 Open Access Article
Open Access ArticleDirect C–H sulfenylation of purines and deazapurines†
Martin
Klečka
ab,
Radek
Pohl
b,
Jan
Čejka
b and
Michal
Hocek
*ab
aDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12843 Prague 2, Czech Republic
bInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead & IOCB Research Center, Flemingovo nam. 2, CZ-16610 Prague 6, Czech Republic. E-mail: hocek@uochb.cas.cz
First published on 12th June 2013
Abstract
A general method for Cu-catalyzed C–H sulfenylation of purines, 7-deaza- and 9-deazapurines with aryl- or alkyldisulfides has been developed. In purines, the reaction occurs at position 8, in 7-deazapurines at position 7 and in 9-deazapurines at position 9, leading to new interesting arylsulfanyl derivatives of purine or deazapurine bases. The resulting 8-arylsulfanylpurines undergo Liebesking–Srogl coupling with arylstannanes or boronic acids, whereas the (arylsulfanyl)deazapurines are not reactive under these conditions.
Tri- and tetrasubstituted purines1 and their analogues show a great variety of biological activities. However, compared to purines, 7-deazapurines (pyrrolo[2,3-d]pyrimidines) and 9-deazapurines (pyrrolo[3,2-d]pyrimidines) have been studied less systematically due to underdeveloped synthesis and substitution chemistry. Among the most important biological effects of deazapurines one should mention compound TWS1192 which directs differentiation of neuronal cells in mice. Several types of 7-deazapurine nucleosides display antibiotic,3 antiviral4 or cytostatic5 effects. In order to synthesize libraries of tri- and tetrasubstituted purines, a combination of inherently orthogonal cross-coupling reactions with C–H activations has been developed in our group.6 6,8,9-Trisubstituted 7-deazapurines were prepared by a “one pot” sequence of C–H borylation followed by Suzuki coupling.7 In order to extend the portfolio of reactions and substituents available for modification of purines and deazapurines, we report here on direct C–H sulfenylations leading to hitherto unknown arylsulfanyl-derivatives and some follow-up reactions.
Direct C–H sulfenylations8 have become quite popular in recent years since they lead to hetarylthioethers suitable for further functional group transformations by Liebeskind–Srogl cross-coupling9 or oxidation and aminations.10 Also some examples of biologically active hetarylthioesters were previously described.11
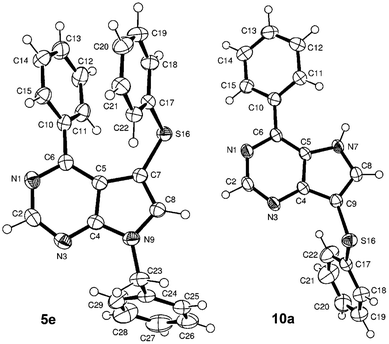
Our project started with the study of C–H sulfenylations of 7-deazapurines which are closely related to indoles. The model starting compound of choice was 6-phenyl-7-deazapurine (1). We started by testing several literature catalytic systems and conditions for direct C–H sulfenylation (Scheme 1).8 The most efficient was the reaction of 1 with disulphides in the presence of copper(I) catalyst (by analogy to the literature8a but replacing DMSO with DMF) giving the desired 7-substituted product 5a in excellent yield (96%, Table 1, entry 1). On a larger scale, a 7,8-bis(phenylsulfanyl) derivative 6a was also isolated as a minor by-product (3%, entry 1). The reaction work-up by EDTA was very important to break up stable complexes of the product with copper (without such a work-up, the isolated yield of 5a was only moderate, ∼50%). These optimised conditions were then used for the synthesis of three other examples, 7-alkyl- or -arylsulfanyl derivatives 5b–d. While the reactions with methyl and methoxyphenyl disulfide gave products 5b, c in good yields (entries 2, 3), the yield of nitrophenylsulfanyl derivative 5d was moderate. The reaction with 9-benzylated 6-phenyl-7-deazapurine 2 gives the 7-substituted product 5e in poor yield (20%, entry 5) due to low conversion. The structure of 5e was confirmed by X-ray (Fig. 1). Apparently, the free NH at position 7 is crucial for the efficiency of this reaction. Another interesting substrate was 6-chloro-7-deazapurine 3 that is suitable for further functional group transformations at position 6. In this case, the C–H sulfenylation also proceeded well to give the desired product 5f in high (90%) yield (entry 6) without any trace of nucleophilic substitution at position 6. Also the reaction of 7-deazaadenine (4) proceeded under the same conditions to give 7-(phenylsulfanyl)-7-deazaadenine (5g) in good yield (entry 7) (Scheme 1).
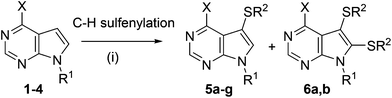 | ||
| Scheme 1 Reagents and conditions: (i) R2S–SR2 (0.75 equiv.), CuI (10%), air, DMF, 110 °C, 18–60 h. | ||
| Entry | Start. compd. | R1 | X | R2 | Product (yield) |
|---|---|---|---|---|---|
| a 5 equiv. of R2S–SR2. b 2.5 equiv. of R2S–SR2 and recovery of the starting compound (71%). | |||||
| 1 | 1 | H | Ph– | Ph– | 5a (96%) + 6a (3%) |
| 2a | 1 | H | Ph– | Me– | 5b (71%) + 6b (15%) |
| 3 | 1 | H | Ph– | 4-MeO–Ph– | 5c (91%) |
| 4 | 1 | H | Ph– | 4-NO2–Ph– | 5d (47%) |
| 5b | 2 | Bn | Ph– | Ph– | 5e (20%)b |
| 6a | 3 | H | Cl– | Ph– | 5f (90%) |
| 7 | 4 | H | NH2– | Ph– | 5g (79%) |
The same C–H sulfenylation protocol was then tested on 9-deazapurines (pyrrolo[3,2-d]pyrimidines, Scheme 2). However, in this case a competitive iodination of the heterocycle by CuI occurred (Table 2, entry 1). The halogenation was suppressed by complexation of the copper catalyst by a 2,2′-dipyridine (bpy) ligand. The reaction of 6-phenyl-9-deazapurine (7) with diphenyl disulfide in the presence of CuI + bpy (entry 2) gave quantitatively the desired 9-phenylsulfanyl derivative 10a (for confirmation of its structure by X-ray, see Fig. 1). The reaction with other disulfides allowed us to synthesize the target 9-alkyl- or -arylsulfanyl derivatives in moderate (10b and 10d, 30% and 55%, respectively, entries 3, 5) or high yields (10c, 85%, entry 4). The reaction with 9-benzyl-6-phenyl-9-deazapurine 8 did not proceed at all (entry 6). The C–H sulfenylation of 6-chloro-9-deazapurine 9 under standard conditions gave a complex mixture of products (TLC, entry 7). Therefore, we tried the reaction in the presence of a more bulky and electron-rich ligand dtbpy (4,4′-di-tert-butyl-2,2′-dipyridine, for more details of the optimization, see ESI†) to give the desired product 10e in good 90% yield (entry 8). The dtbpy ligand was then also tested in the reactions of 7 with diverse disulfides. The phenylsulfenylation proceeded with quantitative conversion (as with bpy) but in the case of other disulfides, the yields of products were lower than with bpy (entries 10–12). Therefore, the dtbpy ligand was only practical for the reaction of 6-chloro derivative 9. On the other hand, using a stoichiometric amount of CuI or CuBr2 in the absence of bpy led to the formation of 9-halogenated products 11a–c in high yields (entries 8–10). The same reaction with CuCl or CuCl2 proceeded as well but only in poor yield. Using the same catalytic system (CuI + bpy) for 7-deazapurine 1 gave the 7-substituted product 5a in poor yield due to low conversion.
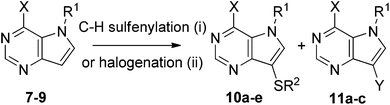 | ||
| Scheme 2 Reagents and conditions: (i) R2S–SR2 (1.5 equiv.), CuI (10%), air, bpy or dtbpy (0.2 equiv.), DMF, 110 °C, 48–90 h; (ii) CuI or CuBr2 (1.1 equiv.), air, DMF, 110 °C, 18 h. | ||
| Entry | Start. compd. | Ligand | R1 | X | R2 (or Y) | Product (yield) |
|---|---|---|---|---|---|---|
| a No bpy added. b 7 equiv. of R2S–SR2. c Recovery of the starting compound (40%). d Condition (ii) applied. | ||||||
| 1a | 7 | — | H | Ph | Ph | 10a (14%) + 11a (9%) |
| 2 | 7 | bpy | H | Ph | Ph | 10a (98%) |
| 3 | 7 | bpy | H | Ph | Me | 10b (30%)c |
| 4 | 7 | bpy | H | Ph | 4-MeO–Ph | 10c (85%) |
| 5 | 7 | bpy | H | Ph | 4-NO2–Ph | 10d (50%) |
| 6 | 8 | bpy | Bn | Ph | Ph | No reaction |
| 7 | 9 | bpy | H | Cl | Ph | Complex mixture |
| 8b | 9 | dtbpy | H | Cl | Ph | 10e (90%) |
| 9 | 7 | dtbpy | H | Ph | Ph | 10a (98%) |
| 10 | 7 | dtbpy | H | Ph | Me | 10b (25%) |
| 11 | 7 | dtbpy | H | Ph | 4-MeO–Ph | 10c (41%) |
| 12 | 7 | dtbpy | H | Ph | 4-NO2–Ph | No reaction |
| 13d | 7 | — | H | Ph | Y = I | 11a (81%) |
| 14d | 7 | — | H | Ph | Y = Br | 11b (75%) |
| 15d | 9 | — | H | Cl | Y = I | 11c (65%) |
Our further efforts focused on the direct C–H sulfenylation of purines. Unfortunately, employing the same catalytic systems as above, no sulfenylation was observed. Using an alternative protocol based on a Lewis acid activation,8e the reaction proceeded to give 8-(phenylsulfanyl)purine 13a in moderate ∼40% yield. Finally, the sulfenylation in the presence of tBuOLi8c in dioxane at 130 °C for 120 h gave the desired product 13a in acceptable 60% yield (Scheme 3, Table 3, entry 1). An analogous reaction with electron-rich bis(methoxyphenyl)disulphide proceeded well to give 13b in 56% (entry 2), whereas the reaction with electron-poor bis(nitrophenyl)disulfide did not work.
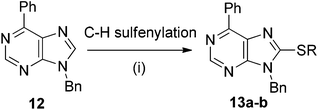 | ||
| Scheme 3 Reagents and conditions: RS–SR (2.5 equiv.), tBuOLi (3 equiv.), 1,4-dioxane, 130 °C, 120 h. | ||
| Entry | X | R | Product (yield) |
|---|---|---|---|
| 1 | Ph– | Ph– | 13a (60%) |
| 2 | Ph– | 4-MeO–Ph– | 13b (56%) |
| 3 | Ph– | 4-NO2–Ph– | No reaction |
Having access to the arylsulfanyl derivatives of purines and deazapurines, we further explored their synthetic applications. The most obvious option was the Liebeskind–Srogl cross-coupling reaction.9 The reactions of the 8-(phenylsulfanyl)purine 13a with phenylboronic acid and diverse stannanes performed under standard conditions proceeded generally well to give the desired 8-aryl products 14a–14c in high yields (57–83%, Scheme 4, Table 4).
 | ||
| Scheme 4 Reagents and conditions: (i) ArSnBu3 (1.2 equiv.), Pd(PPh3)4 (5 mol%), CuMeSal (2.2 equiv.), 50 °C, THF, 17 h; (ii) ArB(OH)2, Pd2(dba)3 (4 mol%), (2-furyl)3P (16 mol%), CuTc (1.3 equiv.), 50 °C, THF, 18 h. | ||


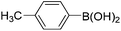
Surprisingly, analogous Liebeskind–Srogl reactions of 7-phenylsulfanyl-7-deazapurine 5a or 9-phenylsulfanyl-9-deazapurine 10a did not proceed at all. Neither stannanes nor boronic acids gave any reaction under a number of different catalytic systems (Cu, Pd, In) and conditions tried (including MW irradiation). This lack of reactivity of arylsulfanyl-deazapurines is probably due to the electron-rich nature of the deazapurine moiety which prevents efficient oxidative addition. Since no literature example of the Liebeskind–Srogl reaction of the related 3-(arylsulfanyl)indole was known, we have tried this reaction under the standard conditions and have confirmed that it does not proceed either. Apparently, this reaction is not applicable for electron-rich indole-type heterocycles.
In conclusion, the Cu-catalyzed C–H sulfenylation of 7- and 9-deazapurines proceeded very well and selectively at position 7 or 9, respectively, to give novel and interesting (arylsulfanyl)deazapurine derivatives. On the other hand, the C–H sulfenylation of purines was less efficient, and the conditions had to be changed. However, the 8-(arylsulfanyl)purines smoothly undergo the Liebeskind–Srogl cross-coupling reactions leading to 8-arylpurines, whereas the 7- and 9-arylsulfanylpurines were not reactive in these reactions. Since all these C–H sulfenylations can be performed with 6-chloro(deaza)purines, there is a potential in combination with classical cross-couplings in the synthesis of libraries of new di- and trisubstituted purines and deazapurine derivatives combining aryl(alkyl)sulfanyl and aryl or amino substituents for biological activity screening. Also there is a further potential in testing other reactivities of the (arylsulfanyl)deazapurines (oxidations, other couplings, etc.). Studies along these lines are under way in our laboratory.
Experimental
For a complete set of experimental procedures and characterization data, see ESI.†Sulfenylation of 7-deazapurines. General procedure
A mixture of 7-deazapurines 1–4 (2 mmol), disulphides (1.5 mmol), and CuI (0.2 mmol, 10 mol%) in DMF (20 mL) was stirred at 110 °C under an air atmosphere for 18 hours until complete consumption of the starting material as monitored by TLC. The solution was then cooled to room temperature, diluted with EtOAc (30 mL), and washed with a 1 M solution of sodium salt of EDTA (20 mL). The aqueous solution was then extracted three times with EtOAc and the combined organic layers were dried over Na2SO4, filtered, and evaporated under vacuum. The crude product was purified by column chromatography on silica gel.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1. Crystallization in hexane–EtOAc gave white needles. M.p. 184–186 °C. 1H NMR (499.8 MHz, DMSO-d6): 6.70 (m, 2H, H-o-SPh); 6.99 (m, 1H, H-p-SPh); 7.06 (m, 2H, H-m-SPh); 7.27 (m, 2H, H-m-Ph); 7.38 (m, 1H, H-p-Ph); 7.53 (m, 2H, H-o-Ph); 8.05 (d, 1H, J6,NH = 2.5, H-6); 8.88 (s, 1H, H-2); 12.86 (bs, 1H, NH). 13C NMR (125.7 MHz, DMSO-d6): 99.90 (C-5); 115.26 (C-4a); 125.25 (CH-p-SPh); 126.04 (CH-o-SPh); 127.29 (CH-m-Ph); 128.80 (CH-m-SPh); 129.23 (CH-p-Ph); 129.86 (CH-o-Ph); 135.69 (CH-6); 137.04 (C-i-Ph); 138.47 (C-i-SPh); 151.53 (CH-2); 153.55 (C-7a); 159.40 (C-4). IR(KBr): 3104, 3059, 2988, 2862, 2818, 1598, 1581, 1551, 1478, 1435, 1322. HRMS (ESI) calculated for C18H14N3S: 304.0902; found: 304.0901. Anal. calculated for C18H13N3S (303.08): C 71.26%, H 4.32%, N 13.85%, S 10.57%; found: C 71.07%, H 4.15%, N 13.57%, S 10.47%.
:1 to 1:1. Crystallization in hexane–EtOAc gave white needles. M.p. 184–186 °C. 1H NMR (499.8 MHz, DMSO-d6): 6.70 (m, 2H, H-o-SPh); 6.99 (m, 1H, H-p-SPh); 7.06 (m, 2H, H-m-SPh); 7.27 (m, 2H, H-m-Ph); 7.38 (m, 1H, H-p-Ph); 7.53 (m, 2H, H-o-Ph); 8.05 (d, 1H, J6,NH = 2.5, H-6); 8.88 (s, 1H, H-2); 12.86 (bs, 1H, NH). 13C NMR (125.7 MHz, DMSO-d6): 99.90 (C-5); 115.26 (C-4a); 125.25 (CH-p-SPh); 126.04 (CH-o-SPh); 127.29 (CH-m-Ph); 128.80 (CH-m-SPh); 129.23 (CH-p-Ph); 129.86 (CH-o-Ph); 135.69 (CH-6); 137.04 (C-i-Ph); 138.47 (C-i-SPh); 151.53 (CH-2); 153.55 (C-7a); 159.40 (C-4). IR(KBr): 3104, 3059, 2988, 2862, 2818, 1598, 1581, 1551, 1478, 1435, 1322. HRMS (ESI) calculated for C18H14N3S: 304.0902; found: 304.0901. Anal. calculated for C18H13N3S (303.08): C 71.26%, H 4.32%, N 13.85%, S 10.57%; found: C 71.07%, H 4.15%, N 13.57%, S 10.47%.
Sulfenylation of 9-deazapurines. General procedure
A mixture of CuI (0.2 mmol, 10 mol%) and 2,2′-bipyridine (0.4 mmol, 20 mol%) in DMF (10 mL) was stirred at rt for 15 minutes and then it was added to a mixture of 9-deazapurines 7–9 (2 mmol) and disulphides (3 mmol) in DMF (20 mL) and then was stirred at 110 °C under an air atmosphere for 48 hours until complete consumption of the starting material as monitored by TLC. The solution was then cooled to room temperature, diluted with EtOAc (30 mL), and washed with a 1 M solution of sodium salt of EDTA (20 mL). The aqueous solution was then extracted three times with EtOAc and the combined organic layers were dried over Na2SO4, filtered, and evaporated under vacuum. The crude product was purified by column chromatography on silica gel.:1 to 1:2. Crystallization in hexane–EtOAc gave white needles. M.p. 210–216 °C. 1H NMR (499.8 MHz, DMSO-d6): 7.10 (m, 3H, H-o,p-SPh); 7.22 (m, 2H, H-m-SPh); 7.61 (m, 1H, H-p-Ph); 7.63 (m, 2H, H-m-Ph); 8.11 (m, 2H, H-o-Ph); 8.29 (s, 1H, H-6); 8.95 (s, 1H, H-2); 12.56 (bs, 1H, NH). 13C NMR (125.7 MHz, DMSO-d6): 101.28 (C-7); 124.83 (C-4a); 125.30 (CH-p-SPh); 126.02 (CH-o-SPh); 128.99 (CH-o-Ph); 129.10, 129.15 (CH-m-Ph, CH-m-SPh); 130.61 (CH-p-Ph); 135.77 (C-i-Ph); 138.63 (C-i-SPh); 140.37 (CH-6); 148.88 (C-4); 151.29 (CH-2); 151.43 (C-7a). IR(KBr): 3066, 2835, 1594, 1542, 1505, 1490, 1480, 1429. HRMS (ESI) calculated for C18H14N3S: 304.0902; found: 304.0902.
Sulfenylation of 9-benzyl-6-phenyl-9H-purine. General procedure
A 20 mL sealable tube equipped with a magnetic stirring bar was charged with all solid reaction components, 9-benzyl-6-phenyl-9H-purine 12 (286 mg, 1 mmol), disulfide (2.5 mmol), tBuOLi (240 mg, 3 mmol) and 1,4-dioxane (2 mL) via a syringe. The vessel was closed by a Teflon-coated screw cap under Ar and was placed in a pre-heated oil bath at 130 °C and stirred until complete consumption of the starting material as monitored by TLC, approx. 130 hours. It was cooled to room temperature and diluted with ethyl acetate (15 mL). The resulting solution was directly filtered through a filter paper and concentrated under reduced pressure.:1 to 1:2. M.p. 101–104 °C. 1H NMR (499.8 MHz, CDCl3): 5.50 (s, 2H, CH2Ph); 7.27–7.35 (m, 5H, H-o,m,p-Bn); 7.37–7.41 (m, 5H, H-m,p-PhS); 7.45–7.50 (m, 3H, H-m,p-Ph); 7.59 (m, 2H, H-o-PhS); 8.74 (m, 2H, H-o-Ph); 8.96 (s, 1H, H-2). 13C NMR (125.7 MHz, CDCl3): 46.59 (CH2Ph); 127.75 (CH-o-Bn); 128.18 (CH-p-Bn); 128.50 (CH-m-Ph); 128.68 (C-i-PhS); 128.82 (CH-m-Bn); 129.03 (CH-p-PhS); 129.37 (CH-m-PhS); 129.68 (CH-o-Ph); 130.78 (CH-p-Ph); 131.16 (C-5); 132.91 (CH-o-PhS); 135.24 (C-i-Bn); 135.54 (C-i-Ph); 151.95 (CH-2); 152.37 (C-6); 152.92 (C-8); 154.46 (C-4). IR(KBr): 2921, 2851, 1580, 1561, 1495, 1459, 1429, 1258, 764. HRMS (ESI) calculated for C24H19N4S: 395.1325; found: 395.1323.
Liebeskind–Srogl cross-coupling of 9-benzyl-6-phenyl-8-(phenylsulfanyl)-9H-purine with stannanes. General procedure
To the mixture of CuMeSal (47 mg, 0.22 mmol, 2.2 equiv.), Pd(PPh3)4 (5.8 mg, 0.005 mmol, 0.05 equiv.) and 9-benzyl-6-phenyl-8-(phenylthio)-9H-purine 13a (39 mg, 0.1 mmol, 1.0 equiv.) and stannane (0.12 mmol, 1.2 equiv.) in THF (2 mL) were added. The reaction mixture was stirred under nitrogen at 50 °C for 18 h, and then 10% aqueous NH4OH (10 mL) was added and the mixture was stirred for an additional 10 min. The reaction mixture was filtered through a plug of Celite, and the filtrate was extracted with ethylacetate (3 × 15 mL). The organic layer was washed with brine (5 mL), dried over NaSO4, and evaporated. The crude product was purified by column chromatography on silica gel.:1 to 2:1. M.p. 135–141 °C. 1H NMR (500.0 MHz, CDCl3): 5.86 (s, 2H, CH2Ph); 6.59 (dd, 1H, J4,3 = 3.6, J4,5 = 1.8, H-4-furyl); 7.22 (m, 2H, H-o-Bn); 7.26 (m, 1H, H-p-Bn); 7.28 (m, 2H, H-m-Bn); 7.29 (dd, 1H, J3,4 = 3.6, J3,5 = 0.8, H-3-furyl); 7.52 (m, 1H, H-p-Ph); 7.58 (m, 2H, H-m-Ph); 7.64 (dd, 1H, J5,4 = 1.8, J5,3 = 0.8, H-5-furyl); 8.88 (m, 2H, H-o-Ph); 9.02 (s, 1H, H-2). 13C NMR (125.7 MHz, CDCl3): 46.96 (CH2Ph); 112.34 (CH-4-furyl); 114.88 (CH-3-furyl);126.85 (CH-o-Bn); 127.84 (CH-p-Bn); 128.62 (CH-m-Ph); 128.76 (CH-m-Bn); 129.79 (CH-o-Ph); 130.82 (CH-p-Ph); 131.05 (C-5); 135.75 (C-i-Ph); 136.16 (C-i-Bn); 144.70 (C-2-furyl); 144.93 (CH-5-furyl); 145.47 (C-8); 152.27 (CH-2); 153.64 (C-6); 154.18 (C-4). IR(KBr): 3068, 1605, 1603, 1562, 1497, 1454, 1334, 1321, 1016. HRMS (ESI) calculated for C22H17ON4: 353.1397; found: 353.1397.
This work was supported by the institutional support of the Charles University and Academy of Sciences of the Czech Republic (RVO: 61388963), by the Czech Science Foundation (P207/12/0205) and by Gilead Sciences, Inc.
Notes and references
- Review: M. Legraverend and D. S. Grierson, Bioorg. Med. Chem., 2006, 14, 3987–4006 CrossRef CAS.
- S. Ding, T. Y. H. Wu, A. Brinker, E. C. Peters, W. Hur, N. S. Gray and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7632–7637 CrossRef CAS.
- K. Anzai, G. Nakamura and S. Suzuki, J. Antibiot., 1957, 10, 201–204 CAS.
- R. Wu, E. D. Smidansky, H. S. Oh, R. Takhampunya, R. Padmanabhan, C. E. Cameron and B. R. Peterson, J. Med. Chem., 2010, 53, 7958–7966 CrossRef CAS.
- (a) P. Nauš, R. Pohl, I. Votruba, P. Džubák, M. Hajdúch, R. Ameral, G. Birkus, T. Wang, A. S. Ray, R. Mackman, T. Cihlar and M. Hocek, J. Med. Chem., 2010, 53, 460–470 CrossRef; (b) P. Spáčilová, P. Nauš, R. Pohl, I. Votruba, J. Snášel, H. Zábranská, I. Pichová, R. Ameral, G. Birkuš, T. Cihlář and M. Hocek, ChemMedChem, 2010, 5, 1386–1396 CrossRef; (c) A. Bourderioux, P. Nauš, P. Perlíková, R. Pohl, I. Pichová, I. Votruba, P. Džubák, P. Konečný, M. Hajdúch, K. M. Stray, T. Wang, A. S. Ray, J. Y. Feng, G. Birkus, T. Cihlar and M. Hocek, J. Med. Chem., 2011, 54, 5498–5507 CrossRef CAS; (d) P. Nauš, P. Perlíková, A. Bourderioux, R. Pohl, L. Slavětínská, I. Votruba, G. Bahador, G. Birkuš, T. Cihlář and M. Hocek, Bioorg. Med. Chem., 2012, 20, 5202–5214 CrossRef.
- (a) I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, Org. Lett., 2006, 8, 5389–5392 CrossRef; (b) I. Čerňa, R. Pohl and M. Hocek, Chem. Commun., 2007, 4729–4730 RSC; (c) I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2008, 73, 9048–9054 CrossRef; (d) I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2010, 75, 2302–2308 CrossRef.
- M. Klečka, R. Pohl, B. Klepetářová and M. Hocek, Org. Biomol. Chem., 2009, 7, 866–868 Search PubMed.
- C–H sulfenylation: (a) Z. Li, J. Hong and X. Z. Li, Tetrahedron, 2011, 67, 3690–3697 CrossRef CAS; (b) G. L. Regina, V. Gatti, V. Famiglini, F. Piscitelli and R. Silvestri, ACS Comb. Sci., 2012, 14, 258–262 CrossRef; (c) L. H. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307–11309 RSC; (d) S. Ranjit, R. Lee, D. Heryadi, C. Shen, J. Wu, P. Zhang, K.-W. Huang and X. Liu, J. Org. Chem., 2011, 76, 8999–9007 CrossRef CAS; (e) Ch. Dai, Z. Xu, F. Huang, Z. Yu and Y.-F. Gao, J. Org. Chem., 2012, 77, 4414–4419 CrossRef CAS; (f) X.-L. Fang, R.-Y. Tang, P. Zhong and J.-H. Li, Synthesis, 2009, 4183–4189 CAS.
- Review: (a) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276–2286 CrossRef ; original paper: ; (b) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS; (c) L. S. Liebeskind and J. Srogl, J. Org. Lett., 2002, 4, 979–981 CrossRef CAS; (d) M. Egi and L. S. Liebeskind, Org. Lett., 2003, 5, 801–802 CrossRef CAS; (e) Z. Zhang and L. S. Liebeskind, Org. Lett., 2006, 8, 4331–4333 CrossRef CAS; (f) A. Aguilar-Aguilar, L. S. Liebeskind and E. Peña-Cabrera, J. Org. Chem., 2007, 72, 8539–8542 CrossRef CAS; (g) E. Lager, J. Liu, A. Aguilar-Aguilar, B. Z. Tang and E. Peña-Cabrera, J. Org. Chem., 2009, 74, 2053–2058 CrossRef CAS; (h) I. J. Arroyo, R. Hu, B. Z. Tang, F. I. López and E. Peña-Cabrera, Tetrahedron, 2011, 67, 7244–7250 CrossRef CAS; (i) F. Pan, H. Wang, P.-X. Shen, J. Zhaob and Z.-J. Shi, Chem. Sci., 2013, 4, 1573–1577 RSC.
- S. Ding, N. S. Gray, Q. Ding and P. G. Schultz, J. Org. Chem., 2001, 66, 8273–8276 CrossRef CAS.
- (a) A. Gangjee, F. Mavandadi, R. L. Kisliuk, J. J. McGuire and S. F. Queener, J. Med. Chem., 1996, 39, 4563–4568 CrossRef CAS; (b) B. G. Ugarkar, J. M. DaRe, J. J. Kopcho, C. E. Browne III, J. M. Schanzer, J. B. Wiesner and M. D. Erion, J. Med. Chem., 2000, 43, 2883–2893 CrossRef CAS; (c) A. Gangjee, X. Lin and S. F. Queener, J. Med. Chem., 2004, 47, 3689–3692 CrossRef CAS; (d) A. Gangjee, X. Lin, R. L. Kisliuk and J. J. McGuire, J. Med. Chem., 2005, 48, 7215–7222 CrossRef CAS; (e) A. Gangjee, H. D. Jain, S. F. Queener and R. L. Kisliuk, J. Med. Chem., 2008, 51, 4589–4600 CrossRef CAS; (f) R. N. Solomyannyi, S. R. Slivchuka, A. N. Vasilenko, E. B. Rusanov and V. S. Brovarets, Russ. J. Gen. Chem., 2012, 82, 317–322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data, copies of NMR spectra. CCDC 926543 and 926544. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob40881g |
| This journal is © The Royal Society of Chemistry 2013 |