 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The relative hydrolytic reactivities of pyrophosphites and pyrophosphates†
Dharmit
Mistry
and
Nicholas
Powles
*
IPOS, The Page Laboratories, Department of Chemical and Biological Sciences, The University of Huddersfield, Queensgate, Huddersfield, HD1 3DH, UK. E-mail: n.t.powles@hud.ac.uk
First published on 16th July 2013
Abstract
The pH-rate profiles for the hydrolysis of pyrophosphate (PP(V)) and pyrophosphite (PP(III), pyro-di-H-phosphonate) are a complex function of pH, reflecting the different ionic species and their relative reactivities. PP(III) is more reactive than PP(V) at all pHs and only PP(III) shows a hydroxide-ion reaction at high pH, so it is 1010-fold more reactive than PP(V) in 0.1 M NaOH. The pKa2 of PP(III) ∼0.44, so the dominant species at pH's > 1 is the di-anion PP(III)2−. Although there is no observable (NMR or ITC) binding of Mg2+ to the PP(III) di-anion there is a modest increase in the rate of hydrolysis of PP(III) by Mg2+. PP(III) is neither a substrate nor an inhibitor of pyrophosphatase, the enzyme that efficiently catalyses the hydrolysis of PP(V).
Introduction
The reactions and stabilities of phosphate mono- and di-esters underpin most life processes such as the storage and manifestation of genetic information, energy transduction, signalling, regulation, differentiation, compartmentalisation, substrate modification to facilitate chemical reactions, and as structural components.1,2 One of the amazing contrasts within this list is the remarkable variation from extreme stability to high reactivity: from half-lives of million of years for the spontaneous hydrolyses of some phosphate esters to milli-second turnovers of their enzyme catalysed reactions.3 A fundamental property of phosphate mono- and di-esters is their negative charge over the normal pH range encountered in living systems, which in turn contributes to their versatility.1 Phosphate mono- and di-esters show remarkable resistance to spontaneous hydrolysis under normal physiological conditions, hence their contribution to the stability of genes. By contrast enzyme-catalysed phosphoryl group transfer reactions are highly efficient and show some of the largest enzymatic rate enhancements4 – up to 1020. The key to understanding enzyme catalysis is the charge and geometric complementarity between substrate and enzyme expressed in the transition state, so a major feature of enzymes catalysing phosphoryl transfer is the neutralisation of the substrate negative charge.5,6Pyrophosphate (PP(V)) is the anhydride of two phosphate ions (Scheme 1) and is important in intermediary metabolism and cell growth.7 The control of intracellular levels of PP(V) during energy metabolism is an essential part of the synthesis of proteins, DNA and RNA.8 The group of enzymes controlling levels of PP(V) are pyrophosphatases (PPases).9 There are two classes of PPases which have been identified – Type I PPases are found in eukaryotes and Escherichia coli and differ from the Type II PPases found in some bacteria.10 These enzymes have remarkably similar active sites but Type I PPases require three Mg2+ ions for activity,11 whereas Type II PPases require four Mn2+ ions.12
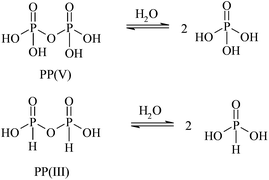 | ||
| Scheme 1 | ||
The P(III) pyrophosphite analogue of pyrophosphate exists as pyro-di-H-phosphonate (PP(III))13 but has only two ionisable protons compared with the four in PP(V) (Scheme 1). We have been interested in H-phosphonates as phosphorylating agents, in particular diethyl pyro-di-H-phosphonate (1).14 Pyrophosphite has also been suggested as a phosphorylating agent in pre-biotic chemistry.15 Herein we report on a comparison of the relative reactivities of the P(III) pyrophosphite (pyro-di-H-phosphonate) and P(V) pyrophosphates towards hydrolysis (Scheme 1) over a wide range of pH and their relative interactions with PPases. The main factor controlling reactivity is the state of ionisation of both species at different pHs.
Results and discussion
pH-rate profiles for the hydrolysis of PP(V) and PP(III)
The rates of hydrolysis of pyrophosphate and pyro-di-H-phosphonate were determined by 31P{H} NMR as a function of pH at 25 °C and ionic strength I = 1.0 M and by auto-titration. The observed first order rate constants were determined at constant pH with different buffer concentrations and the buffer independent rate constant obtained by extrapolation to zero buffer concentration (Fig. 1).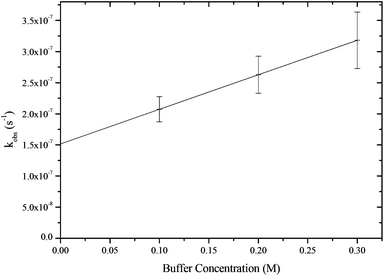 | ||
| Fig. 1 Plot of the observed first-order rate constants for the hydrolysis of PP(III) as a function of MOPS buffer concentration at pH 8.0 at I = 1.0 M (KCl) and 25 °C. | ||
The pKa's of pyrophosphate are known16 but not those for the two ionisable protons of pyro-di-H-phosphonate, which are presumed to be low. However the 31P{H} NMR chemical shift is unchanged from pH 2 to 13.5, suggesting that both pKa's are <1.0 and that the dominant species over this pH range is the di-anion PP(III)2−. However, rapid scanning of 31P{H} NMR spectra at different pH's shows that the chemical shift of PP(III)2− at δ −5.08 ppm does change below pH 2.0 (Fig. 2). Unfortunately PP(III) undergoes acid catalysed hydrolysis at low pH and it is not possible to measure the chemical shift below pH 0.6. Nonetheless, if it is assumed that the observed data is part of a normal sigmoidal titration, it can be fitted to a pKa = 0.44 ± 0.1 as indicated by the solid line (Fig. 2). This value is compatible with analogous systems, which show that P(III) derivatives are generally more acidic than the corresponding P(V) compound. For example, a similar 31P{H} NMR titration with pH for ethyl H-phosphonate (3) yields a pKa = 0.47 which may be compared with that for ethyl phosphate = 1.60.17 The first pKa of P(III) phosphorous acid is 1.0716 and a calculated value is quoted as 0.918 compared with 2.0 for P(V) phosphoric acid.16 The first pKa of isohypophosphoric acid which contains a P(III) and a P(V) phosphorous atom bridged by an oxygen has been estimated to be 0.6.19 A value <0.5 for the pKa of PP(III) is also consistent with the kinetic pH-rate profile reported later.
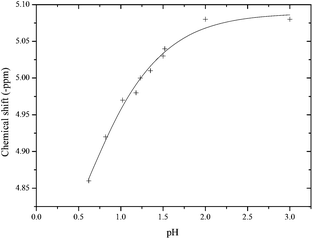 | ||
| Fig. 2 31P{H} NMR chemical shift of PP(III) against pH with external reference to diphenyl phosphate at I = 1.0 M, 25 °C. | ||
The pH-rate profile for the hydrolysis of pyrophosphate and pyro-di-H-phosphonate is a complex function of pH (Fig. 3) reflecting the different ionic species and their relative reactivities. PP(III) is more reactive than PP(V) at all pHs and only the former shows a hydroxide-ion reaction at high pH. The rate of hydrolysis of pyrophosphate PP(V) decreases with increasing pH and the rate constants for the tri- and tetra-anion (Fig. 3) at higher pH are calculated from a recent report.20 The results reported here for the neutral, mono- and di-anionic forms of PP(V) are from our observations using 31P NMR.
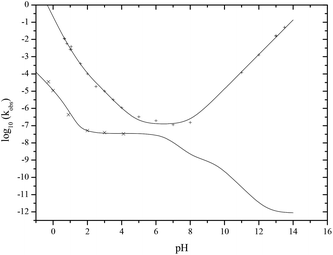 | ||
| Fig. 3 pH rate-profile for the hydrolysis of PP(III) (+) and PP(V) (×) at 25 °C. | ||
The observed pseudo first order rate constant for the hydrolysis of PP(III) shows a first order dependence on H+ concentration between pH 2 and 4, where the dominant species in solution is the di-anion. However, below pH 2 this changes to a second order dependence on H+ concentration, best seen by the enlarged Fig. 4 and changes in values of kobs/[H+] as a function of pH (ESI†). In addition there is a pH independent term for hydrolysis of the PP(III) di-anion between about pH 5 and 8 followed by a rate term that is dependent on hydroxide-ion concentration at higher pH. The observed rate law for the hydrolysis of PP(III) is thus given by eqn (1), where k1, k2, k0 and kOH all refer to the rate constants for the hydrolysis of PP(III) dianion and k2 is that which is first order in H+ concentration, k0 is the pH independent hydrolysis and kOH is the hydroxide-ion catalysed hydrolysis of PP(III)2−. Experimentally, there is no substantial levelling off of the observed pseudo first-order rate constants at low pH or a reversion to a first-order dependence on [H+] which may be expected if the PP(III) mono-anion or undissociated acid become significant species. This is compatible with the 31P{H} NMR data indicating a pKa = 0.44. Consequently, it appears that the k1 term (eqn (1)) refers to a reaction involving PP(III) dianion and [H+]2 or its kinetic equivalent.
| Rate/[PP(III)tot] = kobs = k1[H+]2 + k2[H+] + k0 + kOH[OH−] | (1) |
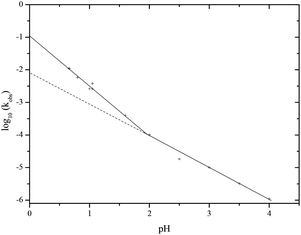 | ||
| Fig. 4 pH rate profile enlarged from Fig. 3 to show the change from first order dependence on H+ (dashed line is a continuation of a first order dependence on H+) to second order dependence on H+. | ||
The line in Fig. 3 is generated from the values of the rate constants given in Table 1. The first two terms in the rate law, k1 and k2 (eqn (1)) have kinetically equivalent expressions, and so, for example, the k1 term could represent the spontaneous hydrolysis of neutral PP(III), and the k2 term could actually reflect either the hydrolysis of the mono-anionic species PP(III)− or even hydroxide-ion attack upon neutral PP(III), although the calculated rate constant for the latter k2Ka1Ka2/Kw is greater than the diffusion controlled rate and so can be excluded.
| Rate constants (eqn (1)) | |
| k 1 | 2.00 × 10−1 (M−2 s−1) |
| k 2 | 9.40 × 10−3 (M−1 s−1) |
| k 0 | 1.20 × 10−7 (s−1) |
| k OH | 1.35 × 10−1 (M−1 s−1) |
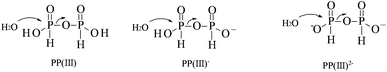
The calculation of these kinetically equivalent mechanisms requires a knowledge of the two pKa's of PP(III). The estimated value of pKa2 = 0.44 is subject to error and there is no simple way to estimate pKa1. Nonetheless it is a worthwhile exercise to enable a comparison with the PP(V) analogues. The third order rate constant k1 is kinetically equivalent to the spontaneous hydrolysis of the undissociated acid with a first order rate constant equal to k1Ka1Ka2. Based on the pH dependence of the 31P NMR chemical shifts (Fig. 2) and the pH-rate profile (Fig. 3) the product Ka1Ka2 is >10−1. The calculated rate constant for the hydrolysis of the undissociated PP(III) is >0.073 s−1. Presumably, the undissociated PP(III) is much more reactive than its mono- and di-anions so that between pH 1 and 2 it is the reactive hydrolytic species even though its concentration is small and falls off with a second order dependence on [H+]. Similarly, between about pH 2 and 5 where the rate of hydrolysis shows a first order dependence on [H+] and the rate law is dominated by the k2 term in eqn (1), hydrolysis is actually occurring through the mono-anion of PP(III) even though, again, the dominant species in solution is the di-anion. The calculated first order rate constant for the hydrolysis of the mono-anion PP(III)− is given by k2Ka1 = 3.41 × 10−3 s−1. The rate constant for the spontaneous hydrolysis of the PP(III) di-anion is 1.20 × 10−7 s−1, so the relative hydrolytic reactivities of the neutral, anionic and di-anionic PP(III) are approximately: 6 × 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 × 104:1.0, respectively. The rates of hydrolysis of the neutral and mono-anionic PP(III) differ by a factor of 20 and presumably both involve nucleophilic attack by water on the more electrophilic neutral P centre although they require the expulsion of different leaving groups, the phosphite mono- and di-anions, respectively (Scheme 2). These relative reactivities are compared later with the hydrolysis of ethyl-H-phosphonate (3) to generate a simple linear free-energy relationship. The much reduced activity of the di-anion of PP(III) reflects the less favourable ease of nucleophilic attack by water on the relatively negatively charged P centre and expulsion of the di-anionic phosphite.
:3 × 104:1.0, respectively. The rates of hydrolysis of the neutral and mono-anionic PP(III) differ by a factor of 20 and presumably both involve nucleophilic attack by water on the more electrophilic neutral P centre although they require the expulsion of different leaving groups, the phosphite mono- and di-anions, respectively (Scheme 2). These relative reactivities are compared later with the hydrolysis of ethyl-H-phosphonate (3) to generate a simple linear free-energy relationship. The much reduced activity of the di-anion of PP(III) reflects the less favourable ease of nucleophilic attack by water on the relatively negatively charged P centre and expulsion of the di-anionic phosphite.
 | ||
| Scheme 2 | ||
In contrast to that for the hydrolysis of PP(III), the observed pseudo first order rate constant for the hydrolysis of PP(V) continues to decrease with increasing pH (Fig. 3) as the state of ionisation increases and forms anions of increasingly lower reactivity. The rate law for the hydrolysis of PP(V) is given by eqn (2). The constants for the tri- and tetra-anions of PP(V) are taken from the recent literature obtained by extrapolation from determinations at elevated temperatures20 and are compatible with other reports.21 The known pKa values (0.79 (I = 1.0 M), 1.72 (I = 1.0 M), 6.6 and 9.4) of PP(V)16 and the rate constants obtained in this work at lower pH (Table 2) have been used to generate the overall pH-rate-profile (Fig. 3).
| Rate = kH[PP(V)][H+] + k0[PP(V)] + k1−[PP(V)−] + k2−[PP(V)2−] + k3−[PP(V)3−] + k4−[PP(V)4−] | (2) |
There is no base-catalysed hydrolysis observed in the hydrolysis of the PP(V) anions in alkaline solutions. This is unlike the behaviour of di-anionic PP(III) which, despite its negative charge, does undergo a hydroxide-ion catalysed hydrolysis and, consequently PP(III) is much more reactive than PP(V) at high pH (Fig. 3) e.g. it is 1010-fold more reactive in 0.1 M NaOH.
Of course, there are kinetically equivalent processes for the hydrolysis of PP(V). For example, if the reported rate constant for the spontaneous hydrolysis of PP(V) tri-anion20 actually reflects the kinetically equivalent hydroxide-ion catalysed hydrolysis of the di-anion, then the corresponding second-order rate constant would be Ka3k3−/Kw = 1.7 × 10−3 M−1 s−1 which can be compared with kOH for PP(III)2− = 1.35 × 10−1 M−2 s−1. This 79-fold higher rate for the pyro-di-H-phosphonate compared with pyrophosphate is of the order expected, so the hydrolysis of the PP(V) tri-anion probably does occur through a mechanism involving hydroxide-ion attack on the di-anion.
Mg2+ catalysed hydrolysis of PP(III)
Many enzymes that catalyse phosphate ester hydrolysis and transesterification require one or more metal-ions as cofactors. It is interesting to note that using 31P{H} NMR or ITC techniques there is no observable binding of Mg2+ to the PP(III) di-anion. Nonetheless, there is a modest increase in the rate of hydrolysis of PP(III) by Mg2+ (Fig. 5) giving a second order rate constant for this process = 2.20 × 10−5 M−1 s−1. If it is assumed that Mg2+ catalysis is due to initial complexation of Mg2+ to PP(III) di-anion followed by nucleophilic attack by water and that metal-ion binding has a weak association constant of <10−2, then the calculated rate constant for water attack on this complex (2) is >2 × 10−3 s−1 which is 4-orders of magnitude greater than that for the uncomplexed PP(III) di-anion. The Mg2+ catalysed hydrolysis of PP(V) at neutral pH is assumed20 to be that of the tetra-anion, i.e. the overall di-anionic MgPP2− is the reactive species with an estimated rate constant at 25 °C of 2.8 × 10−10 s−1, three orders of magnitude greater than that of PP(V)4−. The dissociative or associative nature of phosphoryl transfer reactions is defined by the extent of bond formation between phosphorus and the incoming nucleophile and that between phosphorus and the leaving group in the transition state.3 The processes of phosphorylation and dephosphorylation that occur by associative type mechanisms usually have a penta-coordinate trigonal-bipyramidal geometry in the transition state, whereas in the case of the dissociative pathway it effectively involves a trigonal planar metaphosphate anionic species, PO3−. The 3 orders of magnitude rate enhancement of the hydrolysis of PP(V) by Mg2+ is presumably due to the metal-ion binding to the tetra-anion and so neutralising some of the negative charge and facilitating nucleophilic attack by water. However, the hydrolysis of phosphate monoesters proceeding through the dissociative pathway is not sensitive to the presence of Mg(II) ions in aqueous solution.22 Although metal-ion coordination may enhance the electrophilicity of the phosphorus it will disfavour electron-donation from the non-bridging oxygens thus reducing their ability to assist in expelling the leaving group, which is essential in the dissociative pathway. Similarly, Mg2+ only enhances the rate of hydrolysis of ATP4− by 3-fold and binds preferentially between the β and γ phosphates, making the di-anionic γ phosphate less effective at expelling the ADP leaving group, but does not facilitate an alternative associative pathway.23 An associative transition state can be sensitive to stabilization by Mg2+ ions if coordination occurs to the leaving group.24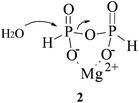
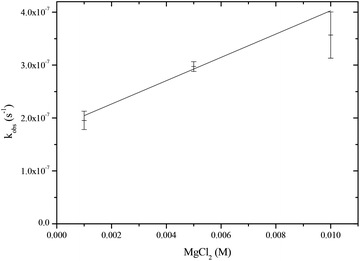 | ||
| Fig. 5 Pseudo first-order rate versus MgCl2 concentration of PP(III) at fixed buffer concentration (0.8 M), pH 7, I = 1.0 M at 25 °C. | ||
Hydrolysis of other P(III) derivatives
The mechanisms of hydrolysis of pyrophosphate mono- and di-esters resemble those for phosphate monoesters,25 except that a phosphate ester or salt is expelled rather than an alcohol. For comparison, the hydrolytic activity of diethyl pyro-di-H-phosphonate (1) H-phosphonate mono- and di-ethyl esters (3 and 4) were investigated.
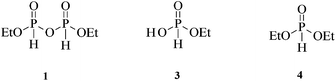
Diethyl-H-phosphonate (4) undergoes hydrolysis to give ethyl-H-phosphonate (3) and shows an acid catalysed hydrolysis with a second order rate constant kH+ = 1.21 × 10−4 M−1 s−1 at 25 °C (I = 1.0 M). It also undergoes a hydroxide-ion catalysed hydrolysis with a second order rate constant kOH = 81.3 M−1 s−1 at 25 °C (I = 1.0 M), obtained from observed pseudo first-order rate constants at constant pH with different buffer concentrations and the buffer independent rate constant obtained by extrapolation to zero buffer concentration.
Ethyl-H-phosphonate (3) is a strong acid and exists predominantly as its mono-anion above pH 1. Its hydrolysis to give phosphorous acid and ethanol at 25 °C (I = 1.0 M) shows a much slower hydroxide-ion catalysed reaction than the di-ester (4) with a second order rate constant kOH = 1.55 × 10−5 M−1 s−1. This corresponds to a rate decrease of 5 × 106 fold due to the additional negative charge and repulsion against the negatively charged nucleophile.
The acid catalysed hydrolysis of the mono-anion shows a second order rate constant kH+ = 6.90 × 10−6 M−1 s−1. The latter reaction is kinetically equivalent to the hydrolysis of the neutral (3) and if it is assumed that the pKa of (3) is ∼0, then the corresponding first order rate constant would be 6.90 × 10−6 s−1. This can then be combined with the data reported earlier for the hydrolysis of other derivatives involving water attack on the neutral H-phosphonates expelling phosphites and ethanol to generate a simple Brønsted plot (Fig. 6) as a function of the pKa of the leaving group XH (5) using the data summarised in Table 3.
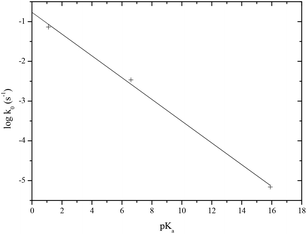 | ||
| Fig. 6 Brønsted plot for the spontaneous pH independent hydrolysis of H-phosphonate derivatives (HO)PH-X at 25 °C as a function of the pKa of the leaving group XH. | ||
| X | pKa XH | k 0 (s−1) |
|---|---|---|
| H2PO3 | 1.1 | 0.073 |
| HPO3− | 6.6 | 3.41 × 10−3 |
| EtO | 15.9 | 6.90 × 10−6 |

Pyrophosphatase catalysed hydrolysis of PP(III)
Inorganic pyrophosphate (PP(V)) or pyrophosphite (PP(III)) may have acted as a phosphoryl group donor in primitive biological systems.15,27 In modern organisms, the enzyme-catalysed hydrolysis of PP(V) is an important part of the metabolism to remove PP(V) that is generated by biosynthetic reactions.7,28 There are two classes of inorganic pyrophosphatase (PPase) that catalyse the hydrolysis of PP(V), both of which have an unusual activity dependence on metal ions but similar active sites.10 Type I PPases (found in eukaryotes and E. coli) require three Mg2+ ions for activity11 and differ significantly from those of Type II PPases (found in some bacteria) which require four Mn2+ ions.12 In both enzymes PP(V) tetra-anion is co-ordinated to metal-ions so that the extremely polar active site neutralizes the negative charges on substrate to facilitate nucleophilic attack by water.29 The metal ions probably also coordinate to the attacking water so decreasing its pKa and providing an activated nucleophile if the Brønsted βnuc for attack by nucleophiles is <1.0.30It is difficult to follow the pyrophosphatase catalysed hydrolysis of PP(V) by 31P{H} NMR because of limited solubility of the substrate. However, the use of isothermal titration calorimetry (ITC) provides a suitably sensitive continuous assay. Experiments were conducted at pH 8.4 and 7.55 as hydrolysis of PP(V) is the slowest step at the higher pH, rather than substrate binding or product release.31 ITC can be used to follow the kinetics of an enzyme catalysed reaction because the amount of heat released is directly proportional to the amount of substrate that has reacted. The instrument output reflects the energy required to balance the heat generated by the reaction to maintain constant temperature (Fig. 7). The initial positive spike represents the endothermic heat of injection/dilution. The exothermic hydrolysis reaction gives a negative displacement from the baseline and as substrate is consumed, the rate at which heat is generated decreases and the output trace returns to the baseline.
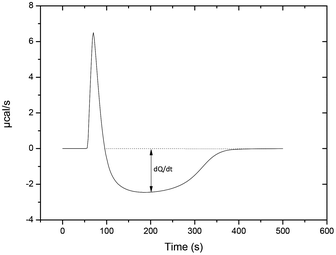 | ||
| Fig. 7 ITC trace of the enzymatic hydrolysis of pyrophosphate (PP(V)) with 1 × 10−9 M E. coli PPase, 3 mM MgCl2, initiated with PP(V) to give a final concentration of 4.11 × 10−5 M at 25 °C. The endothermic peak represents the heat of dilution followed by PP(V) consumption until the heat generated with respect to time (dQ/dt) returns to the baseline. | ||
The total heat released by hydrolysis is represented by the well area under the dashed line and dQ/dt is the difference between the baseline and the heat released at any time t (Fig. 7). The concentration of PP(V) at any time t can be used to give the Michaelis–Menten kinetic parameters kcat and Km as previously described.20
It is of interest to determine if PP(III) is a substrate of PPase and, if not, whether it binds sufficiently to act as an inhibitor of the enzyme. Using the PPase from E. coli (2 × 10−7 M) there was no discernible rate difference from the background hydrolysis of PP(III) at pH 7.55 (300 mM buffer, 50 mM Mg2+, I = 1.0 M), indicating that PP(III) is not a substrate and that kcat/Km is <6 M−1 s−1. This may be due to PP(III) showing no significant binding to Mg2+ and/or having insufficient negative charge to bind to the positively charged active site of PPase. To investigate whether PP(III) binds sufficiently to act as an inhibitor of PPase, the rates of the enzyme catalysed hydrolysis of PP(V) were measured in the absence and presence of PP(III). Above saturation, the kcat values measured for PP(V) at 25 °C at pH 8.40 and 7.55 were 208 s−1 and 82 s−1, respectively. This is in good agreement with a value of kcat = 290 s−1 at pH 8.4 which has been reported.31 The second order rate constant for the enzyme catalysed hydrolysis kcat/Km = 3.68 × 107 M−1 s−1 and 2.89 × 107 M−1 s−1 again at 25 °C, pH 8.40 and 7.55, respectively. These parameters decreased by less than 27% with 1 × 10−3 M PP(III) indicative that the binding of PP(III) to PPase is weak and Ki must be >1 × 10−3 M.
Conclusions
The pH-rate profiles for the hydrolysis of pyrophosphate (PP(V)) and pyrophosphite (PP(III), pyro-di-H-phosphonate) are a complex function of pH. Increasing ionisation with increasing pH steadily decreases the rate of hydrolysis of PP(V). PP(III) is more reactive than PP(V) at all pHs. The pKa2 of PP(III) = 0.44, so the dominant species at pH's > 1 is the di-anion PP(III)2− which shows a hydroxide-ion reaction at high pH, so it is 1010-fold more reactive than PP(V) in 0.1 M NaOH. Although there is no observable (NMR or ITC) binding of Mg2+ to the PP(III) di-anion there is a modest increase in the rate of hydrolysis of PP(III) by Mg2+. PP(III) is neither a substrate nor an inhibitor of pyrophosphatase, the enzyme that efficiently catalyses the hydrolysis of PP(V).Experimental
Materials
PP(III) – samples of PP(III) were initially and kindly provided by T. Kee (University of Leeds) and prepared by dissolving phosphorous acid (16.4 g) and sodium hydroxide (8.0 g) in 100 ml water. The water was removed under reduced pressure. The resulting mixture was heated in a tube furnace with N2 flowing through at 200 °C for three days.Diethyl pyro-di-H-phosphonate (1) and H-phosphonate monoethyl esters (3) were prepared as previously described.14
H-phosphonate di-ethyl ester (>99%) (4) and E. coli pyrophosphatase (lyophilized powder ≥90%, ≥800 units per mg protein) were purchased from Sigma.
Kinetics
Ionic strength of solutions were maintained using KCl. Reactions monitored by 31P{1H} NMR at 25 °C used a 500 MHz Bruker Avance I, and a D2O insert was used for a lock signal and diphenylphosphate as an internal standard. Where auto-titration was used to follow reactions these were carried out using a Metrohm 859.Auto-titration: a jacketed titration vessel aspirated with N2 was used to maintain a constant temperature. The vessel was charged with 50 ml HCl or NaOH at I = 1.0 M and titrated with an appropriate solution to maintain the pH. The titration was initiated by addition of 1.0 mmole PP(III) to the vessel.
Effect of Mg2+ on the hydrolysis of PP(III): samples were run in 0.8 M MOPS buffer at pH 7 and 1.0 M ionic strength containing 10% D2O for a D2O lock and 20 mM diphenylphosphate as an internal standard. Magnesium concentrations were varied by the addition of 0.001 M, 0.005 M and 0.01 M magnesium chloride (MgCl2), to 37.6 mM PP(III).
E. coli Pyrophosphatase catalysed reactions using VP- ITC: the enzyme (1 × 10−9 M) was prepared in 20 mM HEPES buffer pH 8.40 or 20 mM MOPS buffer pH 7.55, with 3 mM magnesium chloride. 10 mM PP(V) in the same buffer, excluding PPase and magnesium chloride was used to initiate the reaction to give a final concentration of 0.0411 mM or 0.0211 mM, I = 0.1 M at 25 °C. Both solutions were degassed for 15 minutes to prevent bubble formation during the experiment. The same experiment was repeated but with the addition of 1 mM PP(III) as a potential inhibitor.
The pKa of PP(III) was monitored by 31P{H} NMR which contained an insert with diphenylphosphate in MOPS buffer, pH 7, I = 1.0 M as an external standard.
Acknowledgements
We thank T. P. Kee for partly initiating this work and for providing an initial sample of pyrophosphite and M. I. Page for helpful discussions.Notes and references
- F. H. Westheimer, Science, 1987, 235, 1173 CAS.
- M. W. Bowler, M. J. Cliff, J. P. Waltho and G. M. Blackburn, New J. Chem., 2010, 34, 784 RSC.
- W. W. Cleland and A. C. Hengge, Chem. Rev., 2006, 106, 3252 CrossRef CAS; A. C. Hengge, Adv. Phys. Org. Chem., 2005, 40, 49 CrossRef , (J. P. Richard (ed.), Academic, New York). J. K. Lassila, J. G. Zalatan and D. Herschlag, Annu. Rev. Biochem., 2011, 80, 669 CrossRef.
- C. Lad, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5607 CrossRef CAS; G. K. Schroeder, C. Lad, P. Wyman, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4052 CrossRef; R. Wolfenden, Chem. Rev., 2006, 106, 3379 CrossRef.
- N. J. Baxter, G. M. Blackburn, J. P. Marston, A. M. Hounslow, M. J. Cliff, W. Bermel, N. H. Williams, F. Hollfelder, D. E. Wemmer and J. P. Waltho, J. Am. Chem. Soc., 2008, 130, 3952 CrossRef CAS.
- M. J. Cliff, M. W. Bowler, A. Varga, J. P. Marston, J. Szabó, A. M. Hounslow, N. J. Baxter, G. M. Blackburn, M. Vas and J. P. Waltho, J. Am. Chem. Soc., 2010, 132, 6507 CrossRef CAS.
- M. Lundin, H. Baltscheffsky and H. Ronne, J. Biol. Chem., 1991, 266, 12168 CAS.
- L. Peller, Biochemistry, 1976, 15, 141 CrossRef CAS.
- T. Shintani, T. Uchiumi, T. Yonezawa, A. Salminen, A. A. Baykov, R. Lahti and A. Hachimori, FEBS Lett., 1998, 439, 263 CrossRef CAS; T. W. Young, N. J. Kuhn, A. Wadeson, S. Ward, D. Burges and G. D. Cooke, Microbiology, 1998, 144, 2563 CrossRef; S. Ahn, A. J. Milner, K. Fütterer, M. Konopka, M. Ilias, T. W. Young and S. A. White, J. Mol. Biol., 2001, 313, 797 CrossRef.
- M. C. Merckel, I. P. Fabrichniy, A. Salminen, N. Kalkkinen, A. A. Baykov, R. Lahti and A. Goldman, Structure, 2001, 9, 289 CrossRef CAS.
- P. Heikinheimo, J. Lehtonen, A. Baykov, R. Lahti, B. S. Cooperman and A. Goldman, Structure, 1996, 4, 1491 CrossRef CAS.
- I. P. Fabrichniy, L. Lehtiö, M. Tammenkoski, A. B. Zyryanov, E. Oksanen, A. A. Baykov, R. Lahti and A. Goldman, J. Biol. Chem., 2006, 282, 1422 CrossRef.
- J. R. Van Wazer, Phosphorus and its Compounds, Interscience Inc., New York, 1958, vol. 1, p. 395 Search PubMed.
- N. T. Powles, J. H. Atherton and M. I. Page, Org. Biomol. Chem., 2012, 10, 5940 CAS.
- D. E. Bryant, K. E. R. Marriott, S. A. Macgregor, C. W. G. Fishwick, M. A. Pasek and T. P. Kee, Chem. Commun., 2010, 46, 3726 RSC.
- H. A. Sober and W. P. Jencks, in Handbook of Biochemistry, ed. H. A. Sober, Chemical Rubber Co., Cleveland, OH, 1968, pp. J150–J189 RSC; R. P. Mitra, H. C. Malhotra and D. V. S. Jain, Trans. Faraday Soc., 1966, 62, 167–172 RSC.
- W. D. Kumler and J. J. Eiler, J. Am. Chem. Soc., 1943, 65, 2355 CrossRef CAS.
- J. P. Guthrie, Can. J. Chem., 1979, 57, 236–239 CrossRef CAS.
- R. L. Carroll and R. E. Mesmer, Inorg. Chem., 1967, 6, 1137 CrossRef CAS.
- R. B. Stocksbridge and R. Wolfenden, J. Biol. Chem., 2011, 286, 18538 CrossRef.
- J. P. Crowther and A. E. R. Westerman, Can. J. Chem., 1954, 32, 42 CrossRef CAS; J. D. McGilvery and J. P. Crowther, Can. J. Chem., 1954, 32, 174 CrossRef; M. Kawabe, O. Ohashi and I. Yamaguchi, Bull. Chem. Soc. Jpn., 1970, 43, 3705 CrossRef.
- I. E. Catrina and A. C. Hengge, J. Am. Chem. Soc., 1999, 121, 2156 CrossRef CAS.
- S. J. Admiraal and D. Herschlag, Chem. Biol., 1995, 2, 729 CrossRef CAS; D. Herschlag and W. P. Jencks, J. Am. Chem. Soc., 1987, 109, 4665 CrossRef.
- N. H. Williams, J. Am. Chem. Soc., 2000, 122, 12023 CrossRef CAS.
- D. L. Miller and F. H. Westheimer, J. Am. Chem. Soc., 1966, 88, 1507 CrossRef CAS; D. L. Miller and T. Ukema, J. Am. Chem. Soc., 1969, 91, 3050 CrossRef.
- M. I. Page and A. Williams, in Organic and Bio-Organic Mechanisms, Longmans, 1997, p. 67 CrossRef CAS; A. Williams, Adv. Phys. Org. Chem., 1992, 27, 1 CrossRef CAS.
- A. Serrano, J. R. Perez-Castineira, H. Baltscheffsky and M. Baltscheffsky, J. Bioenerg. Biomembr., 2004, 36, 127 CrossRef CAS; A. Serrano, J. R. Perez-Castineira, M. Baltscheffsky and H. Baltscheffsky, IUBMB Life, 2007, 59, 76 CrossRef; M. Baltschefsky, A. Schultz and H. Baltschefsky, FEBS Lett., 1999, 452, 121 CrossRef.
- J. Chen, A. Brevet, M. Fromant, F. Lévêque, J.-M. Schmitter, S. Blanquet and P. Plateau, J. Bacteriol., 1990, 172, 5686 CAS.
- L. Yang, R.-Z. Liao, J.-G. Yu and R.-Z. Liu, J. Phys. Chem. B, 2009, 113, 6505 CrossRef CAS.
- S. Bounaga, A. P. Laws, M. Galleni and M. I. Page, Biochem. J., 1998, 331, 703 CAS.
- A. A. Baykov, T. Hyytia, S. E. Volk, V. N. Kasho, A. V. Vener, A. Goldman, R. Lahti and B. S. Cooperman, Biochemistry, 1996, 35, 4655 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob40755a |
| This journal is © The Royal Society of Chemistry 2013 |