 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical-genetic identification of the biochemical targets of polyalkyl guanidinium biocides†
Drew
Bowie
a,
Paria
Parvizi
b,
Dustin
Duncan
ab,
Christopher J.
Nelson
*a and
Thomas M.
Fyles
*b
aDepartment of Biochemistry and Microbiology, University of Victoria, PO Box 3065, Victoria, BC, Canada. E-mail: cjn@uvic.ca; Fax: +1 250-721-8855; Tel: +1 250-853-3889
bDepartment of Chemistry, University of Victoria, PO Box 3065, Victoria, BC, Canada. E-mail: tmf@uvic.ca; Fax: +1 250-721-7147; Tel: +1 250-721-7192
First published on 13th May 2013
Abstract
Alkylated guanidinium compounds exhibit microbiocidal activity in marine environments, yet the mode of action of these compounds has not been defined. A comprehensive chemical-genetic approach in budding yeast was used to define the biological processes affected by these compounds. N-Butyl-N′-decylguanidinium and N-hexyl-N′-(3-hydroxypropyl)-N′′-octylguanidinium chlorides were shown to prevent yeast growth in a dose-dependent manner. All non-essential genes required for tolerance of sub-lethal amounts of these biocides were identified. These unbiased and systematic screens reveal the two related guanidinium compounds have a non-overlapping spectrum of targets in vivo. A functional tryptophan biosynthetic pathway is essential for tolerance of both biocides, which identifies tryptophan amino acid import as one process affected by these compounds. Further analysis of hypersensitive gene lists demonstrates that the substitutions on alkylated guanidiums confer important functional differences in vivo: one derivative renders the ability to generate acidic vacuoles essential, while the other is synthetically lethal with mutants in the transcriptional response to chemical stress. Altogether the results define the common and distinct biological processes affected by biocidal alkylated guanidinium salts.
Any clean surface exposed to the marine environment rapidly develops a complex community of microorganisms (bacteria, diatoms, algae) which is then colonized by larger plants, molluscs, sponges and tubeworms in a process known as biofouling.2 Biofouling affects virtually all marine structures, ranging from sedentary organisms such as sponges which engage in robust chemical warfare with fouling competitors,3 to permanent human installations (wharves, drilling platforms) and shipping. The economic impacts are significant, particularly related to increased fuel costs due to fouling-induced drag on ocean-going ship hulls estimated to run to millions of dollars per ship per year.4,5
Antifouling biocides have been exploited since pre-historic times, resulting in the late 20th century with the development of very effective ablative coatings containing organotin compounds.6 However, the persistence of these organotins in the environment and their serious environmental consequences6 lead to their complete international ban in 2008. Current marine antifouling coatings are based on copper oxide, copper and zinc complexes, and specific antifoulants that act in concert with copper. Assessment of the environmental impact and fate of such additives is on-going but it is clear that there is a need for new approaches, such as compounds that degrade rapidly in seawater before being sequestered in sediment.7 Other promising approaches focus on the use of antifouling natural products,3 inherently antifouling surfaces inspired by nature,8 and on compounds capable of disrupting bacterial quorum sensing, essential to the early stages of biofilm formation.9,10
Our interest in antifouling compounds stems from an serendipitous observation made in a field test of a dissolved oxygen sensor.11,12 As noted above, biofouling of clean surfaces is inevitable in the ocean, so it was surprising to discover that the ion exchange membrane of the sensor remained pristine even while the housing developed the expected fouling layer. Proceeding empirically, we eventually established that the alkyl guanidinium salt of the ion exchanger11 was weakly biocidal, and that polyalkyl guanidinium salts were significantly more effective antifouling agents admixed in coatings.13 Of more significance was the short half-life of such compounds in cool seawater together with the observation of alkyl ureas and alkyl amines as degradation products under sub-lethal conditions.13 We then turned to developing slow-release coatings containing derivatives which showed enhanced antifouling activity as well as an inherent potential to degrade in water in the absence of biota with acceptably short half-life.14 Compounds 1 and 2 are typical of the types of compounds in this class of microbiocides.

Despite good activity in both laboratory and field bioassays, the fact remains that the activity of these compounds was known only empirically. The toxicity of quaternary ammonium and N-alkyl pyridinium salts towards marine biofouling species is reported.15,16 Such compounds act by disruption of the cytoplasmic membrane of bacteria leading to lysis of cellular compartments.17,18 Dodecylguanidinium acetate (dodine) is a licensed fungicide used on soft fruits and appears to have a similar mode of action.19,20 It is certainly possible that polyalkyl guanidinium salts act similarly, although the minimal inhibitory concentrations are several orders of magnitude lower than is usual for quaternary ammonium disinfectants, and the structures of 1, 2, and other active agents in this class13 do not resemble classic detergents. Whether or not there are specific biochemical targets on which the compounds act was entirely unknown.
Chemical-genetic profiling offers the possibility to assess the similarity of a new bioactive agent with no known target to known classes of agents through the effect of the compound on the viability of members of a library of yeast strains bearing specific genetic deletions.21 Gene deletions that render a cell sensitive to a specific compound can be used to infer the pathways that normally protect the cell from such agents. Even if such pathway specific information is not directly evident, the overall pattern of gene–chemical interactions reflects the types of processes affected by an agent. Classes of compounds with a similar mode-of-action result in a similar chemical-genetic profile.21
We therefore undertook a chemical-genetic profiling experiment in order to assess how the biocidal activity of polyalkyl guanidinium salts such as 1 and 2 arises.
Results and discussion
Synthesis and compound stability
The synthesis of polyalkyl guanidinium salts is straightforward (Scheme 1).13,14,22 Alkyl amines react with alkyl isothiocyanates to rapidly produce N,N′-dialkyl thioureas, usually in quantitative yield. Methylation of the sulphur can be achieved with either methyl iodide or somewhat more conveniently, with dimethyl sulphate. Although it is possible to generate a sulphate salt using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in the latter case, it is more convenient to use a 1:1 stoichiometry to produce the methyl sulphate salt. Compound 1 is produced by reaction with excess ammonia in ethanol in a sealed reactor while compound 2 is produced using 3-aminopropanol in ethanol at reflux. If the methyl sulphate salt is used, the excess amine is methylated to produce initially a sulphate salt. Both 1 and 2 are water insoluble/organic soluble, so by-products are readily removed by extraction with water; ion exchange to the chloride salt can be achieved by washing with brine. The synthesis of 2 by the dimethyl sulphate route has been conducted in a single batch in 98% isolated yield (>99% purity) starting with 1 kg of hexyl isothiocyanate. High purity samples for chemical-genetic profiling were converted to the chloride salts with anion exchange chromatography, followed by chromatography on silica to remove trace contaminants.
:1 stoichiometry in the latter case, it is more convenient to use a 1:1 stoichiometry to produce the methyl sulphate salt. Compound 1 is produced by reaction with excess ammonia in ethanol in a sealed reactor while compound 2 is produced using 3-aminopropanol in ethanol at reflux. If the methyl sulphate salt is used, the excess amine is methylated to produce initially a sulphate salt. Both 1 and 2 are water insoluble/organic soluble, so by-products are readily removed by extraction with water; ion exchange to the chloride salt can be achieved by washing with brine. The synthesis of 2 by the dimethyl sulphate route has been conducted in a single batch in 98% isolated yield (>99% purity) starting with 1 kg of hexyl isothiocyanate. High purity samples for chemical-genetic profiling were converted to the chloride salts with anion exchange chromatography, followed by chromatography on silica to remove trace contaminants.
 | ||
| Scheme 1 Synthesis of compounds. | ||
The procedure for the chemical-genetics profiling required the compound to be dispersed in the agar medium, a process that involved a short period of time at a temperature of 40–60 °C, followed by the assay itself at a temperature of 30 °C. Although compound 1 is likely stable under this regime, compound 2 is designed to degrade in water. Direct analysis of the compound within the agar medium was not possible, but the degradation could be followed at 30 °C in aqueous 0.1 M NaCl at the concentration used in the bioassay. Extraction of 2 followed by analysis by ESI-MS gave an apparent half-life of approximately 5 hours under these conditions. Thus it is likely that the compound concentration fell ten-fold within the first ∼16 hours of the assay and the compound was essentially absent after two days.
Chemical-genetic interaction profiling
To determine the biological targets of alkylated guanidinium compounds we utilized a chemical-genetic approach using the yeast Saccharomyces cerevisae. A comprehensive collection of ∼4800 non-essential gene deletion strains in this model eukaryote enables the rapid identification of loss of function mutants that are hypersensitive to a compound of interest. The identification of such gene–compound interactions highlights molecular processes required for growth in the presence of compound. This information has been successful in revealing the mode of action of several drugs and small molecules.21,23We first determined if yeast growth is affected by 1 and 2. To this end, cultures of the laboratory strain BY4741 were plated in duplicate on media containing increasing doses of these compounds. At concentrations above 1 mM, both compounds completely abolished yeast colony formation. Doses of 50 μM of 1 and 100 μM of 2 reduced yeast growth, as assayed by colony forming units, by approximately 50% (Fig. 1). The subtle difference in potency could simply reflect the differential stability of these two alkylated compounds, or may be due to different modes of action.
 | ||
| Fig. 1 Alkylated guanidinium compounds inhibit yeast growth. The BY4741 lab strain of yeast was inoculated on media containing either 1% methanol (0, vehicle control) or the indicated concentrations of 1 and 2. Plates were imaged after incubation at 30 °C for 2 days. | ||
Next, we used an established systematic genetic approach to determine the likely modes of action of these biocides: by replica plating the yeast deletion collection on control media and media with sub-lethal concentrations of guanidinium salts we identified the gene products required for tolerance of alkylated guanidinium biocides. The ∼4800 strain yeast deletion collection, arrayed robotically at 1536 strains per plate, was spotted in triplicate onto rich media containing 30 μM 1, or 100 μM 2 or methanol as a vehicle control (Fig. 2A). After 24 h yeast colonies were imaged and the analysed using the Balony analysis software package (http://code.google.com/p/balony/, generously shared by Drs Christopher Loewen and Barry Young, University of British Columbia)) to generate a ranked list of mutant strains that exhibited reduced growth in the presence of the alkylated guanidinium compound.
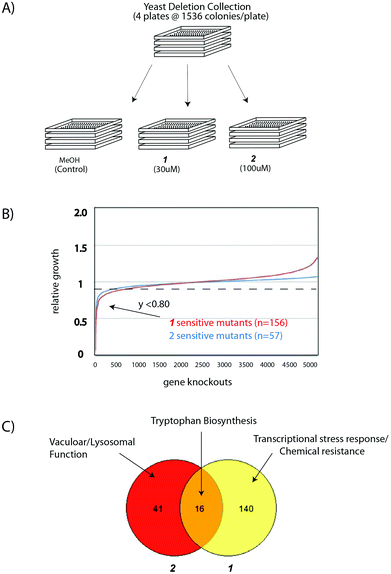 | ||
| Fig. 2 Chemical-genetic interaction profiles of two alkylated guanidinium compounds (A) Experimental scheme for the identification of genes required for tolerance of sub-lethal concentrations of 1 and 2. (B) Results of the chemical genetic screens: setting a threshold of 80% fitness relative to methanol control, 156 and 57 genes were defined as being hyper-sensitive to alkylated guanidinium compounds 1 and 2 respectively. (C) Summary of Gene Ontology (GO) analysis of hypersensitive strains using the FunSpecWebtool. P-values for described ontology categories are provided in ESI Table S1† and the text. | ||
We find that the overwhelming majority of yeast mutants, including those that exhibit well-documented slow growth phenotypes,24 are no more sensitive to guanidinium compounds 1 and 2 than WT yeast. However, 1.1% (n = 57) of yeast mutants exhibited a growth defect on 100 μM 2. Similarly, 3.2% (n = 156) of the non-essential deletion collection is sensitive to 30 μM 1 (Fig. 2B). Thus only a small group of gene products are necessary for tolerance of alkylated guanidinium compounds. This suggests that guanidinium biocides 1 and 2 must affect specific, rather than general, processes in this model eukaryote. The complete lists of chemical genetic interactions for 1 and 2 can be found in ESI Tables S1 and S2.†
Tryptophan biosynthesis is essential in the presence of alkyl guanidinium compounds
A comparison of mutants sensitive to 1 and 2 reveals separate and shared chemical-genetic interactions (Fig. 2C). Gene ontology (GO) analysis using the FunSpecWebtool (Table 1) reveals that mutants sensitive to 1 are highly enriched in transcriptional stress response (p = 1.66 × 10−6) and factors known to mediate resistance to chemical agents (p = 9.59 × 10−5). By contrast yeast strains sensitive to 2 are enriched in vacuolar ATPase complex components that are known to be important in ion homeostasis (p = 3.48 × 10−11) and vacuolor/lysosomal function (p = 1.26 × 10−7). These mutually exclusive GO profiles indicate that a transcriptional response to chemical stress and vacuolar function/ion homeostasis are essential biological processes for the resistance of yeast to 2 and 1 respectively. Significantly, these results indicate that these related guanidinium compounds can affect the biology of this model eukaryote via distinct mechanisms.| GO category | P-value | Genes identified (hits) | # hits | Total gene |
|---|---|---|---|---|
| GO category | ||||
| 1 Hypersensitive | ||||
| Aromatic amino acid family biosynthetic process [GO:0009073] | 2.05 × 10−9 | ARO4 TRP1 ARO1 TRP4 TRP2 ARO2 TRP3 | 7 | 12 |
| Tryptophan biosynthetic process [GO:0000162] | 1.29 × 10−6 | TRP1 TRP4 TRP2 TRP3 | 4 | 5 |
| Negative regulation of transcription from RNA polymerase II promoter [GO:0000122] | 4.75 × 10−6 | SRB8 ARG82 GCN4 PDR1 YGR122W RIM101 SSN8 SKO1 SSN3 | 9 | 57 |
| Ubiquitin-dependent protein catabolism via the multivesicular body sorting pathway [GO:0043162] | 1.46 × 10−5 | SNF7 VPS36 VPS20 SNF8 VPS28 | 5 | 15 |
| Response to drug [GO:0042493] | 1.60 × 10−5 | SNQ2 PDR1 ELM1 CIN5 PDR5 SGE1 | 6 | 25 |
| Regulation of transcription, DNA-dependent [GO:0006355] | 2.54 × 10−5 | CCR4 SRB8 ARG82 NGG1 UPC2 ADA2 GCN4 RIM15 PDR1 DST1 RAD6 GCN5 RIM101 SRB2 ZAP1 SWI6 CDC73 ESC1 SSN8 YAF9 SKO1 POP2 SPT20 CIN5 ISW2 EGD1 SSN3 | 27 | 507 |
| Cellular amino acid biosynthetic process [GO:0008652] | 7.25 × 10−5 | LYS2 ARO4 TRP1 ARO1 TRP4 GCN4 TRP2 ARO2 CYS4 TRP3 | 10 | 98 |
| Transcription, DNA-dependent [GO:0006351] | 7.66 × 10−5 | CCR4 SRB8 ARG82 NGG1 UPC2 ADA2 GCN4 PDR1 DST1 RAD6 GCN5 RIM101 SRB2 ZAP1 CTK1 SWI6 CDC73 ESC1 SSN8 YAF9 SKO1 POP2 SPT20 CIN5 ISW2 EGD1 SSN3 | 27 | 540 |
| 2 Hypersensitive | ||||
| Vacuolar acidification [GO:0007035] | 8.48 × 10−14 | VMA2 VMA1 VMA7 VMA10 VMA22 VPH2 VMA6 STV1 VMA11 | 9 | 26 |
| ATP hydrolysis coupled proton transport [GO:0015991] | 1.42 × 10−9 | VMA2 VMA1 VMA7 VMA6 ST V1 VMA11 | 6 | 17 |
| Proton transport [GO:0015992] | 1.48 × 10−8 | VMA2 VMA1 VMA7 VMA10 VMA6 ST V1 VMA11 | 7 | 41 |
| Ion transport [GO:0006811] | 1.18 × 10−5 | VMA2 VMA1 VMA7 VMA10 VMA6 ST V1 VMA11 | 7 | 107 |
| 1 and 2 Hypersensitive mutants | ||||
| Aromatic amino acid family biosynthetic process [GO:0009073] | 1.79 × 10−14 | TRP1 ARO1 TRP4 TRP2 ARO2 TRP3 | 6 | 12 |
| Tryptophan biosynthetic process [GO:0000162] | 4.51 × 10−11 | TRP1 TRP4 TRP2 TRP3 | 4 | 5 |
| Cellular amino acid biosynthetic process [GO:0008652] | 1.45 × 10−8 | TRP1 ARO1 TRP4 TRP2 ARO2 TRP3 | 6 | 98 |
| Tryptophan metabolic process [GO:0006568] | 1.07 × 10−5 | TRP1 TRP3 | 2 | 3 |
Notably, a comparison of mutants sensitive to both 1 and 2 reveals that 16 mutants are hypersensitive to both guanidinium compounds (Table 1). These genes provide important insight into a biological process affected by the general and shared properties of these related biocides. GO analysis of these 16 genes highlights the tryptophan biosynthetic pathway (p = 1.79 × 10−14). In fact, six of the nine gene products (Aro1–2, Trp1–4) required to convert phosphoenolpyruvate (PEP) to intracellular tryptophan (Fig. 3A) were identified in both screens. The remaining three pathway components are Trp5, which catalyses the final step in tryptophan biosynthesis and Aro3 and Aro4 which together catalyse the conversion of PEP to dihydroxyacetone phosphate (DHAP). These mutants were detected in only one of two screens (which likely indicates a thresholding effect or false negative data from these high-throughput screens).
 | ||
| Fig. 3 Alkylated guanidinium compounds disrupt tryptophan uptake (A) Schematic of tryptophan metabolism in yeast: tryptophan is synthesized from phosphoenolpyruvate (PEP) using the indicated enzymes of the Aro–Trp pathway. Alternatively, when present, extracellular tryptophan is transported into cells using the high affinity Tat2 transporter. The low affinity Tat1 can also exert this function, although at much lower rates.1 Gene products identified in chemical genetic screens of 1 and 2 are indicated in bold. Aro3, Aro4, and Trp5, indicated in italics were identified in only one of the two screens. (B) A subset of yeast mutants are sensitive to 2 because of compromised utilization of exogenous tryptophan. Ten-fold serial dilutions of the indicated 16 hypersensitive mutants were spotted onto the indicated media. Failure of mutants in the Aro–Trp pathway to grow in the presence of 2 can be rescued with exogenous tryptophan. However, the remaining hits are sensitive to 2 independent of tryptophan levels indicating additional processes are affected by this compound. | ||
To validate the results of our robotic screens we confirmed the hypersensitivity of a subset of mutants to 2 in a serial spotting assay (Fig. 3B). This method, which effectively measures the number of viable yeast cells, is intrinsically more sensitive than the genomic screen that simply measures colony size. In this assay 16/17 hits were validated as either synthetically lethal, or sick by at least one order of magnitude. Taken together, these results confirm that a functional tryptophan biosynthetic pathway is needed for yeast to proliferate in the presence of alkylated guanidinium salts. By extension, tryptophan (but not other amino acid) utilization must be compromised by 1 and 2.
Yeast grown in the rich media used in our screen can obtain tryptophan from intracellular biosynthesis via the Aro–Trp pathway, or from extracellular sources using a high affinity Tat2 transporter (Fig. 3A). We reasoned that guanidinium compounds may inhibit yeast growth by reducing tryptophan import capacity. If this is the case, one would expect an excess of tryptophan to rescue the growth defect imposed by 1 and 2. We tested this hypothesis with a collection of 17 hits from the 2 sensitive gene list. As expected, we find that the sensitivity of all mutants in the tryptophan biosynthetic pathway to 2 could be rescued if excess tryptophan was supplied in the medium (Fig. 3B). Of note, exogenous tryptophan does not uniformly obviate the effects of guanidinium compounds: the sensitivity of several mutants to 2 (i.e., erg4, tfp1, tfp3, sac1, rho4, vph2, lem3, nde1, pdr5) is unaffected by the presence of exogenous tryptophan (Fig. 3B). Therefore 2 does not exclusively inhibit tryptophan uptake but tolerance of 2 requires the ability to generate tryptophan.
However, given the nature of the remaining chemical-genetic interactions we predict that loss of tryptophan uptake by 1 and 2 is a consequence of their inherent ability to alter discreet membrane environments as opposed to directly inhibiting the activity of a protein, such as the Tat2 tryptophan importer (see below).
Membrane homeostasis is critical for tolerance of guanidinium compounds
Based on the foregoing, it is likely that alkylated guanidinium compounds compromise tryptophan uptake. This could be the consequence of direct inhibition of Tat2 function. Indeed this effect has been described for the small molecules quinine,25 and phenyl-butyrate.26 It is noteworthy that, unlike our compounds, these agents are structurally similar to tryptophan. Alternatively, guanidinium-mediated reliance on intracellular sources of tryptophan could be the result of mis-localization of Tat2, which migrates through defined lipid environments between the endoplasmic reticulum, secretory vesicles and plasma membrane.27–29 Given their lipidic character, 1 and 2 may compromise the ability of yeast to grow in limiting amounts of tryptophan present in YEPD media by disrupting membrane features linked to Tat2 trafficking. In support of this model, impaired tryptophan import has previously been reported to be a consequence of a mis-localization of the Tat2 transporter protein.29,30 While direct confirmation of this hypothesis requires testing with cell biological methods, several features in the chemical genetic interaction profiles we have generated strongly suggest that alkylated guanidinium compounds affect general membrane lipid homeostasis.First, cdc50 and drs2 mutants were the two most sensitive strains to compound 1 with growth rates less than 10% of wild type yeast (See Table S1†). The products of these genes interact to form the vacuolar cdc50/drs2 complex with endosomal phospholipid flippase activity.31 The Lem3 gene product, a hit in the chemical genetic screen of 2, has a similar flippase function.31,32 Thus, the general ability to regulate membrane leaflet composition is essential in the presence of guanidinium compounds.
Second, erg4 and erg5 strains are hypersensitive to 2. These mutants are deficient in the final steps of ergosterol biosynthesis and are known to be sensitive to several drugs.31–33 While ergosterol is required for Tat2 localization and tryptophan uptake27,30 the sensitivity of erg4 yeast to 2 is not rescued by excess tryptophan (Fig. 3B); in other words erg4 mutants are not hypersensitive to 2 because of reduced tryptophan supply. Instead, the inability to generate ergosterol, and possibly ergosterol-dependent membranes and vesicles, renders yeast unable to tolerate exogenous lipid-like guanidinium compounds. We conclude that the effects of 2 (and likely 1) on yeast affects the activity of multiple membrane locales and by extension, membrane-localized proteins. This includes Tat2, which must be particularly critical under the conditions of our screen.
The third piece of evidence for 1 and 2 affecting general lipid homeostasis is that, while they both genetically interact with the ARO–TRP pathway there are clearly many non-overlapping chemical genetic signatures of each compound (Fig. 2C, and Tables S1 and S2†). Neither compound directly resembles a phospholipid but compound 1, with the guanidinium bearing two dissimilar alkyl groups is more reminiscent of a sphingolipid than 2 which bears not only two similar alkyl groups but an additional polar substituent. Compound 1 appears to elicit a larger chemical-genetic effect than 2, possibly by disrupting a broader spectrum of membrane structures in vivo due a more lipid-like shape. This may explain the stress response/transcription signature of 1: the large number of transcription factors in this hypersensitive mutant list includes members of the SAGA/SLIK (NGG1 ADA2 GCN5 SPT20) and SRB10-mediator (SRB8 SSN8 SSN3) complexes that are known to regulate expression of stress response genes.34–37 By contrast, the chemical-genetic signature of 2 has a much narrower gene interaction signature that focuses on vacuolar function, particularly vacuolar acidification (Fig. 2C and Table S2†). We conclude that 2 may disrupt fewer membrane structures and preferentially target vacuolar membranes, or their acidification. Regardless, the ability to generate acidified vacuoles is essential in the presence of 2.
Conclusions
The foregoing findings establish a number of key features of the microbiocidal activity of compounds 1 and 2. Firstly, these compounds affect specific biochemical pathways and are therefore unlike quaternary ammonium and alkyl pyridinium compounds which act as indiscriminate membrane-disrupting detergents. In keeping with the lipidic character of the compounds, disruption of membrane-related pathways is not surprising, but the specificity is of particular note. The hits in common between the two compounds highlight the common effect of 1 and 2 on a tryptophan uptake pathway via Tat2. The hits unique to each compound are also membrane-related; vacuolar and liposomal acidification and function for 2 and a more deep-seated chemical stress response for 1. The similarities and the differences found will now direct attention to a more focused approach to probing structure–function relationships in this class of compounds.Experimental
Synthesis
Compounds prepared by this method:
N-Butyl-N′-decylurea from decylamine (3.9 g, 5.0 ml, 25 mmol) and butyl isothiocyanate (2.9 g, 3.0 ml, 25 mmol) in 12 ml toluene to yield 6.8 g product (quantitative) spectroscopically identical to previously reported.13
N-Hexyl-N′-octylurea from octylamine (3.2 g, 4.1 ml, 25 mmol) and hexyl isothiocyanate (3.6 g, 3.8 ml, 25 mmol) in 12 ml toluene to yield 6.8 g product (quantitative) spectroscopically identical to previously reported.14
Yeast assays
:4500 dilution of an overnight culture of BY4741. Plates were incubated 2 days at 30 °C and photographed.
Acknowledgements
The discovery and development of polyalkyl guanidinium biocides was achieved through the efforts of Christina Pinelli, Dean Amantea, Ryan Bonfield, Avril Brett, Sarah Mooi, and especially Bob Rowe over many years. The sustained support of the Natural Sciences and Engineering and Research Council of Canada (NSERC) is gratefully acknowledged. Research in the CJN lab is support by operating grants from NSERC and the Canadian Cancer Society Research Institute (CCSRI).Notes and references
- A. Schmidt, M. N. Hall and A. Koller, Mol. Cell. Biol., 1994, 14, 6597–6606 CAS
.
- M. Wahl, Mar. Ecol.: Prog. Ser., 1989, 58, 175–189 CrossRef
- P.-Y. Qian, Y. Xu and N. Fusetani, Biofouling, 2010, 26, 223–234 CrossRef CAS
- R. L. Townsin, Biofouling, 2003, 19, 9–15 CrossRef
- M. P. Schultz, J. A. Bendick, E. R. Holm and W. M. Hertel, Biofouling, 2011, 27, 87–98 CrossRef CAS
- I. Omae, Appl. Organomet. Chem., 2003, 17, 81–103 CrossRef CAS
- K. V. Thomas and S. Brooks, Biofouling, 2010, 26, 73–88 CrossRef CAS
- A. J. Scardino and R. de Nys, Biofouling, 2011, 27, 73–86 CrossRef CAS
- S. Dobretsov, M. Teplitski, M. Bayer, S. Gunasekera, P. Proksch and V. J. Paul, Biofouling, 2011, 27, 893–905 CrossRef CAS
- S. Dobretsov, M. Teplitski and V. J. Paul, Biofouling, 2009, 25, 413–427 CrossRef CAS
- T. M. Fyles, J.-J. Lee, R. D. Rowe and G. D. Robertson, J. Membr. Sci., 2008, 321, 31–36 CrossRef CAS
-
R. D. Rowe, T. M. Fyles and G. D. Robertson, Ion exchange membrane for dissolved gas sensor, U. S. Patent, 6,391,174 B1, 2002 Search PubMed
-
T. M. Fyles and R. D. Rowe, Microbiocidal properties of poly-substituted guanidinium salts, U. S. Patent, 6,518,309 B1, 2003 Search PubMed
-
T. M. Fyles, Antifouling polymeric agent for marine applications, U. S. Patent, 8,076,390 B2, 2011 Search PubMed
- M. J. Smith, G. Adma, H. J. Duncan and M. J. Cowling, Estuarine, Coastal Shelf Sci., 2002, 55, 361–367 CrossRef CAS
- H. Sutterlin, R. Alexy and K. Kummerer, Ecotoxicol. Environ. Saf., 2008, 71, 498–505 CrossRef CAS
-
W. Hugo and G. Snow, Biochemistry of antibacterial action, Chapman and Hall Ltd., London, 1981 Search PubMed
- G. McDonnell and A. D. Russell, Clin. Microbiol. Rev., 1999, 12, 147–179 CAS
- J.-Y. Maillard, J. Appl. Microbiol. Symp. Suppl., 2002, 92, 16S–27S CrossRef
- J. P. S. Cabral, Can. J. Microbiol., 1992, 38, 115–123 CrossRef CAS
- A. B. Parsons, A. Lopez, I. E. Givoni, D. E. Williams, C. A. Gray, J. Porter, G. Chua, R. Sopko, R. L. Brost, J. Wang, T. Ketala, C. Brenner, J. A. Brill, G. E. Fernandez, T. C. Lorenz, G. S. Payne, S. Isihihara, Y. Ohya, B. Andrews, T. R. Hughes, B. J. Frey, T. R. Graham, R. J. Andresen and C. Boone, Cell, 2006, 126, 611–625 CrossRef CAS
- B. Dietrich, T. M. Fyles, J. M. Lehn, L. G. Pease and D. L. Fyles, J. Chem. Soc., Chem. Commun., 1978, 934–936 RSC
- A. Lopez, A. B. Parsons, C. Nislow, G. Giaver and C. Boone, Prog. Drug Res., 2008, 66, 237–271 Search PubMed
- G. Giaever, A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis and M. Johnston, Nature, 2002, 418, 387–391 CrossRef CAS
- C. Khozoie, R. J. Pleass and S. V. Avery, J. Biol. Chem., 2009, 284, 17968–17974 CrossRef CAS
- M. Liu, W. S. Brusilow and R. Needleman, Curr. Genet., 2004, 46, 256–268 CrossRef CAS
- K. Umebayashi and A. Nakano, J. Cell Biol., 2003, 161, 1117–1131 CrossRef CAS
- M. Okamoto, T. Yoko-o, M. Umemura, K. Nakayama and Y. Jigami, J. Biol. Chem., 2006, 281, 4013–4023 CrossRef CAS
- K. Daicho, N. Makino, T. Hiraki, M. Ueno, M. Uritani, F. Abe and T. Ushimaru, FEMS Microbiol. Lett., 2009, 298, 218–227 CrossRef CAS
- K. Daicho, H. Maruyama, A. Suzuki, M. Ueno, M. Uritani and T. Ushimaru, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 1681–1690 CrossRef CAS
- K. Saito, K. Fujimura-Kamada, N. Furuta, U. Kato, M. Umeda and K. Tanaka, Mol. Biol. Cell, 2004, 15, 3418–3432 CrossRef CAS
- P. K. Hanson, L. Malone, J. L. Birchmore and J. W. Nichols, J. Biol. Chem., 2003, 278, 36041–36050 CrossRef CAS
- B. A. Skaggs, J. F. Alexander, C. A. Pierson, K. S. Schweitzer, K. T. Chun, C. Koegel, R. Barbuch and M. Bard, Gene, 1996, 169, 105–109 CrossRef CAS
- K. L. Huisinga and B. F. Pugh, Mol. Cell, 2004, 13, 573–585 CrossRef CAS
- F. C. Holstege, E. G. Jennings, J. J. Wyrick, T. I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander and R. A. Young, Cell, 1998, 95, 717–728 CrossRef CAS
- Y. Chi, M. J. Huddleston, X. Zhang, R. A. Young, R. S. Annan, S. A. Carr and R. J. Deshaies, Genes Dev., 2001, 15, 1078–1092 CrossRef CAS
- C. Nelson, S. Goto, K. Lund, W. Hung and I. Sadowski, Nature, 2003, 421, 187–190 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Excel spreadsheet of all chemical-genetic interactions of compounds in yeast and gene-ontology analysis. See DOI: 10.1039/c3ob40593a |
| This journal is © The Royal Society of Chemistry 2013 |