by MALDI MS/MS analysis following a previously published procedure.33
Standard dot blot assay was used to verify the dissociation constant of hit BPBAs. In particular, one peptide (BPBA1) stood out for its low micromolar binding affinity (1.4 μM).13 Further modifications were made wherein the boronic acid was removed (BPBA1.1, 8.2 μM) or fluorine was placed ortho to the boronic acid at position A4 (BPBA1.2, 0.8 μM) to probe the role of the boron atom in RNA binding (see ESI† for details). The results suggested that the boronic acid functional group had involvement in the interaction with RRE IIB, and that the Lewis acidity of the boronic acid had a significant effect on binding affinity. These results demonstrate that incorporation of boronic acid side chains into peptides can provide increased binding affinities to structured RNA targets as well as introduce a unique binding mode that increases the gamut of RNA-binding motifs.
BPBA1 binds RRE IIB via all branches of the peptide
To determine whether branching in our peptides contributes to the binding affinity toward RRE IIB (Table 1), control linear peptide variants of BPBA1 were synthesized. To minimize perturbations from the parent structure of the peptide, glycine was used in place of the branching lysine residue between positions A3 and A4 (LPBA1, LPBA4, and LPBA5). Truncation of the N-terminal branching peptide fragment linked to either the α- or ε-nitrogen of lysine resulted in peptidesLPBA3 and LPBA1, respectively, with a dramatic loss (>50 fold) of binding affinity compared to BPBA1. Moreover, when the C-terminal fragment was removed to generate LPBA2, a similar decrease (>50 fold) in binding affinity was observed. Collectively, these results suggest that all “branches” of the branched peptide are required for high affinity binding. In addition to probing the effects of branching, modifications to the structure of BPBA1 simultaneously probed the contribution of electrostatics to the binding of RRE. For example, a marked decrease in affinity was observed for LPBA2, which is isoelectronic with BPBA1. Peptide derivatives LPBA1 and LPBA3 with two fewer lysine residues than BPBA1 and decreased net positive charge also demonstrated significant loss in binding affinity (Kd > 75 μM). To investigate whether the change in binding affinity could be a consequence of decreased electrostatic interactions, the WKK branching fragment was installed on either the N-terminus or C-terminus of LPBA1 to provide LPBA4 and LPBA5, respectively. Consistent with previous reports that electrostatic interactions have a significant role in boosting binding affinity and not necessarily selectivity,1,11 the Kd values of these peptides (∼8 μM) were improved compared to LPBA2 but remained at least 5-fold weaker than BPBA1. Gratifyingly, when a branched peptide containing a scrambled sequence of BPBA1 was tested (RPBA1), the Kd value increased to >75 μM suggesting that the sequence of BPBA1 is essential for tight binding to RRE IIB. Taken together, these results indicated that binding occurs through interactions with the three branches of BPBA1, and that the sequence of the branched peptide plays a significant role in binding.| Compound | Scheme | Sequencea | K d (μM) |
|---|---|---|---|
| a * = Lysine branching unit. b α-Nitrogen of lysine is acetylated and KKW is attached to the ε-nitrogen. c α-Nitrogen of tyrosine is acetylated and KWKBBA is attached to the ε-nitrogen of branching lysine. Each value is an average of at least three experiments. | |||
| BPBA1 |

|
(WKK)2*KBBAYWY | 1.4 ± 0.4 |
| LPBA1 |

|
WKKGKBBAYWY | >75 |
| LPBA2 |

|
(WKK)2* | >75 |
| LPBA3 |

|
Ac(WKK)*KBBAYWYb | >75 |
| LPBA4 |

|
WKKWKKGKBBAYWY | 7 ± 2 |
| LPBA5 |

|
WKKGKBBAYWYWKK | 9 ± 2 |
| RPBA1 |

|
AcYKW*KWKY (KWKBBA)c | >75 |
Selectivity of BPBA1 toward RRE IIB tertiary structure
Several mutations were made to RRE IIB to determine whether BPBA1 could discriminate between the native tertiary structure of the target RNA and closely related structural analogs. RRE IIB RNA is composed of two stems, two internal loops, a single nucleotide bulge and an apical loop, which are expected to contribute to its tertiary structure (Fig. 3). We synthesized ‘hexaloop RNA’, where the size of the trinucleotide apical loop (AAU) was increased to a hexa-nucleotide loop (AUGGCC) (Fig. 3A). The measured Kd for this variant using dot blot assay increased by ∼3-fold to 4.4 μM compared to the wild type (Fig. 3B and C). We next removed the 4 base pairs in the upper stem to generate ‘stem B deleted RNA’ and obtained a similar Kd of 3.7 μM. The small change in Kd with these two mutant RRE RNA structures suggests that BPBA1 has a minor interaction with the apical loop and stem B region of RRE IIB. Hill analysis of the hexaloop RNA and stem B deleted RNA provides coefficients (n) of 1.7 and 1.6, respectively, and suggest cooperative binding of BPBA1 to these variant structures (see ESI† for details). | ||
| Fig. 3 (A) Sequence and structure of RRE IIB and variants, (B) titration curves and (C) dissociation constants of BPBA1 with indicated RNAs. | ||
We also probed regions toward the lower stem by generating ‘loop B/bulge A deleted RNA’, and observed 6-fold lower affinity (Kd = 8.7 μM). The RRE IIB variants ‘loop A(A–G)/B/bulge A deleted’, which contained a smaller loop A from the removal of the A–G mismatch, and ‘stem A/loop A deleted RNA’ were synthesized to determine whether the affected region of the RNA is the potential site for binding BPBA1. These mutant RNAs demonstrated >10 fold lower affinity with BPBA1 (Fig. 3B and C) and suggested major interaction with the nucleotides deleted from RRE IIB. We suspected that changes or removal of sequences in loop A, loop B, and bulge A would result in an altered tertiary structure that minimizes interaction with BPBA1. When these structural elements were eliminated to produce ‘loop A/B/bulge A deleted RNA’ as a stem loop with a significantly altered tertiary structure, a dramatic loss (>75 fold) in binding affinity was observed. These results support our hypothesis that the exact tertiary structure of RRE IIB is required for optimal binding with BPBA1, where BPBA1 likely makes contacts primarily with nucleotides present in loops A and B. Hill analysis of the dot blot data from ‘loop B/bulge A deleted RNA,’ ‘stem A/loop A deleted RNA,’ ‘loop A/B/bulge A deleted RNA,’ and ‘loop A(A–G)/B/bulge A deleted RNA’ generated Hill coefficients of 0.9, 1.3, 1.3, and 1.3, respectively, indicating noncooperative binding of BPBA1 (see ESI† for details).
To further characterize the selectivity against other RNA structures, binding affinities between BPBA1 and RRE IIB were measured by dot blot in the presence of excess bacterial tRNA. Initially, a 10-fold molar excess of competing tRNA relative to RRE IIB was included during incubation with the peptide, which resulted in an observed Kd (2.1 μM) that was within experimental error with that obtained in the absence of tRNA (Fig. 4). Encouraged by this result, we increased the amount of competing tRNA to a 1000-fold molar excess. Gratifyingly, the observed Kd was 5.3 μM, representing a minute shift in RRE IIB binding. These results suggest that peptideBPBA1 is selective for RRE IIB in the presence of a competing complex mixture of tRNA structures. It is also important to note that these results are quite improved from our previous generation of branched peptides that did not feature boronic acids, as peptides from the previous library demonstrated a dramatic shift (10-fold) in binding affinity in the presence of only a 10-fold molar excess of tRNAs.11 We next explored the selectivity of BPBA1 against DNA composed of the same RRE IIB sequence by dot blot assay. The high dissociation constant (31.8 ± 6.6 μM) confirms our hypothesis that BPBAs confer selectivity toward RNA (23-fold). A change in the tertiary structure, loss of the 2′-OH groups, or a combination of both could account for the dramatic shift in binding affinity.
 | ||
| Fig. 4 Titration curves of BPBA1 with WT RRE IIB, RRE IIB DNA, and in the presence of competing tRNA. | ||
Determination of the BPBA1 binding site by RNase protection assay
The results of our binding selectivity experiments with RRE IIB mutants allowed the evaluation of structural elements required for binding of BPBA1. To better determine regions of specific nucleotide contacts with RRE IIB, we performed ribonuclease protection assays. Hence, 5′-32P-labeled RRE IIB was incubated with varying concentrations (up to 20 μM) of BPBA1 in the presence of RNase T1, RNase A, or RNase V1 (Fig. 5). Potential binding sites were confirmed based on the ability of the peptide to protect the RNA from enzymatic cleavage. While there were no noticeable changes in band intensity with RNase T1, the most prominent cleavage bands over all the experiments conducted were observed between A16 through G19 using RNase V1, which preferentially cleaves double stranded regions of RNA. As shown in Fig. 5, there is a concentration dependent protection (0.2 μM to 20 μM) of this region in the presence of BPBA1, which suggests that this portion of RRE IIB serves a potential binding site. Interestingly, this region is also the site wherein the native protein partner Rev binds.34,35BPBA1 also visibly protected U7, C21, and U36 from cleavage by RNase A, which hydrolyzes at the 3′ side of unpaired pyrimidine bases. In particular, U7 and U36 are potential contact points located in the internal loop region of RRE IIB. Indeed, this same internal loop region, when deleted, caused dramatically reduced binding affinities with BPBA1 (vide supra). Taken together, the RNA footprinting assay indicated that the binding site for BPBA1 encompasses a large surface area constituting stem B and loops A and B. Hill analysis (n = 1) supported a noncooperative, possibly 1:1 binding stoichiometry between BPBA1 and RRE IIB. This may indicate that BPBA1 is bound in a folded groove saddled between the internal loop regions and upper stem bases of RRE IIB, as this compound is interacting with these structural elements. Although the precise nature of the RNA–peptide interaction is currently unknown, protection from enzymatic cleavage of these regions could result either from steric blockade or remodeling of its tertiary structure.
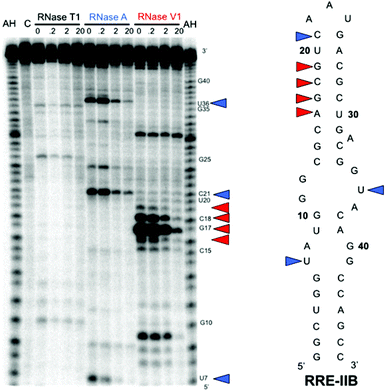 | ||
| Fig. 5 RNase protection assay of RRE IIB. The gel depicts the autoradiogram of alkaline hydrolysis (AH) and RNase protection experiments using RNases T1, A, and V1 with increasing concentration of BPBA1. Colored triangles highlight bases protected from cleavage by RNase A (blue) and RNase V1 (red). | ||
Cell permeability and cytotoxicity of branched peptideboronic acids
Our previous generation of branched peptides that featured Arg-rich N-termini exhibited excellent cell permeability and non-cytotoxic properties.11 The key feature of the current library is the incorporation of unnatural amino acids with boronic acid functional groups and the elimination of Arg in place of Lys—it was unclear whether these changes would have an impact on cell permeability. Despite these changes, however, we suspected that BPBAs would be cell permeable in part because they maintained a medium molecular weight (∼1000–2000 Da), and contained multiple basic residues.36 Therefore, cellular uptake was assayed in both HeLa and A2780 cells by incubation with the FITC-labeled version of BPBA1 (FBPBA1) using an established procedure in tissue culture medium for 4 h at 37 °C.11 After washing, cells were fixed with 4% (w/v) paraformaldehyde in PBS followed by incubation with BSA and then DAPI stain. Cells were imaged using a confocal microscope. HeLa and A2780 cells incubated with FBPBA1 showed fluorescence evenly distributed throughout the cytoplasm and nucleus suggesting that the boronic acid moiety did not have a negative impact on the membrane permeability of BPBAs (Fig. 6A and B). Further, both cell lines were viable in an MTT assay after incubation with BPBA1, indicating that this branched peptideboronic acid was nontoxic at a concentration range of 100 nM to 30 μM (Fig. 6C, ESI†). However, increasing the concentration of BPBA1 to 60 μM and above showed a dramatic reduction in cell viability. | ||
| Fig. 6 Cellular uptake of 1 μM FBPBA1 into (A) HeLa and (B) A2780 cells. Top left: fluorescence image of cells; top right: DAPI staining of the nucleus; bottom left: DIC image; bottom right: overlay of the three images. (C) MTT cell toxicity assay of 30 μM BPBA1 at 24 h of exposure. | ||
Conclusions
The unique aspect of our study is the use of boronic acid groups that introduce an alternative mode of binding to a well-folded, therapeutically relevant target RNA. To our knowledge, this is the first instance of boronic acid side chains being utilized in peptidic RNA ligands. In this work, we characterized the peptideBPBA1 that was selected from a 3.3.4-branched peptideboronic acid library. Our results demonstrated that this medium-sized peptideboronic acid selectively binds RRE IIB RNA against competitor tRNAs, six RRE IIB related structural variant RNAs, and an RRE IIB DNA analogue. We also show that BPBA1 is cell permeable and poses minimal cytotoxicity in two eukaryotic cell lines. Finally, we provided evidence that BPBA1 binding to RRE IIB spans a large surface area in the upper stem and internal loops of the RNA. This work demonstrates complex, multiple intermolecular interactions along a large surface area as a key feature in developing selective binders of unique RNA tertiary structures. These results highlight the potential of this novel class of RNA binding compounds to be further refined for use in therapeutic development as well as the utility of this method as a general platform for RNA ligand discovery. Current efforts are aimed to further improve the binding affinity and selectivity of BPBAs and demonstrate their inhibition activity in cell-based assays. Lessons learned from this study will inform our ongoing efforts to target highly structured RNAs with high affinity and selectivity.Experimental
Peptide synthesis, purification and characterization
Unlabeled and fluorescein-5-isothiocyanate (FITC) (Sigma) labeled peptides were synthesized on Rink amide MBHA resin (100–200 mesh) (Novabiochem). In the preparation of the FITC-labeled peptides, Fmoc-Lys(ivDde)-OH (Novabiochem) was used as the branching unit.11Acetic anhydride in DMF (1:1 v/v) with 10 equivalents of DIEA was used to cap the first N-terminus. Then, ivDde was removed by treatment with 2% hydrazine in DMF for 1 h, and the second N-terminus was synthesized through the ε-N of the Lys side chain. Fmoc-6-Ahx-OH (AnaSpec) was coupled to the N-terminal amino acid to provide a linker for FITC, which prevents autocleavage of FITC under acidic conditions.11,37 All subsequent steps were protected from light. FITC (5 equiv.) was reacted with the deprotected N-terminus of the peptides for 6 h using DIEA (14 equiv.). The boronic acid deprotection was performed as previously described.38 The supernatant was dried under reduced pressure, and the crude peptide was triturated from cold diethyl ether. The peptides were purified using a Jupiter 4 μm Proteo 90 Å semiprep column (Phenomenex) using a solvent gradient composed of 0.1% TFA in Milli-Q water and HPLC grade acetonitrile. Peptide purity was determined using a Jupiter 4 μm Proteo 90 Å analytical column (Phenomenex), and peptide identity was confirmed by MALDI-TOF analysis. Unlabeled peptide concentrations were measured in nuclease free water at 280 nm using their calculated extinction coefficients. FITC-labeled peptide concentrations were monitored at 495 nm using the extinction coefficient of FITC at 77000 mol−1 cm−1 in 100 mM glycine, pH 9.0.
Preparation of 32P-labeled RNA and DNA
Wild-type and mutant RRE-IIB RNAs were transcribed in vitro by T7 polymerase with the Ribomax T7 Express System (Promega) using previously reported techniques.39 The antisense templates, sense complementary strand (5′-ATGTAATACGACTCACT ATAGG-3′) and RRE IIB reverse PCR primer (5′-GGCTGGCCT GTAC-3′) were purchased from Integrated DNA Technologies. Antisense templates were used as follows: RRE IIB RNA 5′-GGCTGGCCTGTACCGTCAGCGTCATTGACGCTGCGCCCA TACCAGCCCTATAGTGAGTCGTATTACAT-3′; HexaLoop 5′-GGCTGGCCTGTACGTCAGCGTCGG CATTGACGCTGCGCCCATACCAGCCCTATAGTGAGTCGTATTACAT-3′; Stem A/Loop A Deleted 5′-GGCGTACCGTCAGCGTCATTGACGCTGCGCCCGCCCTATAGTGAGTCGTATTACAT-3′; Stem B Deleted 5′-GGCTGGCCTGTACCGTCACATTGTGCGCCCATACCAGCCCTATAGTGAGTCGTAT TACAT-3′; Loop A(A–G)/B/Bulge A Deleted 5′-GGCTGGCCAGCGTCATTGACGCTGACCAGCCCT ATAGTGAGTCGTATTACAT-3′; Loop B/Bulge A Deleted 5′-GGCTGGCCTGCAGCGTCATTGACGC TGCATACCAGCCCTATAGTGAGTCGTATTACAT-3′; Loop A/B/Bulge A Deleted 5′-GGCTGGCAGC GTCATTGACGCTGCCAGCCCTATAGTGAGTCGTATTACAT-3′. TetraLoop and RRE IIB were both PCR amplified using HotstarTaq DNA polymerase (Qiagen) followed by a clean-up procedure using a spin column kit (Qiagen). For the preparation of all other sequences, the antisense DNA template was annealed with the sense DNA complementary strand in reaction buffer at 95 °C for 2 min then cooled on ice for 4 min. T7 transcription proceeded at 42 °C for 1.5 h. After transcription, DNA templates were degraded with DNase at 37 °C for 45 min, and the RNA was purified by a 12% polyacrylamide gel containing 7.5 M urea. The band corresponding to the RNA of interest was excised from the gel and eluted overnight in 1× TBE buffer at 4 °C. The sample was desalted using a Sep-Pak syringe cartridge (Waters Corporation) and lyophilized. The products, along with bacterial tRNA, were dephosphorylated with calf intestinal phosphatase (CIP) in NEBuffer 3 (New England Biolabs) according to manufacturer's protocol. The product was recovered by a standard phenolextraction followed by ethanolprecipitation. Purified RNA was stored as a pellet at −80 °C. RRE IIB DNA (5′-GGCTGGTATGGGCGCAGCGTCAATGACGCTGACGGTACAGGCCAGCC-3′) was purchased from Integrated DNA Technologies and stored at −20 °C.HIV-1 RRE IIB RNA as well as the mutant RNA sequences, tRNA, and RRE IIB DNA were labeled at the 5′-end by treating 10 pmol of dephosphorylated RNA/DNA with 20 pmol of [γ-32P] ATP (111 TBq mol−1) and 20 units of T4 polynucleotide kinase in 70 mM Tris·HCl, 10 mM MgCl2, and 5 mM dithiothreitol, pH 7.6. The mixture was incubated at 37 °C for 30 min, and then at rt for 20 min. The kinase was heat-inactivated at 65 °C for 10 min. The product was recovered by ethanolprecipitation, and the purity was examined using 12% denaturing PAGE followed by autoradiography.
Dot blot assay
Dot blot assays were performed at rt using a Whatman Minifold I 96 well Dot Blot system and Whatman 0.45 μm pore size Protran nitrocellulose membranes. To determine the binding affinities, 0.04 nM radiolabeled RNA was titrated with peptide (0.001–100 μM). First, a solution of 0.08 nM 32P-labeled RNA was refolded in 2× phosphate buffer (20 mM potassium phosphate, 200 mM KCl, 1 mM MgCl2, 40 mM NaCl, pH 7.0) by heating at 95 °C for 3 min and then slowly cooling at rt for 20 min. Next, 25 μL of the [32P]-RNA solution was added to 25 μL of peptide in nuclease free water and incubated at rt for 4 h. The 50 μL mixtures were filtered through the nitrocellulose membrane, which was immediately followed by two consecutive 50 μL washes with 1× phosphate buffer. Peptide binding was visualized by autoradiography using a storage phosphor screen (GE Healthcare) and a Typhoon Trio phosphorimager (GE Healthcare). Densitometry measurements were quantified using ImageQuant TL (Amersham Biosciences). Binding curves were generated using a four parameter logistic equation with Kaleidagraph (Synergy Software): y = m1 + (m2 − m1)/(1 + 10log(m3)−x); m1 = 100; m2 = 1; m3 = 0.000003, where y = percentage of RNA binding, x = log[peptide], m1 = percentage of RNA binding affinity at infinite concentration (nonspecific binding), m2 = percentage of RNA binding affinity at zero concentration, m3 = peptide concentration at 50% binding (Kd). Each experiment was performed in triplicate and error bars represent the standard deviation calculated over three replicates.Nuclease protection assay
RNA was first refolded by heating a solution of 5′-32P-labeled RRE-IIB (10 nM) and excess unlabeled RRE-IIB (200 nM) at 95 °C for 3 min and then snap cooling on ice. The refolded RNA was incubated on ice for 4 h in a solution containing the BPBAs and buffer composed of 10 mM Tris, pH 7, 100 mM KCl, and 10 mM MgCl2. RNase (Ambion) was then added to the solution, which was further incubated on ice for 10 min (0.002 Units RNase V1), or 1 h (1 Unit RNase T1; 20 ng RNase A). Inactivation/precipitationbuffer (Ambion) was added to halt digestion, and the RNA was pelleted by centrifugation at 13200 rpm for 15 min. Pelleted RNA was redissolved into tracking dye and run through a 12% PAGE containing 7.5 M urea and imaged by autoradiography.
Cellular internalization of peptides and MTT toxicity assay
HeLa cells were grown and plated for peptide internalization using previously established methods.11 A2780 cells were grown and plated in RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% L-glutamine, 100 units per mg penicillin, 100 μg mL−1streptomycin, and 0.25 μg mL−1 amphotericin (Invitrogen). Both cell lines were maintained in a 37 °C incubator containing a 5% CO2 atmosphere and were subcultured once per week. Cell samples were prepared in 35 mm poly-lysine treated glass-bottom dishes containing a no. 1.5 coverglass (MatTek) using the manufacturer's protocol. The dishes were pre-equilibrated with media for 15 min at 37 °C in 5% CO2 atmosphere. Media was aspirated from the dishes and the cells were plated at 1.5 × 104 cells per dish in a 500 μL media suspension and incubated for 1 h to allow for initial cell adherence. Additional media (2 mL) was added and the cells were incubated for a total of 48 h. After the removal of medium and washing of cells with PBS, 600 μL of FITC-labeled branched peptideboronic acid (FBPBA1, 1 μM) in Opti-MEM (Invitrogen/Gibco) was added to the dish; cells were incubated with the peptide for 4 h. Control cell samples were incubated in 600 μL of Opti-MEM for 4 h. After incubation and following each subsequent step, a 15 min PBS wash was applied to all cell samples at rt. Cells were fixed with 4% (w/v) paraformaldehyde (Acros) in PBS for 15 min, and incubated with 3% (v/v) BSA (New England Biolabs) in PBS for 30 min. Nucleus staining was performed with the addition of 150 μL of 600 nM DAPI to the cell samples for 4 min. Finally, cells were submersed in PBS, and the dishes were sealed with parafilm and stored at 4 °C in the dark until imaged. Cells were imaged using a 40× water-immersion objective (N.A. = 1.2) on an LSM 510 confocal system mounted on an Axiovert 100 inverted microscope (Zeiss). For each cell line, identical acquisition parameters were used for both peptide and control samples. Brightness and contrast were adjusted for the image processing of DAPI and DIC channels using AxioVisionLE software (Zeiss).For the MTT toxicity assays, 96-well plates (Nunc) were incubated with 100 μL of poly-lysine (Sigma) for 24 h at 4 °C. The poly-lysine was removed and the wells were rinsed ten times with sterile nuclease-free water. HeLa and A2780 cells were plated in the poly-lysine treated wells and analyzed via an MTT toxicity assay using a previously published procedure.11
Acknowledgements
We thank K. Ray and R. Helm in the VT mass spectrometry incubator for their assistance with MALDI-TOF analysis, and K. DeCourcy at the Fralin Life Science Institute for her assistance with cell microscopy. This research was supported by NIGMS of the National Institutes of Health under award number RO1 GM093834. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.Notes and references
- L. Guan and M. D. Disney, ACS Chem. Biol., 2012, 7, 73–86 CrossRef CAS.
- J. R. Thomas and P. J. Hergenrother, Chem. Rev., 2008, 108, 1171–1224 CrossRef CAS.
- S. J. Seedhouse, L. P. Labuda and M. D. Disney, Bioorg. Med. Chem. Lett., 2010, 20, 1338–1343 CrossRef CAS.
- M. M. Lee, A. Pushechnikov and M. D. Disney, ACS Chem. Biol., 2009, 4, 345–355 CrossRef CAS.
- A. C. Stelzer, A. T. Frank, J. D. Kratz, M. D. Swanson, M. J. Gonzalez-Hernandez, L. Janghyun, I. Andricioaei, D. M. Markovitz and H. M. Al-Hashimi, Nat. Chem. Biol., 2011, 7, 553–559 CrossRef CAS.
- A. V. Filikov, V. Mohan, T. A. Vickers, R. H. Griffey, P. D. Cook, R. A. Abagyan and T. L. James, J. Comput. Aided Mol. Des., 2000, 14, 593–610 CrossRef CAS.
- F. Aboul-ela, Fut. Med. Chem., 2010, 2, 93–119 CrossRef CAS.
- G. Galicia-Vázquez, L. Lindqvist, X. Wang, I. Harvey, J. Liu and J. Pelletier, Anal. Biochem., 2009, 384, 180–188 CrossRef.
- L. Yen, M. Magnier, R. Weissleder, B. R. Stockwell and R. C. Mulligan, RNA, 2006, 12, 797–806 CrossRef CAS.
- A. Czerwoniec, S. Dunin-Horkawicz, E. Purta, K. H. Kaminska, J. M. Kasprzak, J. M. Bujnicki, H. Grosjean and K. Rother, Nucleic Acids Res., 2009, 37, D118–D121 CrossRef CAS.
- D. I. Bryson, W. Zhang, P. M. McLendon, T. M. Reineke and W. L. Santos, ACS Chem. Biol., 2012, 7, 210–217 CrossRef CAS.
- D. I. Bryson, W. Zhang, W. K. Ray and W. L. Santos, Mol. BioSyst., 2009, 5, 1070–1073 RSC.
- W. Zhang, D. I. Bryson, J. B. Crumpton, J. Wynn and W. L. Santos, Chem. Commun., 2013, 49, 2436–2438 RSC.
- A. I. Dayton, J. G. Sodroski, C. A. Rosen, W. C. Goh and W. A. Haseltine, Cell, 1986, 44, 941–947 CrossRef CAS.
- K. Hamasaki and A. Ueno, Bioorg. Med. Chem. Lett., 2001, 11, 591–594 CrossRef CAS.
- M. L. Zapp, D. W. Young, A. Kumar, R. Singh, D. W. Boykin, W. D. Wilson and M. R. Green, Bioorg. Med. Chem., 1997, 5, 1149–1155 CrossRef CAS.
- W. D. Wilson and K. Li, Curr. Med. Chem., 2000, 7, 73–98 CrossRef CAS.
- Y. Lee, S. Hyun, H. J. Kim and J. Yu, Angew. Chem., Int. Ed., 2008, 47, 134–137 CrossRef CAS.
- M. L. Zapp, D. W. Young, A. Kumar, R. Singh, D. W. Boykin, W. D. Wilson and M. R. Green, Bioorg. Med. Chem., 1997, 5, 1149–1155 CrossRef CAS.
- G. Xiao, A. Kumar, K. Li, C. T. Rigl, M. Bajic, T. M. Davis, D. W. Boykin and W. D. Wilson, Bioorg. Med. Chem., 2001, 9, 1097–1113 CrossRef CAS.
- N. W. Luedtke, Q. Liu and Y. Tor, Biochemistry, 2003, 42, 11391–11403 CrossRef CAS.
- K. Li, T. M. Davis, C. Bailly, A. Kumar, D. W. Boykin and W. D. Wilson, Biochemistry, 2001, 40, 1150–1158 CrossRef CAS.
- R. A. Hart, J. N. Billaud, S. J. Choi and T. R. Phillips, Virology, 2002, 304, 97–104 CrossRef CAS.
- E. S. DeJong, C. E. Chang, M. K. Gilson and J. P. Marino, Biochemistry, 2003, 42, 8035–8046 CrossRef CAS.
- C. E. Prater, A. D. Saleh, M. P. Wear and P. S. Miller, Bioorg. Med. Chem., 2007, 15, 5386–5395 CrossRef CAS.
- M. Legiewicz, C. S. Badorrek, K. B. Turner, D. Fabris, T. E. Hamm, D. Rekosh, M. L. Hammarskjold and S. F. Le Grice, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14365–14370 CrossRef CAS.
- T. C. Lee, B. A. Sullenger, H. F. Gallardo, G. E. Ungers and E. Gilboa, New Biol., 1992, 4, 66–74 CAS.
- T. L. Symensma, S. Baskerville, A. Yan and A. D. Ellington, J. Virol., 1999, 73, 4341–4349 CAS.
- L. Giver, D. P. Bartel, M. L. Zapp, M. R. Green and A. D. Ellington, Gene, 1993, 137, 19–24 CrossRef CAS.
- L. Chaloin, F. Smagulova, E. Hariton-Gazal, L. Briant, A. Loyter and C. Devaux, J. Biomed. Sci., 2007, 14, 565–584 CrossRef CAS.
- A. Friedler, D. Friedler, N. W. Luedtke, Y. Tor, A. Loyter and C. Gilon, J. Biol. Chem., 2000, 275, 23783–23789 CrossRef CAS.
- N. L. Mills, M. D. Daugherty, A. D. Frankel and R. K. Guy, J. Am. Chem. Soc., 2006, 128, 3496–3497 CrossRef CAS.
- J. B. Crumpton, W. Zhang and W. L. Santos, Anal. Chem., 2011, 83, 3548–3554 CrossRef CAS.
- J. Kjems, B. J. Calnan, A. D. Frankel and P. A. Sharp, EMBO J., 1992, 11, 1119–1129 CAS.
- J. Kjems, M. Brown, D. D. Chang and P. A. Sharp, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 683–687 CrossRef CAS.
- S. H. Park, J. Doh, S. I. Park, J. Y. Lim, S. M. Kim, J. I. Youn, H. T. Jin, S. H. Seo, M. Y. Song, S. Y. Sung, M. Kim, S. J. Hwang, J. M. Choi, S. K. Lee, H. Y. Lee, C. L. Lim, Y. J. Chung, D. Yang, H. N. Kim, Z. H. Lee, K. Y. Choi, S. S. Jeun and Y. C. Sung, Gene Ther., 2010, 17, 1052–1061 CrossRef CAS.
- M. Jullian, A. Hernandez, A. Maurras, K. Puget, M. Amblard, J. Martinez and G. Subra, Tetrahedron Lett., 2009, 50, 260–263 CrossRef CAS.
- S. J. Coutts, J. Adams, D. Krolikowski and R. J. Snow, Tetrahedron Lett., 1994, 35, 5109–5112 CrossRef CAS.
- J. F. Milligan, D. R. Groebe, G. W. Witherell and O. C. Uhlenbeck, Nucleic Acids Res., 1987, 15, 8783–8798 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Structure of boronic acids, Hill plot analyses, cytotoxicity assay, and HPLC/MALDI MS of peptides. See DOI: 10.1039/c3ob41053f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |