 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxynucleoside triphosphates bearing histamine, carboxylic acid, and hydroxyl residues – synthesis and biochemical characterization†
Marcel Hollenstein*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: hollenstein@dcb.unibe.ch; Fax: (+41) 31 631 3422; Tel: (+41) 31 631 4372
First published on 6th June 2013
Abstract
Modified nucleoside triphosphates (dAHsTP, dUPOHTP, and dCValTP) bearing imidazole, hydroxyl, and carboxylic acid residues connected to the purine and pyrimidine bases through alkyne linkers were prepared. These modified dN*TPs were excellent substrates for various DNA polymerases in primer extension reactions. Moreover, the combined use of terminal deoxynucleotidyl transferase (TdT) and the modified dNTPs led to efficient tailing reactions that rival those of natural counterparts. Finally, the triphosphates were tolerated by polymerases under PCR conditions, and the ensuing modified oligonucleotides served as templates for the regeneration of unmodified DNA. Thus, these modified dN*TPs are fully compatible with in vitro selection methods and can be used to develop artificial peptidases based on DNA.
Introduction
The selective scission of the amide bonds of proteins and peptides is of crucial importance for numerous biochemical and biotechnological applications1 and plays a decisive role in physiological processes.2,3 Considering the inertness of the peptide bond (t1/2 nonenz ∼ 500 years),4,5 proficient proteolytic enzymes with high catalytic efficiencies (kcat/KM) need to be used in order to degrade proteins or peptides into smaller fragments or for promoting site-specific cleavage. However, in terms of practical applications, proteolytic enzymes are not always applicable since they often lead to the generation of rather short fragmentation products, lack sequence-specificity, and cleave at multiple sites. Thus, artificial proteases based on synthetic reagents that could favor the selective cleavage of proteins under mild conditions are highly sought after. In this context, transition metal complexes were suggested as prime candidates to serve as biomimetic chemical proteases since they are often more effective than organic residues at promoting the scission of inert bonds.1,6,7 Moreover, metal centers are often combined with reagents such as β-cyclodextrins and porphyrins that facilitate the recognition and the binding to the target site and thus reach appreciable catalytic efficiencies for the scission of amide linkages in peptides and even proteins.8–11 However, even though some of these artificial, transition metal-based proteases achieve cleavage with multiple turnover12 and with some site selectivity,8,13 they suffer from severe drawbacks such as side-reactions, low kcat values, and in general perform rather poorly when compared to protein enzymes.DNA enzymes or DNAzymes have emerged as a new and prominent class of biomolecular catalysts, and have been selected to accelerate an increasing number of chemical processes.14–18 Indeed, application of SELEX and related combinatorial methods of in vitro selection19,20 resulted in the identification of nucleic acid enzymes catalyzing an abundance of reactions ranging from the formation of C–C,21–24 C–N,25 C–S,26 and P–O,27,28 bonds to the scission of ribophosphodiester linkages.29,30 Surprisingly, the scission of amide linkages is a reaction that has eluded catalytic nucleic acids so far.31 Indeed, in an early effort towards this goal, Joyce et al. initially reported on the selection of a ribozyme capable of cleaving a substrate containing an embedded 3′-NH–C(O)–CH2-5′ linkage32 but later realized that the reaction proceeded through a different mechanism leading to scission of a phosphodiester bond adjacent to the target site rather than the intended amide bond.33 Similarly, Silverman et al. set out to select a DNAzyme capable of the selective hydrolysis of amide bonds of a tripeptide sequence,34 which culminated in the serendipitous discovery of a class of very potent DNA-cleaving DNAzymes.35,36
Recently, blending of the chemical functionalities found in the active sites of enzymes together with tools of in vitro selection resulted in the identification of potent DNAzymes.37,38 In particular, this approach allowed for the development of highly functionalized DNA-based RNase A mimics that operate either in the presence of cofactors39–41 or in an M2+-independent regime.42–47 Moreover, all the functional groups encompassed in these modified DNAzymes seem to act in synergy and in a cooperative manner, one of the fundaments in the design of de novo enzyme mimics and catalysts in general.48 Finally, this methodology is applicable to other reactions than the cleavage of ribophosphodiester bonds25,49 provided that the functional groups can easily be introduced into DNA, i.e. that the modified nucleoside triphosphates (dN*TPs) are compatible with methods of in vitro selection.
In this context, the enzymatic polymerization of dN*TPs has advanced as a versatile platform for the inclusion of functional groups into nucleic acids.50 Indeed, dN*TPs have been used to introduce a myriad of functionalities including amino acids,51–57 boronic acids,58,59 ligands for transition metals,60–65 thiols,66–68 diamondoid-like residues,69 bile acids,70 side chains capable of organocatalysis,71 and even oligonucleotides.72
Herein, I report on the synthesis and biochemical characterization of modified dN*TPs bearing the side chains found in the active site of serine proteases.31,73,74 Indeed, dAHsTP (7-propargylamido-histamine-dA) 1, dUPOHTP (5-pentynol-dU) 2, and dCValTP (5-valeric acid-dC) 3 (Fig. 1) are equipped with imidazole, hydroxyl, and carboxylic acid residues, respectively, that are reminiscent of the side chains of the amino acids His, Ser, and Asp that form the catalytic triad of serine proteases. These modified dN*TPs are good substrates for a variety of DNA polymerases in the context of primer extension reactions. In addition, when used in conjunction with the terminal deoxynucleotidyl transferase (TdT), efficient tailing reactions that rival those of the natural counterparts could be observed. Finally, these modified dN*TPs were readily incorporated into DNA by the polymerase chain reaction (PCR), further underscoring their compatibility with in vitro selection techniques.
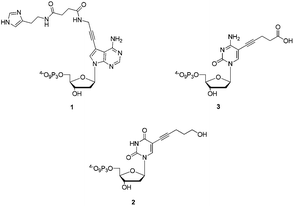 | ||
| Fig. 1 Chemical structures of dAHsTP 1, dUPOHTP 2, and dCValTP 3. | ||
Results and discussion
Synthesis of the modified nucleoside triphosphates
The nucleoside analogues considered herein all bear modifications either at the C5 position of the pyrimidine nucleobase or at the N7 of 7-deaza-2′-deoxyadenosine. Indeed, these anchoring sites were chosen for synthetic purposes (the modifications can easily be introduced via palladium-catalyzed coupling reactions) and because of their minimal disturbance and impact on substrate acceptance and duplex stability.53,71,75–77The adenosine analogue 1 adorned with a histamine residue was obtained in a 6-step sequence that started with the DMTr-protection of the known propargylamino modified 7-deaza-2′-deoxyadenosine 4 (Scheme 1).53,75,78 After removal of the TFA masking group, nucleoside 6 was converted into derivative 7 in 64% yield under standard 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-mediated amide bond-formation conditions using the suitably protected carboxylic acid S2 (ESI†). The resulting intermediate 7 was then 3′-O-acetylated under standard conditions and in good yield (84%). Following a global acid-mediated deprotection step (quantitative yield), nucleoside analogue 9 could be converted into the corresponding triphosphate dAHsTP 1 in acceptable yields (17%) by application of the one-pot four-step triphosphorylation method developed by Ludwig and Eckstein.79
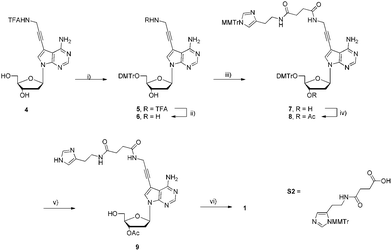 | ||
| Scheme 1 Synthesis of modified dAHsTP 1. Reagents and conditions: (i) DMTrCl, pyridine, rt, 15 h, 75%; (ii) NaOH, MeOH, H2O, rt, 12 h, 84%; (iii) S2, EDC, HOBt, NMM, DMF, rt, 12 h, 64%; (iv) Ac2O, DMAP, NEt3, pyridine, 0 °C, 2.5 h, 86%; (v) DCAA, CH2Cl2, rt, 40 min, quant.; (iv) 1. 2-chloro-1,3,2-benzodioxaphosphorin-4-one, pyridine, dioxane, rt, 45 min; 2. (nBu3NH)2H2P2O7, DMF, nBu3N, rt, 45 min; 3. I2, pyridine, H2O, rt, 30 min; 4. NH3(aq.), rt, 1.5 h, 17% (4 steps). | ||
The synthetic path leading to dUPOHTP 2 commenced with the Sonogashira cross-coupling reaction of the DMTr-protected deoxyuridine analogue 11 (which was easily obtained by tritylation of the commercially available 5-iodo-2′-deoxyuridine 10, ESI†) and free 4-pentyn-1-ol as outlined in Scheme 2. After an uneventful acetylation–detritylation sequence, precursor 14 could be converted to the corresponding triphosphate 2 by application of the Ludwig and Eckstein conditions and was obtained in 14% yield after a thorough RP-HPLC purification step.79
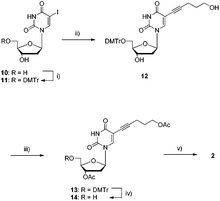 | ||
| Scheme 2 Synthesis of modified dUPOHTP 2. Reagents and conditions: (i) DMTrCl, pyridine, rt, 12 h, 93%; (ii) 4-pentyn-1-ol, Pd(PPh3)4, CuI, NEt3, DMF, rt, 12 h, 91%; (iii) Ac2O, DMAP, NEt3, pyridine, rt, 12 h, 86%; (iv) DCAA, CH2Cl2, rt, 40 min, 87%; (v) 1. 2-chloro-1,3,2-benzodioxaphosphorin-4-one, pyridine, dioxane, rt, 45 min; 2. (nBu3NH)2H2P2O7, DMF, nBu3N, rt, 45 min; 3. I2, pyridine, H2O, rt, 30 min; 4. NH3(aq.), rt, 1.5 h, 14% (4 steps). | ||
The synthesis of the modified dC*TP analogue 3 proceeded in an analogous manner as highlighted in Scheme 3. Indeed, commercially available 5-iodo-2′-deoxycytidine 15 was converted into the ester functionalized nucleoside 16 in good yield (90%) using a Sonogashira cross-coupling reaction with 4-pentynoic acid methyl ester.80 The free 5′-hydroxyl residue of 16 was then protected with a 4,4′-dimethoxytrityl group in moderate yields. The ensuing protected nucleoside 17 was then acetylated yielding a mixture of the desired 3′-O-monoacetylated 18 (61%) and diacetylated product S4 (ESI†). Unfortunately, all attempts at N-4-deacetylating S4 using ZnBr2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 67 only led to detritylation (data not shown). Finally, following removal of the DMTr masking group under acidic conditions, intermediate 19 could be converted to the corresponding triphosphate 3 in 9% yield (4 steps), again by application of the Ludwig–Eckstein protocol.79
67 only led to detritylation (data not shown). Finally, following removal of the DMTr masking group under acidic conditions, intermediate 19 could be converted to the corresponding triphosphate 3 in 9% yield (4 steps), again by application of the Ludwig–Eckstein protocol.79
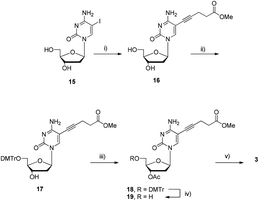 | ||
| Scheme 3 Synthesis of triphosphate dCValTP 3. Reagents and conditions: (i) 4-pentynoic acid methyl ester, Pd(PPh3)4, CuI, NEt3, DMF, rt, 12 h, 90%; (ii) DMTrCl, DMAP, NEt3, pyridine, rt, 18 h, 45%; (iii) Ac2O, DMAP, NEt3, pyridine, 0 °C, 1 h, 61%; (iv) DCAA, CH2Cl2, rt, 40 min, 92%; (v) 1. 2-chloro-1,3,2-benzodioxaphosphorin-4-one, pyridine, dioxane, rt, 45 min; 2. (nBu3NH)2H2P2O7, DMF, nBu3N, rt, 45 min; 3. I2, pyridine, H2O, rt, 30 min; 4. NaOH, H2O, 0 °C, 5 min, 9% (4 steps). | ||
Primer extension reactions
An important prerequisite for modified dN*TPs to be applicable in selection experiments is that they serve as substrates for DNA polymerases in primer extension reactions.71 Furthermore, not only do primer extension reactions constitute the first step of an in vitro selection experiment, but they represent the main tool for gauging the enzymatic recognition of modified dN*TPs in general. Consequently, I explored the substrate acceptance of the modified dN*TPs 1–3 with different DNA polymerases under primer extension reaction conditions (Fig. 2 and ESI†). Therefore, a variety of DNA polymerases (Vent (exo−), Pwo, 9°Nm, and the Klenow fragment of E. coli DNA polymerase I) were used in conjunction with the 98-mer long template T1 and the 25-mer primer P1 (see ESI† for the sequence compositions).70,71 The modified dN*TPs were then investigated as to whether they could act as surrogates for their natural counterparts, either as lone modifications (lanes 1–3, Fig. 2), as combinations of two modified and two natural dNTPs (lanes 4–6, Fig. 2), or in conjunction with the lone natural dGTP (lane 7, Fig. 2). Vent (exo−) was revealed to be highly proficient at extending primer P1 to full length since no faster running bands were apparent in all the combinations that were investigated. Moreover, 16 dAHs1 and 21 dUPOH2 that were incorporated in the nascent chain led to a significant and expected gel retardation as compared to the natural control sequence (compare lanes 1 and 2 with lane 8, Fig. 2). Surprisingly, the inclusion of 18 dCVal3 had little impact on the gel mobility of the resulting modified DNA (lane 3 vs. lane 8, Fig. 2).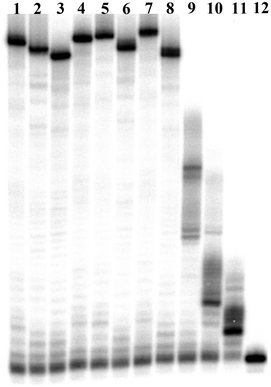 | ||
| Fig. 2 Gel image (PAGE 15%) of primer extension reactions with primer P1 and template T1 using Vent (exo−) DNA polymerase. Lane 1: dAHsTP 1; lane 2: dUPOHTP 2; lane 3: dCValTP 3; lane 4: dAHsTP 1 and dCValTP 3; lane 5: dAHsTP 1 and dUPOHTP 2; lane 6: dUPOHTP 2 and dCValTP 3; lane 7: dAHsTP 1, dUPOHTP 2, and dCValTP 3; lane 8: natural dNTPs; lane 9: natural dNTPs without dATP; lane 10: natural dNTPs without dTTP; lane 11: natural dNTPs without dCTP; lane 12: primer P1. | ||
Furthermore, besides Vent (exo−), all the other DNA polymerases that were tested in primer extension reactions showed very high substrate tolerance for all the dN*TPs and led to fully extended products (Fig. S1–S3, ESI†). Interestingly, the Klenow fragment, a prominent member of the family A polymerases, was as efficient at incorporating the modified dN*TPs as the more tolerant family B polymerases (Fig. S2, ESI†).53,81,82
TdT-mediated polymerization
The terminal deoxynucleotidyl transferase (TdT) is a Co2+-dependent family X DNA polymerase that catalyzes the random and template-independent polymerization of nucleoside triphosphates at the 3′-OH termini of single-stranded oligonucleotides.83,84 Recently, TdT-mediated reactions using modified dN*TPs have been employed for the generation of 3′-fluorescently labelled oligonucleotides,85,86 to confer exonuclease stability to specific sequences,87 and to synthesize ssDNAs containing stretches of nucleotides bearing redox active tags.88 However, the tailing reaction was never used for the incorporation of nucleotides bearing side chains that potentially could confer catalytic activity to the ensuing modified ssDNA. Consequently, the modified dN*TPs 1–3 were assayed for their capacity to act as substrates in TdT-mediated tailing reactions. Thus, the 5′-32P-labelled 15 nt long primer P3 (ESI†) was incubated at 37 °C for 60 min in the presence of TdT and varying concentrations of the modified dN*TPs (Fig. 3). | ||
| Fig. 3 Gel image (PAGE 15%) of the TdT-catalyzed extension reactions. Lane 1: dAHsTP 1; lane 2: dUPOHTP 2; lane 3: dCValTP 3; lane 4: dATP; lane 5: dTTP; lane 6: dCTP; lane 7: primer P3. | ||
The 7-substituted 7-deaza-deoxyadenosine derivative dAHsTP 1 was revealed to be the best substrate for TdT, since the tailing efficiencies are better than those of its natural counterpart (compare lanes 1 and 4, Fig. 3). Indeed, at a concentration as low as 10 μM, mainly 7–15 additional dAHs nucleotides were incorporated along with some longer tailed sequences, while the natural dATP led to the appendage of a rather narrow 4–17 nucleotide distribution with little polydisperse longer-sized oligonucleotides. Furthermore, at a concentration of 50 μM several tens of both the dAHs and dA nucleotides were appended. Moreover, dUPOHTP 2 (lanes 2 in Fig. 3) led to the best tailing efficiencies amongst the pyrimidine triphosphates, a tendency that is opposite to that observed with natural dNTPs (vide infra). Indeed, the TdT-mediated homopolymerization was rather ineffective at 10 μM since only 2–5 additional dUPOH units were appended, but at higher concentrations (>50 μM) only highly modified slow-running products could be observed. Unlike its natural counterpart, dCValTP 3 stifles the TdT and only a few additional residues (∼3 to 15 dCValMPs) are added, even at 200 μM (lanes 3 in Fig. 3).
Polymerase chain reactions (PCR)
An additional and important part of the biochemical characterization of modified dN*TPs is the assessment of their substrate acceptance by DNA polymerases under PCR conditions. Consequently, the dN*TPs 1–3 were evaluated for their capacity to amplify the 98-mer template T170,71 flanked by a 20 nt long forward and a 25 nt reverse primer using four different DNA polymerases (Fig. 4 and ESI†).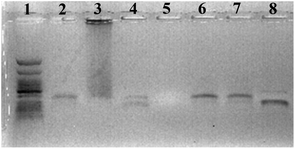 | ||
| Fig. 4 Agarose gel (2%) stained with ethidium bromide, showing the PCR products with the 98-mer template T1, a dNTP mixture composed of dAHsTP 1, dUPOHTP 2, dCValTP 3, and natural dGTP and different enzymes and conditions. Lane 1: ladder; lane 2: Vent (exo−); lane 3: 9°Nm polymerase; lane 4: Pwo polymerase; lane 5: Klenow fragment of E. coli DNA polymerase I; lane 6: Vent (exo−) with 500 μM dN*TPs; lane 7: Vent (exo−) with 500 μM dN*TPs and 7 mM Mg2+; lane 8: natural dNTPs and Vent (exo−). | ||
Surprisingly, unlike what had been observed for primer extension reactions, under standard PCR conditions (2 mM Mg2+, 200 μM dNTPs, 30 PCR cycles), only the Vent (exo−) DNA polymerase was capable of faithfully amplifying the template, albeit with a slightly reduced efficiency as compared to the control experiment with natural dNTPs (lane 2 versus lane 8, Fig. 4). Indeed, the 9°Nm polymerase led to a marked smearing pattern (lane 3), the Pwo polymerase only partially accepted the dN*TPs under these conditions and yielded a mixture of a full length product and a shorter product (lane 4), and no amplicon could be observed in the case of the Klenow fragment (lane 5). However, when the dNTP concentration was raised from 200 to 500 μM, an efficient amplification could be observed with Vent (exo−) (lane 6). Furthermore, additional Mg2+ (lane 7) had no positive impact on the amplification efficiency. Good amplifications were observed as well when only one of the natural dNTPs was substituted with a modified one (data not shown).
In terms of in vitro selections, it is of primordial importance that single-stranded modified DNAs can be transformed into wild-type oligonucleotides that can serve as templates for subsequent rounds of selection.30,44 Thus, in a different PCR experiment a 5′-phosphorylated 79-mer long template T2 (ESI†) was amplified using the 19- and 20-mer primers P4 and P5 in the presence of Vent (exo−) and 500 μM dNTPs. The resulting modified dsDNA was then λ-exonuclease digested, and used as a template for a subsequent PCR using natural dNTPs, the same set of primers, and Vent (exo−) (Fig. S5, ESI†).71 This experiment clearly underscores the compatibility of the modified dN*TPs 1–3 with methods of in vitro selection since a modified oligonucleotide could serve as a template to regenerate natural DNA (Fig. S5, ESI†).
Finally, in order to assess the fidelity of the PCR amplification with the modified dN*TPs 1–3, a sequencing experiment based on the dideoxynucleoside triphosphate (ddNTP) method developed by Sanger et al. was carried out (Fig. S6 and S7, respectively, ESI†).52,89 Briefly, the 5′-phosphorylated template T2 and primer P4 were used in a primer extension reaction with Vent (exo−). The unmodified template was then λ-exonuclease digested, and the resulting modified ssDNA was used as a template for PCR with each one of the 5′-biotinylated primers P4B and P5B. Following immobilization of the duplexes on streptavidin coated magnetic beads, the unbiotinylated oligonucleotides were eluted with hydroxide washes and then sequenced. Comparison of the sequencing patterns of both the forward and reverse directions (Fig. S6 and S7, ESI†) clearly reveals that no loss of sequence information occurs upon converting a heavily modified ssDNA into its complementary natural sequence, and that the fidelity of PCR with the modified dN*TPs is high.
Conclusions
Three modified dN*TPs adorned with side-chains reminiscent of the Ser, His, and Asp residues found in the active site of serine proteases could easily be accessed in 5 or 6 steps starting from known and/or commercially available precursors. The functionalities were anchored on the C5 of pyrimidines and the N-7 of 7-deaza-adenosine in order to minimize the destabilization of duplexes and disruption of Watson–Crick base pairing, and concomitantly, to maximize the substrate acceptance. All of the modified dN*TPs were found to be excellent substrates for all four DNA polymerases, including the Klenow fragment of DNA polymerase I, in primer extension reactions. It is noteworthy that the polymerases readily accept and tolerate the presence of the negative charge of the carboxylic acid of dCValTP 3 and allow for the incorporation of up to 3 consecutive dCVal residues.Moreover, the substrate acceptance for TdT is heavily dependent on the nature and position of the substituent and followed the order dAHsTP > dUPOHTP > dCValTP. This trend is opposite to what is observed for natural dNTPs, since TdT utilizes dGTP and dCTP much more efficiently than dTTP and more markedly, dATP.90 The additional negative charge on dCValTP might account for its limited substrate acceptance by TdT.
Finally, these modified dN*TPs were shown to be good substrates for the Vent (exo−) DNA polymerase under PCR conditions. However, unlike what was observed for primer extension reactions, only optimized conditions and the sole Vent (exo−) DNA polymerase led to clean and robust amplifications. The resulting modified oligonucleotides could serve as a template to regenerate natural DNA, which is an important prerequisite for the use of these dN*TPs in selection experiments. Moreover, PCR amplification using the modified dN*TPs and the subsequent conversion into natural DNA was shown to proceed with high fidelity.
Thus, these three nucleoside triphosphate analogues, equipped with functionalities that are reminiscent of the residues found in the active site of serine proteases, could be shown to be fully compatible with methods of in vitro selection. Consequently, their simultaneous polymerization will considerably increase the chemical landscape that can be covered in the course of a selection experiment for the generation of DNAzymes that can promote the catalytic scission of amide bond linkages, which is a long-standing goal in the field of nucleic acid enzymes. Such an artificial peptidase would be an invaluable synthetic and biochemical tool, and selection for such an enzyme mimic is currently under way.
Experimental part
General method for the Sonogashira coupling reactions
The nucleoside was dissolved in dry DMF (5 mL mmol−1). CuI (0.1 eq.) and NEt3 (2 eq.) were then added and the reaction mixture was stirred for 10 min at room temperature. The alkyne (4 eq.) and Pd(PPh3)4 (0.05 eq.) were then added in turn. The reaction mixture was cooled down to −78 °C and thoroughly degassed. After stirring at room temperature overnight, the DMF was removed under reduced pressure. The blackened crude product was then dissolved in CH2Cl2 (50 mL) and washed with brine (50 mL). Following drying (MgSO4) and removal of the solvent in vacuo, the residue was purified by flash column chromatography.General method for detritylation
The nucleoside analogue was dissolved in dry CH2Cl2 (10 mL) and dichloroacetic acid (0.1%, 0.1 mL) was added. The orange-coloured reaction mixture was stirred at room temperature for 40 min. MeOH (3 mL) was added and the solution was stirred for another 5 min at room temperature. Following removal of the solvent under reduced pressure, the residue was purified by flash column chromatography.:5), yielding 5 as a pale yellow foam (0.46 g, 75%). Rf: 0.46 (CH2Cl2–MeOH 9:1 +1% NEt3). 1H-NMR (400 MHz, CDCl3): δ 2.41–2.52 (m, 2H), 3.37 (d, 2H, J = 4.4 Hz), 3.76 (s, 6H), 4.05 (q, 1H, J = 4.0 Hz), 4.35–4.39 (m, 2H), 4.58 (q, 1H, J = 4.9 Hz), 5.64 (br s, 2H), 6.62 (t, 1H, J = 6.2 Hz), 6.81 (dd, 4H, J = 2.0, 8.8 Hz), 7.20–7.32 (m, 7H), 7.39–7.46 (m, 3H), 8.25 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 30.9, 41.2, 53.6, 63.7, 72.1, 78.3, 83.6, 85.3, 85.7, 86.9, 94.9, 103.6, 113.6, 126.7, 127.2, 128.2, 130.3, 136.2, 136.3, 144.4, 149.8, 153.3, 157.5, 158.7. 19F-NMR (376.5 MHz, CDCl3): δ −75.67. HR MS: m/z: calcd for C37H35O6N5F3 ([M + H]+): 702.2534, found: 702.2524.:1) yielded 6 as a white foam (0.69 g, 84%). Rf: 0.33 (CH2Cl2–MeOH 9:1). 1H-NMR (400 MHz, CDCl3): δ 2.29–2.51 (m, 2H), 3.30–3.40 (m, 2H), 3.64 (s, 2H), 3.77 (s, 6H), 4.03 (q, 1H, J = 3.1 Hz), 4.56 (q, 1H, J = 4.9 Hz), 5.57 (br s, 2H), 6.62 (t, 1H, J = 6.4 Hz), 6.81 (d, 4H, J = 8.7 Hz), 7.17–7.32 (m, 8H), 7.38–7.43 (m, 2H), 8.22 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 32.6, 40.9, 55.5, 64.0, 72.6, 76.0, 83.5, 85.5, 86.8, 92.7, 96.2, 103.7, 113.5, 125.6, 127.1, 128.1, 128.4, 130.2, 130.3, 136.0, 144.8, 149.9, 153.2, 157.5, 158.8. HR MS: m/z: calcd for C35H36O5N5 ([M + H]+): 606.2711, found: 606.2704.:5 to 9:1. The histamine modified derivative 7 was obtained as a white foam (0.40 g, 64%). Rf: 0.55 (CH2Cl2–MeOH 9:1). 1H-NMR (400 MHz, CDCl3): δ 2.39–2.49 (m, 7H), 2.64 (t, 2H, J = 6.4 Hz), 3.30–3.36 (m, 2H), 3.45 (q, 2H, J = 6.2 Hz), 3.75 (s, 6H), 3.77 (s, 3H), 4.03 (q, 1H, J = 3.7 Hz), 4.12 (d, 1H, J = 5.2 Hz), 5.55 (br s, 2H), 6.55 (d, 1H, J = 0.8 Hz), 6.59 (t, 1H, J = 6.4 Hz), 6.77–6.83 (m, 7H), 7.00–7.02 (m, 3H), 7.07–7.13 (m, 2H), 7.18–7.32 (m, 12H), 7.35–7.40 (m, 3H), 8.18 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 27.6, 30.5, 31.9, 39.8, 40.9, 55.5, 64.0, 72.3, 83.4, 85.5, 86.8, 88.1, 95.9, 103.8, 113.5, 118.8, 125.2, 128.1, 128.2, 128.4, 129.8, 130.3, 131.3, 134.6, 136.1, 138.8, 139.2, 142.9, 144.8, 149.9, 153.3, 157.7, 158.7, 159.3, 172.1, 172.9. HR MS: m/z: calcd for C64H63O8N8 ([M + H]+): 1071.4763, found: 1071.4750.:4). The fully protected analogue 8 was obtained as a white foam (0.35 g, 86%). Rf: 0.63 (CH2Cl2–MeOH 95:5 +1% NEt3). 1H-NMR (400 MHz, CDCl3): δ 2.06 (s, 3H), 2.45–2.53 (m, 5H), 2.56–2.62 (m, 1H), 2.65 (t, 2H, J = 6.0 Hz), 3.35 (t, 2H, J = 3.2 Hz), 3.46 (q, 2H, J = 6.0 Hz), 3.76 (s, 6H), 3.78 (s, 3H), 4.12 (d, 2H, J = 5.6 Hz), 4.16 (q, 1H, J = 3.2 Hz), 5.37–5.40 (m, 1H), 5.87 (br s, 2H), 6.55 (d, 1H, J = 1.2 Hz), 6.62 (q, 1H, J = 4.7 Hz), 6.78–6.83 (m, 6H), 7.01 (d, 2H, J = 8.8 Hz), 7.07–7.11 (m, 4H), 7.17–7.22 (m, 2H), 7.23–7.26 (m, 3H), 7.27–7.31 (m, 10H), 7.37–7.40 (m, 2H), 8.18 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 21.3, 27.6, 30.5, 31.9, 38.4, 39.8, 55.5, 64.0, 75.3, 83.4, 83.7, 96.3, 103.8, 113.5, 118.8, 124.9, 127.14, 128.1, 128.2, 128.3, 128.5, 129.8, 130.3, 130.4, 131.3, 134.6, 135.9, 136.0, 138.9, 139.3, 142.9, 144.7, 150.2, 153.5, 157.7, 158.8, 159.3, 170.6, 172.1, 172.9. HR MS: m/z: calcd for C66H65O9N8 ([M + H]+): 1113.4869, found: 1113.4872.:1 to give 9 as a white foam (0.17 g, quant.). Rf: 0.16 (CH2Cl2–MeOH 9:1). 1H-NMR (400 MHz, DMSO-d6): δ 2.09 (s, 3H), 2.30–2.38 (m, 5H), 2.45–2.50 (m, 2H), 2.60 (t, 2H, J = 7.4 Hz), 2.65–2.72 (m, 1H), 3.23 (q, 2H, J = 6.8 Hz), 3.60 (d, 2H, J = 1.6 Hz), 4.02 (q, 1H, J = 1.9 Hz), 4.09 (d, 2H, J = 5.2 Hz), 5.31 (d, 1H, J = 6.0 Hz), 6.46 (dd, 1H, J = 5.6, 9.2 Hz), 6.75 (br s, 1H), 7.50 (s, 1H), 7.69 (s, 1H), 7.88 (t, 1H, J = 3.6 Hz), 8.10 (s, 1H), 8.44 (t, 1H, J = 5.2 Hz). 13C-NMR (101 MHz, DMSO-d6): δ 20.9, 29.2, 30.5, 30.6, 36.9, 54.9, 61.7, 74.8, 75.1, 83.3, 84.8, 89.3, 95.0, 102.3, 125.7, 134.5, 149.3, 152.7, 157.5, 170.0, 171.0, 171.6. HR MS: m/z: calcd for C25H31O6N8 ([M + H]+): 539.2361, found: 539.2375.:0 +1% NEt3 to 95:5) to give 12 as a white foam (0.51 g, 91%). Rf: 0.32 (CH2Cl2–MeOH 95:5 +1% NEt3). 1H-NMR (400 MHz, CDCl3): δ 1.43–1.49 (m, 2H), 2.19 (t, 2H, J = 6.8 Hz), 2.23–2.28 (m, 1H), 2.48 (ddd, 1H, J = 2.8, 5.6, 13.6 Hz), 3.28 (dd, 1H, J = 3.4, 10.6 Hz), 3.40 (dd, 1H, J = 3.0, 10.6 Hz), 3.47 (t, 2H, J = 6.0 Hz), 3.77 (s, 6H), 4.06 (q, 1H, J = 2.9 Hz), 4.49–4.53 (m, 1H), 6.31 (dd, 1H, J = 5.8, 7.4 Hz), 6.82 (d, 4H, J = 9.2 Hz), 7.24–7.33 (m, 7H), 7.41 (d, 2H, J = 7.2 Hz), 8.00 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 16.3, 30.9, 41.7, 55.5, 61.5, 63.7, 71.5, 72.5, 85.8, 86.7, 87.2, 95.0, 101.1, 113.6, 127.2, 128.2, 128.3, 130.1, 130.2, 135.8, 135.9, 141.7, 144.7, 149.4, 158.8, 162.2. HR MS: m/z: calcd for C35H37O8N2 ([M + H]+): 613.2544, found: 613.2537.:1). The fully protected compound 13 was obtained as a white foam (0.30 g, 86%). Rf: 0.34 (CH2Cl2–MeOH 95:5 +1% NEt3). 1H-NMR (400 MHz, CDCl3): δ 1.34–1.41 (m, 2H), 1.91 (s. 3H), 1.99 (s, 3H), 2.06 (t, 2H, J = 7.0 Hz), 2.25–2.33 (m, 1H), 2.43–2.48 (m, 1H), 3.28–3.37 (m, 2H), 3.70 (s, 6H), 3.81 (t, 2H, J = 6.4 Hz), 4.05 (d, 1H, J = 1.6 Hz), 5.30 (d, 1H, J = 6.0 Hz), 6.25 (dd, 1H, J = 5.6, 8.8 Hz), 6.75 (d, 4H, J = 8.8 Hz), 7.11–7.27 (m, 7H), 7.32–7.36 (m, 2H), 7.98 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 16.5, 21.1, 21.2, 27.6, 29.9, 38.8, 55.4, 63.3, 63.8, 71.3, 75.2, 84.7, 85.3, 87.5, 94.1, 101.4, 113.6, 127.2, 128.1, 128.3, 130.2, 135.6, 135.7, 136.2, 141.7, 144.6, 149.4, 149.7, 158.9, 161.7, 170.5, 171.1. HR MS: m/z: calcd for C39H41O10N2 ([M + H]+): 697.2756, found: 697.2763.:1 to give 9 as a pale yellow solid (0.21 g, 87%). Rf: 0.16 (CH2Cl2–MeOH 9:1). 1H-NMR (400 MHz, DMSO-d6): δ 1.76–1.83 (m, 2H), 2.01 (s, 3H), 2.06 (s, 3H), 2.25–2.32 (m, 2H), 2.45 (t, 2H, J = 7.2 Hz), 3.63 (d, 2H, J = 2.8 Hz), 4.01 (q, 1H, J = 2.8 Hz), 4.09 (t, 2H, J = 6.4 Hz), 5.20–5.22 (m, 1H), 6.14 (apparent t, 1H, J = 7.2 Hz), 8.13 (s, 1H), 11.72 (s, 1H). 13C-NMR (101 MHz, DMSO-d6): δ 15.6, 20.7, 20.8, 27.3, 37.1, 61.1, 62.7, 63.4, 73.1, 74.6, 84.4, 84.9, 92.3, 99.2, 142.6, 149.5, 161.6, 165.5, 170.0, 170.4. HR MS: m/z: calcd for C18H23O8N2 ([M + H]+): 395.1449, found: 395.1441.:4 to 9:1). Rf: 0.20 (CH2Cl2–MeOH 9:1). 1H-NMR (400 MHz, DMSO-d6): δ 1.93–2.00 (m, 1H), 2.10–2.16 (m, 1H), 2.60–2.67 (m, 4H), 3.52–3.61 (m, 2H), 3.62 (s, 3H), 3.78 (q, 1H. J = 3.3 Hz), 4.17–4.22 (m, 1H), 5.04 (t, 1H, J = 5.2 Hz), 5.18 (d, 1H, J = 4.0 Hz), 6.10 (t, 1H, J = 6.6 Hz), 6.68 (br s, 1H), 7.71 (br s, 1H), 8.06 (s, 1H). 13C-NMR (101 MHz, DMSO-d6): δ 15.0, 32.4, 45.7, 51.9, 61.0, 70.2, 72.5, 85.3, 87.4, 90.0, 94.3, 143.5, 153.5, 164.4, 172.2. HR MS: m/z: calcd for C15H20O6N3 ([M + H]+): 338.1347, found: 338.1344.:4 to give 17 as a pale yellow solid (0.23 g, 45%). Rf: 0.45 (CH2Cl2–MeOH 9:1 +1% NEt3). 1H-NMR (400 MHz, CDCl3): δ 2.08–2.15 (m, 1H), 2.25–2.31 (m, 2H), 2.33–2.39 (m, 2H), 2.63–2.68 (m, 1H), 3.20–3.28 (m, 2H), 3.58 (s, 3H), 3.69 (s, 6H), 4.06 (q, 1H, J = 3.3 Hz), 4.40–4.43 (m, 1H), 5.89 (br s, 1H), 6.24 (t, 1H, J = 6.4 Hz), 6.42 (br s, 1H), 6.73 (dd, 4H, J = 1.2, 8.8 Hz), 7.09–7.20 (m, 3H), 7.23–7.27 (m, 4H), 7.35 (d, 2H, J = 7.2 Hz), 8.01 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 15.5, 33.0, 46.2, 52.1, 55.4, 63.8, 72.3, 72.5, 86.6, 87.0, 87.1, 91.4, 94.5, 113.5, 127.1, 128.1, 128.2, 130.1, 130.2, 135.9, 136.0, 143.4, 144.8, 154.9, 158.8, 165.2, 172.7. HR MS: m/z: calcd for C36H38O8N3 ([M + H]+): 640.2653, found: 640.2647.:5 +1% NEt3, Rf (starting material): 0.38; Rf (18): 0.5; Rf (S2): 0.86) revealed the complete disappearance of the starting material. MeOH (3 mL) was then added and the solution was stirred for an additional 5 min at 0 °C. The solvents were removed under reduced pressure and the crude product was purified by flash column chromatography on silica gel (gradient from CH2Cl2–MeOH 98:2 to 95:5), yielding 18 (0.23 g, 61%) and S4 (0.12 g, 30%) both as white foams. Characterization for 18: 1H-NMR (400 MHz, CDCl3): δ 2.04 (s, 3H), 2.20–2.28 (m, 1H), 2.35–2.40 (m, 2H), 2.43–2.48 (m, 2H), 2.69 (ddd, 1H, J = 1.5, 5.5, 14.1 Hz), 3.06–3.12 (m, 1H), 3.31–3.39 (m, 2H), 3.66 (s, 3H), 3.77 (s, 6H), 4.16 (q, 1H, J = 2.5 Hz), 5.34 (apparent d, 1H, J = 6.0 Hz), 6.10 (br s, 1H), 6.20 (br s, 1H), 6.31 (dd, 1H, J = 5.4, 8.2 Hz), 6.81 (dd, 4H, J = 1.8, 9.0 Hz), 7.16–7.33 (m, 8H), 7.40 (apparent d, 1H, J = 7.2 Hz), 8.07 (s, 1H). 13C-NMR (101 MHz, CDCl3): δ 15.6, 21.2, 33.0, 39.7, 52.2, 55.5, 63.8, 72.2, 75.4, 84.6, 86.8, 87.3, 91.6, 94.8, 113.5, 127.4, 128.1, 128.2, 130.1, 130.2, 135.7, 135.8, 143.2, 144.7, 154.3, 158.9, 164.9, 170.6, 172.7. HR MS: m/z: calcd for C38H40O9N3 ([M + H]+): 682.2759, found: 682.2750.:0 to 94:6), to give 9 as a white solid (0.11 g, 92%). Rf: 0.31 (CH2Cl2–MeOH 95:5 +% NEt3). 1H-NMR (400 MHz, DMSO-d6): δ 2.06 (s, 3H), 2.11–2.19 (m, 1H), 2.25–2.32 (m, 1H), 2.63–2.67 (m, 4H), 3.56–3.60 (m, 1H), 3.64 (s, 3H), 4.01 (q, 1H, J = 2.8 Hz), 5.19 (apparent d, 2H, J = 6.0 Hz), 6.15 (dd, 1H, J = 5.8, 7.8 Hz), 6.83 (br s, 1H), 7.73 (br s, 1H), 8.06 (s, 1H). 13C-NMR (101 MHz, DMSO-d6): δ 15.0, 20.8, 32.4, 37.8, 51.5, 61.2, 72.3, 74.7, 84.9, 85.2, 90.5, 94.5, 143.3, 153.5, 164.4, 170.0, 172.2. HR MS: m/z: calcd for C17H22O7N3 ([M + H]+): 380.1452, found: 380.1443.Triphosphorylation of nucleosides
HPLC of triphosphates
The purification of the various triphosphates was carried out on a Phenomenex Jupiter semi-preparative RP-HPLC column (see ESI†) at a flow rate of 3.5 mL min−1, using a triethylammonium bicarbonate (TEAB) buffer system (eluent I: 50 mM TEAB, pH 7.7; eluent II: 50 mM TEAB in CH3CN–H2O 1:1), and one of the programs highlighted in Table 1.
| Time (min) | Program A (% II) | Program B (% II) | Time (min) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 8 | 0 | 0 | 8 |
| 50 | 50 | 17 | 31 |
| 52 | 100 | 80 | 33 |
| 54 | 100 | 80 | 35 |
| 56 | 0 | 0 | 37 |
Acknowledgements
This work was supported by the Swiss National Science Foundation (grants no. PZ00P2_126430/1 and PZ00P2_144595). I would like to gratefully acknowledge Prof. C. Leumann for providing me the lab space and equipment, as well as for his constant support. Dr Alessandro Calabretta is acknowledged for his precious help and his suggestions. I thank Ms. Christine Smith for a critical reading of the manuscript. M.H. thanks Mr Luís ‘Lucho’ Lara for fruitful discussions.Notes and references
- J. Suh, Acc. Chem. Res., 2003, 36, 562–570 CrossRef CAS.
- H. Neurath and K. A. Walsh, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3825–3832 CrossRef CAS.
- B. Turk, Nat. Rev. Drug Discovery, 2006, 5, 785–799 CrossRef CAS.
- A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105–6109 CrossRef CAS.
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS.
- J. Suh and W. S. Chei, Curr. Opin. Chem. Biol., 2008, 12, 207–213 CrossRef CAS.
- D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef CAS.
- N. M. Milović, J. D. Badjić and N. M. Kostić, J. Am. Chem. Soc., 2004, 126, 696–697 CrossRef.
- N. M. Milović, L.-M. Dutcă and N. M. Kostić, Chem.–Eur. J., 2003, 9, 5097–5106 CrossRef.
- C. E. Yoo, P. S. Chae, J. E. Kim, E. J. Jeong and J. Suh, J. Am. Chem. Soc., 2003, 125, 14580–14589 CrossRef CAS.
- K. Stroobants, E. Moelants, H. G. T. Ly, P. Proost, K. Bartik and T. N. Parac-Vogt, Chem.–Eur. J., 2013, 19, 2848–2858 CrossRef CAS.
- J. Suh and S.-J. Moon, Inorg. Chem., 2001, 40, 4890–4895 CrossRef CAS.
- L.-M. Dutcă, K.-S. Ko, N. L. Pohl and N. M. Kostić, Inorg. Chem., 2005, 44, 5141–5146 CrossRef.
- D. A. Baum and S. K. Silverman, Cell. Mol. Life Sci., 2008, 65, 2156–2174 CrossRef CAS.
- K. Schlosser and Y. Li, Chem. Biol., 2009, 16, 311–322 CrossRef CAS.
- S. K. Silverman, Chem. Commun., 2008, 3467–3485 RSC.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- S. K. Silverman, Angew. Chem., Int. Ed., 2010, 49, 7180–7201 CrossRef CAS.
- G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791–836 CrossRef CAS.
- G. F. Joyce, Angew. Chem., Int. Ed, 2007, 46, 6420–6436 CrossRef CAS.
- B. Seelig and A. Jäschke, Chem. Biol., 1999, 6, 167–176 CrossRef CAS.
- S. Fusz, A. Eisenfuhr, S. G. Srivatsan, A. Heckel and M. Famulok, Chem. Biol., 2005, 12, 941–950 CrossRef CAS.
- M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2008, 130, 2936–2937 CrossRef CAS.
- U. Mohan, R. Burai and B. R. McNaughton, Org. Biomol. Chem., 2013, 11, 2241–2244 CAS.
- T. W. Wiegand, R. C. Janssen and B. E. Eaton, Chem. Biol., 1997, 4, 675–683 CrossRef CAS.
- G. Sengle, A. Eisenführ, P. S. Arora, J. S. Nowick and M. Famulok, Chem. Biol., 2001, 8, 459–473 CrossRef CAS.
- P. I. Pradeepkumar, C. Höbartner, D. A. Baum and S. K. Silverman, Angew. Chem., Int. Ed., 2008, 47, 1753–1757 CrossRef CAS.
- O. Y. Wong, P. I. Pradeepkumar and S. K. Silverman, Biochemistry, 2011, 50, 4741–4749 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS.
- S. Ameta and A. Jäschke, Chem. Sci., 2013, 4, 957–964 RSC.
- X. Dai, A. De Mesmaeker and G. F. Joyce, Science, 1995, 267, 237–241 CrossRef CAS.
- G. F. Joyce, X. Dai and A. De Mesmaeker, Science, 1996, 272, 18–19 CrossRef CAS.
- M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 CrossRef CAS.
- Y. Xiao, M. Chandra and S. K. Silverman, Biochemistry, 2010, 49, 9630–9637 CrossRef CAS.
- V. Dokukin and S. K. Silverman, Chem. Sci., 2012, 3, 1707–1714 RSC.
- M. Kuwahara and N. Sugimoto, Molecules, 2010, 15, 5423–5444 CrossRef CAS.
- M. Hollenstein, Molecules, 2012, 17, 13569–13591 CrossRef CAS.
- S. W. Santoro, G. F. Joyce, K. Sakthivel, S. Gramatikova and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2433–2439 CrossRef CAS.
- M. Hollenstein, C. Hipolito, C. Lam, D. Dietrich and D. M. Perrin, Angew. Chem., Int. Ed., 2008, 47, 4346–4350 CrossRef CAS.
- C. H. Lam, C. J. Hipolito, M. Hollenstein and D. M. Perrin, Org. Biomol. Chem., 2011, 9, 6949–6954 CAS.
- D. M. Perrin, T. Garestier and C. Hélène, J. Am. Chem. Soc., 2001, 123, 1556–1563 CrossRef CAS.
- A. V. Sidorov, J. A. Grasby and D. M. Williams, Nucleic Acids Res., 2004, 32, 1591–1601 CrossRef CAS.
- M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, Nucleic Acids Res., 2009, 37, 1638–1649 CrossRef CAS.
- M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, ChemBioChem, 2009, 10, 1988–1992 CrossRef CAS.
- C. J. Hipolito, M. Hollenstein, C. H. Lam and D. M. Perrin, Org. Biomol. Chem., 2011, 9, 2266–2273 CAS.
- M. Hollenstein, C. J. Hipolito, C. H. Lam and D. M. Perrin, ACS Comb. Sci., 2013, 15, 174–182 CrossRef CAS.
- L. Baltzer and J. Nilsson, Curr. Opin. Biotechnol., 2001, 12, 355–360 CrossRef CAS.
- T. M. Tarasow, S. L. Tarasow and B. E. Eaton, Nature, 1997, 389, 54–57 CrossRef CAS.
- M. Hocek and M. Fojta, Org. Biomol. Chem., 2008, 6, 2233–2241 CAS.
- M. Kuwahara, K. Hanawa, K. Ohsawa, R. Kitagata, H. Ozaki and H. Sawai, Bioorg. Med. Chem., 2006, 14, 2518–2526 CrossRef CAS.
- S. Jäger and M. Famulok, Angew. Chem., Int. Ed., 2004, 43, 3337–3340 CrossRef.
- S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef.
- O. Thum, S. Jäger and M. Famulok, Angew. Chem., Int. Ed., 2001, 40, 3990–3993 CrossRef CAS.
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eaton, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS.
- K. Sakthivel and C. F. Barbas, Angew. Chem., Int. Ed., 1998, 37, 2872–2875 CrossRef CAS.
- V. Raindlová, R. Pohl and M. Hocek, Chem.–Eur. J., 2012, 18, 4080–4087 CrossRef.
- Y. Cheng, C. Dai, H. Peng, S. Zheng, S. Jin and B. Wang, Chem.–Asian J., 2011, 6, 2747–2752 CrossRef CAS.
- N. Lin, J. Yan, Z. Huang, C. Altier, M. Li, N. Carrasco, M. Suyemoto, L. Johnston, S. Wang, Q. Wang, H. Fang, J. Caton-Williams and B. Wang, Nucleic Acids Res., 2007, 35, 1222–1229 CrossRef CAS.
- L. Kalachova, R. Pohl and M. Hocek, Org. Biomol. Chem., 2012, 10, 49–55 CAS.
- L. Kalachova, R. Pohl, L. Bednárová, J. Fanfrlík and M. Hocek, Org. Biomol. Chem., 2013, 11, 78–89 CAS.
- T. Rühl and E. Stulz, Supramol. Chem., 2010, 22, 103–108 CrossRef.
- B. Alpha-Bazin, H. Bazin, S. Guillemer, S. Sauvaigo and G. Mathis, Nucleosides Nucleotides, 2000, 19, 1463–1474 CAS.
- T. Ehrenschwender, A. Barth, H. Puchta and H.-A. Wagenknecht, Org. Biomol. Chem., 2012, 10, 46–48 CAS.
- H. Weizman and Y. Tor, J. Am. Chem. Soc., 2002, 124, 1568–1569 CrossRef CAS.
- H. A. Held and S. A. Benner, Nucleic Acids Res., 2002, 30, 3857–3869 CrossRef CAS.
- A. Roychowdhury, H. Illangkoon, C. L. Hendrickson and S. A. Benner, Org. Lett., 2004, 6, 489–492 CrossRef CAS.
- H. Macíčková-Cahová, R. Pohl, P. Horáková, L. Havran, J. Špaček, M. Fojta and M. Hocek, Chem.–Eur. J., 2011, 17, 5833–5841 CrossRef.
- Y. Wang, B. A. Tkachenko, P. R. Schreiner and A. Marx, Org. Biomol. Chem., 2011, 9, 7482–7490 CAS.
- S. Ikonen, H. Macíčková-Cahová, R. Pohl, M. Šanda and M. Hocek, Org. Biomol. Chem., 2010, 8, 1194–1201 CAS.
- M. Hollenstein, Chem.–Eur. J., 2012, 18, 13320–13330 CrossRef CAS.
- A. Baccaro, A.-L. Steck and A. Marx, Angew. Chem., Int. Ed., 2012, 51, 254–257 CrossRef CAS.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4523 CrossRef CAS.
- L. Polgár, Cell. Mol. Life Sci., 2005, 62, 2161–2172 CrossRef.
- T. Gourlain, A. V. Sidorov, N. Mignet, S. J. Thorpe, S. E. Lee, J. A. Grasby and D. M. Williams, Nucleic Acids Res., 2001, 29, 1898–1905 CrossRef CAS.
- P. Čapek, H. Cahová, R. Pohl, M. Hocek, C. Gloeckner and A. Marx, Chem.–Eur. J., 2007, 13, 6196–6203 CrossRef.
- A. Baccaro and A. Marx, Chem.–Eur. J., 2010, 16, 218–226 CrossRef CAS.
- F. Seela, M. Zulauf, M. Sauer and M. Deimel, Helv. Chim. Acta, 2000, 83, 910–927 CrossRef CAS.
- J. Ludwig and F. Eckstein, J. Org. Chem., 1989, 54, 631–635 CrossRef CAS.
- H. Shimotahira, S. Fusazaki, I. Ikeda and Y. Ozoe, Bioorg. Med. Chem. Lett., 2011, 21, 1598–1600 CrossRef CAS.
- Y. Cheng, H. Peng, W. Chen, N. Ni, B. Ke, C. Dai and B. Wang, Chem.–Eur. J., 2013, 19, 4036–4042 CrossRef CAS.
- M. Kuwahara, J.-I. Nagashima, M. Hasegawa, T. Tamura, R. Kitagata, K. Hanawa, S.-I. Hososhima, T. Kasamatsu, H. Ozaki and H. Sawai, Nucleic Acids Res., 2006, 34, 5383–5394 CrossRef CAS.
- A. M. Michelson and S. H. Orkin, J. Biol. Chem., 1982, 257, 14773–14782 CAS.
- A. J. Rattray and J. N. Strathern, Annu. Rev. Genet., 2003, 37, 31–66 CrossRef CAS.
- Y. Cho and E. T. Kool, ChemBioChem, 2006, 7, 669–672 CrossRef CAS.
- M. Hollenstein, F. Wojciechowski and C. J. Leumann, Bioorg. Med. Chem. Lett., 2012, 22, 4428–4430 CrossRef CAS.
- M. Kuwahara, S. Obika, H. Takeshima, Y. Hagiwara, J.-i. Nagashima, H. Ozaki, H. Sawai and T. Imanishi, Bioorg. Med. Chem. Lett., 2009, 19, 2941–2943 CrossRef CAS.
- P. Horáková, H. Macíčková-Cahová, H. Pivoňková, J. Špaček, L. Havran, M. Hocek and M. Fojta, Org. Biomol. Chem., 2011, 9, 1366–1371 Search PubMed.
- F. Sanger, S. Nicklen and A. R. Coulson, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5463–5467 CrossRef CAS.
- E. A. Motea and A. J. Berdis, Biochim. Biophys. Acta, 2010, 1804, 1151–1166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob40842f |
| This journal is © The Royal Society of Chemistry 2013 |