 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of the origin and synthetic application of the pseudodilution effect for Pd-catalyzed macrocyclisations in concentrated solutions with immobilized catalysts†
Elisabeth
Brehm
ab and
Rolf
Breinbauer
*abc
aMax-Planck-Institute of Molecular Physiology, Otto-Hahn-Str. 11, D-44227 Dortmund, Germany
bFachbereich Chemie, Technische Universität Dortmund, Otto-Hahn-Str. 6, D-44227 Dortmund, Germany
cInstitute of Organic Chemistry, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria. E-mail: breinbauer@tugraz.at; Fax: +43 316 873 32402; Tel: +43 316 873 32400
First published on 25th June 2013
Abstract
Immobilized Pd-complexes allowed macrocyclisations via the Tsuji–Trost-reaction in concentrated solutions. Systematic studies suggest that the origin of this pseudodilution effect is neither film diffusion nor gel diffusion, but the reduction in conformational freedom of intermediates and intramolecular prenucleophile activation. In contrast a pseudodilution effect could not be observed for Sonogashira- and Suzuki-macrocyclisations.
The conformational restriction caused by the macrocyclic nature of many natural products and several drug molecules is believed to be of superior importance for their biological activity.1 The synthesis of such macrocyclic structures is a challenging task as any cyclisation reaction of a difunctional substrate has to compete with concurring intermolecular oligomerization reactions.2 According to the high dilution principle of Ziegler and Ruggli, concentrations of 10−3 M or less are applied to favor intramolecular reactions.3 In 1982 Trost and Warner reported a stunning observation: with a polymer-bound metal complex macrocyclisations via Pd-catalyzed allylation of vinylepoxides succeeded in good yields at 0.1–0.5 M concentrations, while using a homogeneous variant of the catalyst at the same concentrations delivered only oligomerization products.4 Besides another report of Tsuji–Trost allylation,5 the only other examples of a similar effect have been documented for macrocyclisations via Stille-coupling6 and Cu-catalyzed Huisgen-cycloaddition.7 The dramatic improvements in macrocyclisation products using an immobilized catalyst have been interpreted as originating from a pseudodilution effect. This effect is well documented in solid phase synthesis for immobilized substrates at low substrate loadings, which favors intramolecular reactions through site isolation.8 We report now a systematic investigation to study several parameters which might be involved in the performance of immobilized catalysts. In particular we were interested in the following questions: (1) How general is this effect? Can it be extended to other metal-catalyzed reactions? (2) Is the substrate transport to and across the catalyst-bead an influencing factor?9 (3) Do other resin properties (loading, flexibility, hydrophilicity/hydrophobicity) of the polymer support play a role?10
One prerequisite for our studies was a reliable access for well characterized polymer bound phosphine ligands. A limitation of previous studies which have relied on polystyrene (PS) phosphine is that this ligand is not only difficult to prepare but due to its monodentacity it might lead to several different catalytically active species. We identified the bis(phosphinomethyl)amine moiety11 as a convenient and suitable ligand unit for our purposes, as it can be easily made from readily accessible H2N-functionalized resins in quantitative yield.12 The bidentate ligand should warrant more defined Pd-coordination, which through the chelating effect should also diminish leaching. We synthesized a series of polymer-supported bis(phosphinomethyl)amine ligands 1a–1g, in which we varied loading, cross linking, bead size and hydrophilicity of the support (Scheme 1, Table 1). All polymer ligands were characterized by 31P-NMR showing only a single signal for the P(III)-species. The ligands were converted to the Pd-complexes 1a–g/Pd by stirring the resin with Pd[PPh3]4 followed by extensive washing.
![Synthesis of immobilized phosphine ligands 1a–g and the conversion to the corresponding Pd-complexes. (i) (1) Ph2PH, HCHO, MeOH, 60 °C, (2) resin, toluene, 105 °C, 2d; (ii) Pd[PPh3]4.](/image/article/2013/OB/c3ob41020j/c3ob41020j-s1.gif) | ||
| Scheme 1 Synthesis of immobilized phosphine ligands 1a–g and the conversion to the corresponding Pd-complexes. (i) (1) Ph2PH, HCHO, MeOH, 60 °C, (2) resin, toluene, 105 °C, 2d; (ii) Pd[PPh3]4. | ||
For a first series of experiments in the Tsuji–Trost-macrocyclisation we needed a suitable substrate which can be synthesized in a convergent manner and would give ready access to differently sized analogs. Similar to the methodology reported before, substrate 8 was prepared from vinylepoxide 5, which resulted from a reaction of sulfur ylide 4 with aldehyde 3.13 After silyl deprotection, alcohol intermediate 5 was esterified with carboxylic acid 7 to furnish cyclisation substrate 8 in 21% overall yield for the 6 step synthesis in the longest linear sequence. Adapting the conditions of Fürstner and Weintritt established for their total synthesis of roseophilin,5 we achieved the Tsuji–Trost-macrocyclisation (10% Pd[PPh3]4, 20% dppe, THF, reflux, 0.0015 M) of 8 to form 17-membered lactone 9 in 64% yield (Scheme 2).
![Synthesis of cyclisation substrate 8. (i) NaH, TBDMSCl, 67%; (ii) (COCl)2, DMSO, Et3N, DCM, −78 °C to rt, 88%; (iii) MeOH–H2O (9/1); (iv) 3, DCM–10 M NaOH (1/1), −20 °C to rt, 1 h, 80%; (v) NH4F, MeOH, reflux, 8 h, 65%; (vi) MeOH, H2SO4, 89%; (vii) (PhSO2)2CH2, NaH, DMF, 81%; (viii) LiOH, THF–MeOH–H2O (3/3/1), quant.; (ix) EDC, DMAP, DCM, RT, 12 h, 68%; (x) 10% Pd[PPh3]4, 20% dppe, syringe pump, 0.0015 M, THF, reflux, 64%.](/image/article/2013/OB/c3ob41020j/c3ob41020j-s2.gif) | ||
| Scheme 2 Synthesis of cyclisation substrate 8. (i) NaH, TBDMSCl, 67%; (ii) (COCl)2, DMSO, Et3N, DCM, −78 °C to rt, 88%; (iii) MeOH–H2O (9/1); (iv) 3, DCM–10 M NaOH (1/1), −20 °C to rt, 1 h, 80%; (v) NH4F, MeOH, reflux, 8 h, 65%; (vi) MeOH, H2SO4, 89%; (vii) (PhSO2)2CH2, NaH, DMF, 81%; (viii) LiOH, THF–MeOH–H2O (3/3/1), quant.; (ix) EDC, DMAP, DCM, RT, 12 h, 68%; (x) 10% Pd[PPh3]4, 20% dppe, syringe pump, 0.0015 M, THF, reflux, 64%. | ||
In order to evaluate the performance of polymer-supported catalysts for the same macrocyclisation of 8, we used catalyst 1a/Pd as a standard, as it featured the most popular resin properties (PS, 1% DVB, 200–400 mesh) used in solid phase organic synthesis. Using 10% 1a/Pd under reflux conditions in THF, we varied the substrate concentration from 0.002 to 0.2 M (Fig. 1). To our delight, we could observe that immobilized catalyst 1a/Pd produced macrocycle 9 even at the high concentration of 0.2 M in good yields (45%), confirming the previously reported effect of catalyst immobilization in Tsuji–Trost-cyclizations. The best yields were observed at 0.04 M (63%) (Fig. 1).
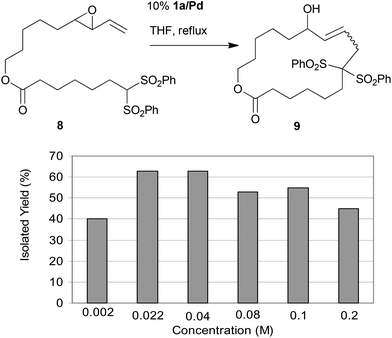 | ||
| Fig. 1 Influence of the substrate concentration on the product yield for the macrocyclisation of substrate 8 with immobilized catalyst 1a/Pd. | ||
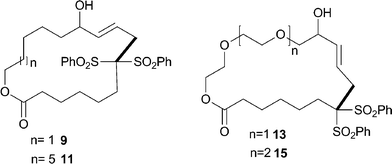 | ||
| Fig. 2 Tsuji–Trost-macrocyclisation products. The bond formed in the cyclisation step is indicated in bold. | ||
We used the concentration of 0.04 M for the next experiment in which we varied the reaction temperature. While at RT the yield decreased to 59% yield, at 50 °C it improved to 67%. As these experiments had confirmed that the immobilization effect was also valid for our substrate, we were considering the hypothesis that diffusion through the laminar layer surrounding the bead (film diffusion) resp. diffusion inside the bead (pore/gel diffusion) might be rate limiting. In such a scenario a locally lower substrate concentration inside the bead would be created, potentially equivalent to high dilution concentrations.9,14 In order to validate this hypothesis, we performed two sets of experiments. To test for potential film diffusion at first, we varied the stirring rate of the magnetic stirrer, which should influence the thickness of the laminar film layer around the beads. No influence of film diffusion could be detected, as the yields for a reaction at 0.022 M remained almost indistinguishable at the highest stirring rate (52%) from the one where no stirring was applied (51%) and values in between. Secondly, we screened our set of Pd-catalysts produced from polymer-supported phosphine resins 1a–g featuring different bead sizes and degrees of cross linking (Table 2).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Size [mesh] | 9 Yield [%] |
| 1 | 1a/Pd | 200–400 | 67 |
| 2 | 1b/Pd | 100–200 | 67 |
| 3 | 1c/Pd | 70–90 | 59 |
| 4 | 1d/Pd | 70–100 | 62 |
| 6 | 1e/Pd | ∼120 | 70 |
| 7 | 1f/Pd | ∼20 | 51 |
| 8 | 1g/Pd | ∼100 | 44 |
When comparing the similarly structured catalysts 1a/Pd, 1b/Pd, 1c/Pd, 1d/Pd, which differ primarily in their bead size, consistently good yields (59–67%) could be observed. Catalyst 1e/Pd, which is in bead size similar to catalyst 1b/Pd but has only half its loading, performs equally well (70%). As the larger beads (1c and 1d) exhibited similar or slightly diminished yields, the hypothesis that pore/gel diffusion might have any (or even a positive) effect on macrocycle yields is not supported. For comparison, the homogeneous catalyst Pd[PPh3P]4 gave under otherwise identical conditions only 29% yield of 9. A resin which significantly performed differently, in this case worse than others, was catalyst 1g made from a highly crosslinked resin, which does not swell in solvents. Interestingly, Tentagel-derived catalyst 1f which is most distinct from all other resins in terms of bead size, ligand flexibility, loading and hydrophilicity performed quite well with test substrate 8. From these two series of experiments we conclude that we have no evidence that limitation of substrate transport to and across the beads has an influence on macrocyclisation yields for the Pd-catalyzed Tsuji–Trost-macrocyclisation of our test substrate. To explore other substrates we limited our investigations to three catalysts: our standard catalyst 1a/Pd, its close analog 1b/Pd, and Tentagel-derived catalyst 1f/Pd, and compared them with the homogeneous catalyst Pd[PPh3]4.
When comparing catalyst performance, it can be seen that 1a/Pd and 1b/Pd gave consistently good yields also for other substrates, whereas the Tentagel-based catalyst 1f/Pd gave results similar to the homogeneous catalyst with the previously investigated standard substrate 8 as an exception. The almost congruent behaviour between homogeneous and Tentagel-immobilized catalysts matches well the perception that Tentagel-resin through its long and flexible spacer units emulates solution-phase behaviour, which has been used advantageously for several applications.15 Together these data led us to conclude that the observed effect, that catalyst immobilization on cross-linked polystyrene resins leads to improved yields for Tsuji–Trost-cyclisations, can be explained by two factors: (a) a pseudodilution effect, which results from the intermittent immobilization of the substrate as a Pd–allyl complex within a polymeric bead, thereby restricting the conformational reach of its reactive ends, and (b) the special case of the internal base activation mechanism via intramolecular deprotonation of the prenucleophile by the alkoxide resulting from vinylepoxide opening by the Pd-catalyst, which positions the nucleophile in close proximity to the electrophilic Pd–allyl complex (Fig. 3).4
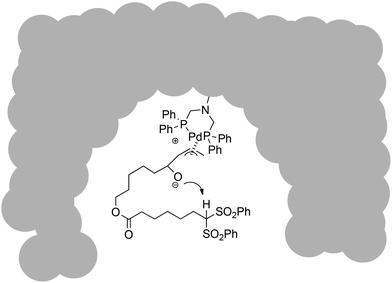 | ||
| Fig. 3 Mechanistic proposal for intramolecular cyclization at the active site within a catalyst bead. | ||
We next set out to explore if the catalyst-immobilization effect can be extended to other Pd-catalyzed transformations. In particular, we were interested in the Sonogashira- and Suzuki-macrocyclisations. Despite its frequent application in synthesis these macrocyclisation reactions have not been performed with immobilized Pd catalysts yet. For the Sonogashira-macrocyclisation16 a set of eleven different substrates was synthesized, which would result in 14- to 25-membered rings. The synthetic strategy is based on the modular assembly of terminal alkyne building blocks with various iodoarenes. As an example the synthesis of 19 is described in Scheme 3 (for all other substrates see ESI†) (Table 3, Fig. 2).
![Synthesis of Sonogashira-cyclisation substrate 19. (i) TBMSCl, imidazole, 50%; (ii) EDC, DMAP, 83%; (iii) NH4F, quant.; (iv) 2-iodobenzoic acid, EDC, DMAP, 83%; (v) 20% Pd[PPh3]4, 20% CuI, 0.04 M, 1,4-dioxane–piperidine (2/1), 60 °C, 15 h, 22%.](/image/article/2013/OB/c3ob41020j/c3ob41020j-s3.gif) | ||
| Scheme 3 Synthesis of Sonogashira-cyclisation substrate 19. (i) TBMSCl, imidazole, 50%; (ii) EDC, DMAP, 83%; (iii) NH4F, quant.; (iv) 2-iodobenzoic acid, EDC, DMAP, 83%; (v) 20% Pd[PPh3]4, 20% CuI, 0.04 M, 1,4-dioxane–piperidine (2/1), 60 °C, 15 h, 22%. | ||
12-Hydroxy undecanoic acid (16) was silyl-protected allowing the esterification with 5-hexyn-1-ol to produce ester 18. Fluoride-mediated deprotection of the TBDMS-group quantitatively furnished the free alcohol which underwent carbodiimide-mediated esterification with 2-iodo benzoic acid to produce macrocyclisation substrate 19 in 34% yield for the entire four step synthetic sequence. The homogeneous catalyst Pd[PPh3]4 and the standard polymer supported catalyst 1a/Pd were compared at 0.04 M substrate concentration for the transformation of 19 to cyclisation product 20. Both catalysts produced the 23-membered ring in 22% yield. In order to gain a more representative view of the Sonogashira-macrocyclisation, more substrates were investigated to produce the macrocycles depicted in Fig. 4 (Table 4).
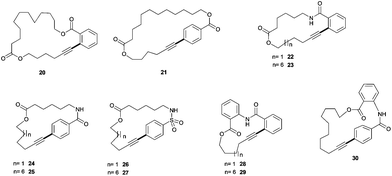 | ||
| Fig. 4 Sonogashira-macrocyclisation products. The bond formed in the cyclisation step is indicated in bold. | ||
| Entry | Product | Ring size | Yield with Pd[PPh3]4 | Yield with 1a/Pd |
|---|---|---|---|---|
| Conditions: 20% catalyst, 20% CuI, 0.04 M, 1,4-dioxane/piperidine (2/1), 60 °C, 15 h. | ||||
| 1 | 20 | 23 | 22% | 22% |
| 2 | 21 | 25 | — | 25% |
| 3 | 22 | 17 | 40% | 50% |
| 4 | 23 | 22 | 29% | 27% |
| 5 | 24 | 19 | 0% | 0% |
| 6 | 25 | 24 | 9% | 13% |
| 7 | 26 | 19 | 6% | 12% |
| 8 | 27 | 24 | 40% | 38% |
| 9 | 28 | 14 | 0% | 0% |
| 10 | 29 | 19 | 35% | 33% |
| 11 | 30 | 21 | 14% | 14% |
The experimental results allow two important conclusions: (1) due to the conformational constraints imposed by the large number of sp2- and sp-hybridized carbon atoms in our substrates the yield depends strongly on the particular substrate ranging from 0% for highly constrained rings 24 and 28 (entries 5 and 9) up to 50% (entry 3) for product 22, which is less constrained. (2) The yields from the reactions with the immobilized catalyst and the homogeneous reaction are very similar with only slight variations. We interpret these data that we cannot observe a catalyst immobilization effect for the Sonogashira-macrocyclisation of the investigated substrates. Apparently, the pseudodilution effect stemming from the transient immobilization of a reaction intermediate as it would occur during oxidative addition of the substrate is not sufficient to produce an improved cyclisation yield for the Sonogashira reaction.
The Suzuki-reaction has achieved considerable attention for macrocyclisations.17 However, to the best of our knowledge no efforts have been made to explore the use of immobilized catalysts in the context of this transformation. For substrate synthesis we had to adapt our modular synthetic strategy, as a direct transformation of our alkyne substrates for Sonogashira-macrocyclisations via hydroboration failed because of substrate decomposition. Our preferred synthetic route is illustrated for substrate 34 in Scheme 4 (for all other Suzuki substrates, see ESI†).
![Synthesis of Suzuki-macrocyclisation substrate 34. (i) TBDMSCl, Et3N, DMAP, DCM, overnight, quant.; (ii) catecholborane, 75 °C, 6 h; (iii) H2O, 1 h, then CH2O, 1 h, then pinacol, overnight; (iv) NH4F, MeOH, 6 h, 49% (3 steps); (v) DIC, DMAP, DCM, overnight, 68%; (vi) 10% Pd[PPh3]4, 0.04 M DME–H2O–DMF (7/3/2), 2 eq. Cs2CO3, 80 °C, 15 h, 60%.](/image/article/2013/OB/c3ob41020j/c3ob41020j-s4.gif) | ||
| Scheme 4 Synthesis of Suzuki-macrocyclisation substrate 34. (i) TBDMSCl, Et3N, DMAP, DCM, overnight, quant.; (ii) catecholborane, 75 °C, 6 h; (iii) H2O, 1 h, then CH2O, 1 h, then pinacol, overnight; (iv) NH4F, MeOH, 6 h, 49% (3 steps); (v) DIC, DMAP, DCM, overnight, 68%; (vi) 10% Pd[PPh3]4, 0.04 M DME–H2O–DMF (7/3/2), 2 eq. Cs2CO3, 80 °C, 15 h, 60%. | ||
Alkynol 31 was converted via hydroboration by catecholborane followed by in situ hydrolysis and transesterification to vinyl pinacol boron ester 32. This intermediate underwent carbodiimide-mediated esterification with carboxylic acid 33 to furnish cyclisation substrate 34 in 33% yield for the five-step reaction sequence. All in all, we had six substrates in hand which upon Suzuki-reaction would form 17- to 24-membered rings (Fig. 5, Table 5).
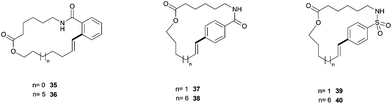 | ||
| Fig. 5 Suzuki-macrocyclisation products. The bond formed in the cyclisation step is indicated in bold. | ||
When performing the cyclisations with immobilized catalyst 1a/Pd and homogeneous catalyst Pd[PPh3]4 at 0.04 M substrate concentration, we again saw a great substrate dependence originating from the conformational strain of the formed compounds. Importantly, we also observed again a largely coherent efficiency between heterogeneous and homogeneous catalysts, showing better yields with the homogeneous catalyst for substrates 35 (entry 1) and 38 (entry 4), which led us to conclude that no substrate immobilization effect can be observed for the Suzuki-macrocyclisation of our tested substrates.
In conclusion, we have achieved the macrocyclisation of suitable substrates via Tsuji–Trost-macrocyclisation, and for the first time the Sonogashira-macrocyclisation and Suzuki-macrocyclisation using polymer-bound Pd-complexes.18 Our study has revealed that the catalyst-immobilization effect is not of general applicability to Pd-catalyzed macrocyclisations. For a set of eleven Sonogashira- and six Suzuki-macrocyclisation substrates we could not observe any evidence for such an effect. However, we could consistently see such an effect for Pd-catalyzed Tsuji–Trost-allylation of vinylepoxides. In contrast to previous intermolecular alkylation reactions catalyzed by solid-supported phase transfer catalysts,9 we could show that for the intramolecular Tsuji–Trost-allylation, substrate transport to and across the resin beads does not play a major role. Instead we believe that the combination of restricted freedom of the intermittently immobilized substrate and the idiosyncratic intramolecular activation mechanism through the formation of an alcoholate base and subsequent deprotonation of the prenucleophile plays an important role in this reaction. The flexibility of Tentagel resins does obscure the immobilization effect. It remains to be seen that other macrocyclisation reactions can be designed in which the effect of catalyst immobilization will allow macrocyclisations even in concentrated solutions,19 as has now been well exemplified for the intramolecular Tsuji–Trost-allylation with vinylepoxide substrates.
Notes and references
- Leading reviews: (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS; (b) C. M. Madsen and M. H. Clausen, Eur. J. Org. Chem., 2011, 3107–3115 CrossRef CAS; (c) A. Isidro-Llobet, T. Murillo, P. Bello, A. Cilibrizzi, J. T. Hodgkinson, W. R. J. D. Galloway, A. Bender, M. Welch and D. R. Spring, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6793–6798 CrossRef CAS; (d) C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS ; and leading original works: ; (e) Y.-k. Kim, M. A. Arai, T. Arai, J. O. Lamenzo, E. F. Dean III, N. Patterson, P. A. Clemons and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 14740–14745 CrossRef CAS; (f) M. T. Burger and P. A. Bartlett, J. Am. Chem. Soc., 1997, 119, 12697–12698 CrossRef CAS.
- Reviews: (a) B. Dietrich, P. Viout and J.-M. Lehn, Macrocyclic Chemistry, VCH, Weinheim, 1992 Search PubMed; (b) Q. Meng and M. Hesse, Top. Curr. Chem., 1991, 161, 107–176 CrossRef; (c) C. J. Roxburgh, Tetrahedron, 1995, 51, 9767–9822 CrossRef CAS.
- Reviews: (a) P. Knops, N. Sendhoff, H.-B. Mekelburger and F. Vögtle, Top. Curr. Chem., 1991, 161, 1–36 CrossRef; (b) K. Ziegler, Methoden der Organischen Chemie (Houben-Weyl), 4th edn, 1955, Vol. 4/II, pp. 729–822 Search PubMed.
- (a) B. M. Trost and R. W. Warner, J. Am. Chem. Soc., 1982, 104, 6112–6114 CrossRef CAS; (b) B. M. Trost and R. W. Warner, J. Am. Chem. Soc., 1983, 105, 5940–5942 CrossRef CAS; (c) B. M. Trost, Angew. Chem., Int. Ed., 1999, 28, 2817–2825 Search PubMed.
- A. Fürstner and H. Weintritt, J. Am. Chem. Soc., 1998, 120, 2817–2825 CrossRef.
- J. K. Stille, H. Su, D. H. Hill, P. Schneider, M. Tanaka, D. L. Morrison and L. S. Hegedus, Organometallics, 1991, 10, 1993–2000 CrossRef CAS.
- A. R. Kelly, J. Wei, S. Kesavan, J.-C. Marie, N. Windmon, D. W. Young and L. A. Marcaurelle, Org. Lett., 2009, 11, 2257–2260 CrossRef CAS.
- (a) M. Fridkin, A. Patchornik and E. Katchalski, J. Am. Chem. Soc., 1965, 87, 4646–4648 CrossRef CAS; (b) S. Mazur and P. Jayalekshmy, J. Am. Chem. Soc., 1979, 101, 677–683 CrossRef CAS; (c) J. Rebek Jr. and J. E. Trend, J. Am. Chem. Soc., 1979, 101, 737 CrossRef; (d) H. Franzyk, M. K. Christensen, R. M. Jorgensen, M. Meldal, H. Cordes, S. Mouritsen and K. Bock, Bioorg. Med. Chem., 1997, 5, 21–40 CrossRef CAS; (e) C. Herb and M. E. Maier, J. Org. Chem., 2003, 68, 8129–8135 CrossRef CAS; (f) E. Gonthier and R. Breinbauer, Mol. Diversity, 2005, 9, 51–62 CrossRef CAS.
- The influence of transport processes on intermolecular reactions on beads has been demonstrated before, see: (a) S. L. Regen and J. J. Besse, J. Am. Chem. Soc., 1979, 101, 4059–4063 CrossRef CAS; (b) M. Tomoi and W. T. Ford, J. Am. Chem. Soc., 1980, 102, 7140–7141 CrossRef CAS; (c) M. Tomoi and W. T. Ford, J. Am. Chem. Soc., 1981, 103, 3821–3828 CrossRef CAS; (d) M. Tomoi and W. T. Ford, J. Am. Chem. Soc., 1981, 103, 3828–3837 CrossRef CAS; (e) B. Kim, P. Kirszensztejn and S. L. Regen, J. Am. Chem. Soc., 1983, 105, 1567–1571 CrossRef CAS.
- For studies on diffusion and reactivity phenomena on polymeric beads, see: (a) W. Li, X. Xiao and A. W. Czarnik, J. Comb. Chem., 1999, 1, 127–129 CrossRef CAS; (b) A. R. Vaino and K. D. Janda, J. Comb. Chem., 2000, 2, 579–596 CrossRef CAS; (c) J. Rademann, M. Barth, R. Brock, H.-J. Egelhaaf and G. Jung, Chem.–Eur. J., 2001, 7, 3884–3889 CrossRef CAS; (d) T. Groth, M. Grotli and M. Meldal, J. Comb. Chem., 2001, 3, 461–468 CrossRef CAS; (e) J. Kress, R. Zanaletti, A. Rose, J. G. Frey, W. S. Brocklesby and M. Bradley, J. Comb. Chem., 2003, 5, 28–32 CrossRef CAS.
- (a) A. M. LaPointe, J. Comb. Chem., 1999, 1, 101–104 CrossRef CAS; (b) M. T. Reetz, G. Lohmer and R. Schwickardi, Angew. Chem., Int. Ed. Engl., 1997, 36, 1526–1529 CrossRef CAS; (c) M. T. Reetz and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 1997, 36, 865–867 CrossRef CAS.
- (a) Y. Uozumi and Y. Nakami, Org. Lett., 2002, 4, 2997–3000 CrossRef CAS; (b) E. Gonthier and R. Breinbauer, Synthesis, 2003, 1049–1051 CAS; (c) M. Westhus, E. Gonthier, D. Brohm and R. Breinbauer, Tetrahedron Lett., 2004, 45, 3141–3142 CrossRef CAS.
- M. Rosenberger, C. Newkom and E. R. Aig, J. Am. Chem. Soc., 1983, 105, 3656–3661 CrossRef CAS.
- For authoritative reviews about diffusion phenomena in heterogeneous catalysis, see: (a) G. Emig and R. Dittmeyer, in Handbook of Heterogeneous Catalysis, ed, G. Ertl, H. Knözinger and J. Weitkamp, VCH, Weinheim, 1997, pp. 1209–1252 Search PubMed; (b) M. Baerns, H. Hofmann and A. Renken, Chemische Reaktionstechnik, 2, Aufl. Thieme, Stuttgart, 1992 Search PubMed; (c) F. Kapteijn and J. A. Moulijn, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, VCH, Weinheim, 1997, 1359–1376 Search PubMed; (d) R. J. Madon and M. Boudart, Ind. Eng. Chem. Fundam., 1982, 21, 438–447 CrossRef CAS.
- See for example: (a) K. Kuramochi, T. Haruyama, R. Takeuchi, T. Sunoki, M. Watanabe, M. Oshige, S. Kobayashi, K. Sakaguchi and F. Sugawara, Bioconjugate Chem., 2005, 16, 97–104 CrossRef CAS; (b) B. Lehr, H.-J. Egelhaaf, H. Fritz, W. Rapp, E. Bayer and D. Oelkrug, Macromolecules, 1996, 29, 7931–7936 CrossRef CAS; (c) P. Wright, D. Lloyd, W. Rapp and A. Andrus, Tetrahedron Lett., 1993, 34, 3373–3376 CrossRef CAS.
- For selected examples of Sonogashira-macrocyclisations, see: (a) M. Schmittel and H. Ammon, Synlett, 1999, 750–752 CrossRef CAS; (b) A. C. Spivey, J. McKendrick, R. Srikaran and B. A. Helm, J. Org. Chem., 2003, 68, 1843–1851 CrossRef CAS; (c) T. Harada, S.-I. Matsui, T. M. T. Tuyet, M. Hatsuda, S. Ueda, A. Oku and M. Shiro, Tetrahedron: Asymmetry, 2003, 14, 3879–3884 CrossRef CAS; (d) S. Odermatt, J. L. Alonso-Gomez, P. Seiler, M. M. Cid and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 5074–5078 CrossRef CAS; (e) J. Krauss, D. Unterreitmeier, C. Neudert and F. Bracher, Arch. Pharm., 2005, 338, 605–608 CrossRef CAS; (f) V. Balraju, D. S. Reddy, M. Periasamy and J. Iqbal, J. Org. Chem., 2005, 70, 9626–9628 CrossRef CAS; (g) J. Sakamoto and D. A. Schlüter, Eur. J. Org. Chem., 2007, 2700–2712 CrossRef CAS; (h) G. Chen, L. Wang, D. W. Thompson and Y. Zhao, Org. Lett., 2008, 10, 657–660 CrossRef CAS.
- For selected examples of Suzuki-macrocyclisations, see: (a) R. Ostaszewski, W. Verboom and D. N. Reinhoudt, Synlett, 1992, 354–356 CrossRef CAS; (b) W. Li and K. Burgess, Tetrahedron Lett., 1999, 40, 6527–6530 CrossRef CAS; (c) N. C. Kallan and R. L. Halcomb, Org. Lett., 2000, 2, 2687–2690 CrossRef CAS; (d) V. Lobregat, G. Alcaraz, H. Bienayme and M. Vaultier, Chem. Commun., 2001, 817–818 RSC; (e) J. D. White, R. Hanselmann, R. W. Jackson, W. J. Porter, J. Ohba, T. W. Tille and S. Wang, J. Org. Chem., 2001, 66, 5217–5231 CrossRef CAS; (f) M. Bauer and M. E. Maier, Org. Lett., 2002, 4, 2205–2208 CrossRef CAS; (g) J. T. Njardarson, K. Biswas and S. J. Danishefsky, Chem. Commun., 2002, 2759–2761 RSC; (h) P. J. Mohr and R. L. Halcomb, J. Am. Chem. Soc., 2003, 125, 1712–1713 CrossRef CAS; (i) G. A. Molander and F. Dehmel, J. Am. Chem. Soc., 2004, 126, 10313–10318 CrossRef CAS; (j) T. Esumi, M. Wada, E. Mizushima, N. Sato, M. Kodama, Y. Asakawa and Y. Fukuyama, Tetrahedron Lett., 2004, 45, 6941–6945 CrossRef CAS; (k) B. Wu, Q. Liu and G. A. Sulikowski, Angew. Chem., Int. Ed., 2004, 43, 6673–6675 CrossRef CAS; (l) D. Deng, B. Wang, A. J. Augatis and N. I. Totah, Tetrahedron Lett., 2007, 48, 4605–4607 CrossRef; (m) J. M. W. Chan and T. M. Swager, Tetrahedron Lett., 2008, 49, 4912–4914 CrossRef CAS; (n) M. Tortosa, N. A. Yakelis and W. R. Roush, J. Org. Chem., 2008, 73, 9657–9667 CrossRef CAS; (o) A. B. Smith, M. A. Foley, S. Dong and A. Orbin, J. Org. Chem., 2009, 74, 5987–6001 CrossRef CAS; (p) Z. Wang, M. Bois-Choussy, Y. Jia and J. Zhu, Angew. Chem., Int. Ed., 2010, 49, 2018–2022 CrossRef CAS; (q) J. Dufour, L. Neuville and J. Zhu, Chem.–Eur. J., 2010, 16, 10523–10534 CrossRef CAS; (r) W. Huang, M. Wang, C. Du, Y. Chen, R. Qin, L. Su, C. Zhang, Z. Liu, C. Li and Z. Bo, Chem.–Eur. J., 2011, 17, 440–444 CrossRef CAS; (s) A. Afonso, L. Feliu and M. Planas, Tetrahedron, 2011, 67, 2238–2245 CrossRef CAS; (t) M. Dieckmann, M. Kretschmer, P. Li, S. Rudolph, D. Herkommer and D. Menche, Angew. Chem., Int. Ed., 2012, 51, 5667–5670 CrossRef CAS; (u) F.-M. Meyer, J. C. Collins, B. Borin, J. Bradow, S. Liras, C. Limberakis, A. M. Mathiowetz, L. Philippe, D. Price, K. Song and K. James, J. Org. Chem., 2012, 77, 3099–3114 CrossRef CAS.
- Reviews about the synthesis and application of polymer-bound Pd complexes: (a) N. E. Leadbeater, Curr. Med. Chem., 2002, 9, 2147 CrossRef CAS; (b) N. End and K. U. Schöning, Top. Curr. Chem., 2004, 242, 241 CrossRef CAS; (c) Y. Uozumi, Top. Curr. Chem., 2004, 242, 77–112 CrossRef CAS; (d) M. Guino and K. K. Hii, Chem. Soc. Rev., 2007, 36, 608–617 RSC; (e) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS; (f) A. Molnar, Chem. Rev., 2011, 111, 2251–2320 CrossRef CAS; (g) A. C. Albeniz and N. Carrera, Eur. J. Inorg. Chem., 2011, 2347–2360 CrossRef CAS.
- Recently, the ring closing metathesis with Ru-complexes immobilized in silica nanopores has been reported to occur with improved yields for macrocyclisation products: J. E. Jee, J. L. Cheong, J. Lim, C. Chen, S. H. Hong and S. S. Lee, J. Org. Chem., 2013, 78, 3048–3056 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob41020j |
| This journal is © The Royal Society of Chemistry 2013 |