 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum catalysed hydrosilylation of propargylic alcohols†
Catherine A.
McAdam
a,
Mark G.
McLaughlin
a,
Adam J. S.
Johnston
a,
Jun
Chen
a,
Magnus W.
Walter
b and
Matthew J.
Cook
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, Northern Ireland, UK. E-mail: m.cook@qub.ac.uk; Fax: +44 (0) 28 90976524; Tel: +44 (0) 28 90974682
bEli Lilly and Company Limited, Erl Wood Manor, Windlesham, Surrey GU20 6PH, UK
First published on 10th May 2013
Abstract
A facile and user-friendly protocol has been developed for the selective synthesis of E-vinyl silanes derived from propargylic alcohols using a PtCl2/XPhos catalyst system. The reaction is generally high yielding and provides a single regioisomer at the β-position with E-alkene geometry. The reaction is extremely tolerant of functionality and has a wide scope of reactivity both in terms of alkynes and silanes used. The catalyst loading has been investigated and it is found that good reactivity is observed at extremely low catalyst loadings. This methodology has also been extended to a one-pot hydrosilylation Denmark–Hiyama coupling.
Introduction
Vinyl silanes are useful intermediates which can undergo a wide range of chemical transformations.1 The use of silanes as a protecting group for oxygen functionalities is, of course, well documented, and much interest has focused on their use in low cost, non-toxic routes to the formation of carbon–carbon bonds. These include Sakurai allylations,2 fluoride mediated Hiyama cross-coupling,3 and the fluorine-free Denmark–Hiyama cross-coupling variation4 and a number of copper mediated oxygen–carbon silicon migrations have been reported in the last decade.5 They have also found use in the formation of carbon–oxygen bonds through Tamao–Fleming oxidation6 and the development of an epoxidation–oxidation reaction sequence.7,8There are many methods for the preparation of vinyl silanes.9 The most widely used of these methods centre around the use of either hydrosilylation or other silylmetallation reactions. The majority of existing methods do not tolerate the incorporation of further functionality in either the substrate or the silane used, thus severely limiting their utility in synthesis. Such methods include the use of silylcuprates, developed by Fleming and later modified by Lipshutz, with each isomer of the vinyl silane being formed selectively by altering the nature of the Cu(I) species involved.10–13 Organochromium species have proven to be of great use in the formation of vinyl silanes from aldehydes, which react selectively over ketones.14–17 Further molecular complexity of these substrates can be achieved through Mizoroki–Heck type cross-coupling reactions18 and Peterson olefinations.19
The hydrosilylation of alkynes is a powerful method of forming vinyl silanes in a stereocontrolled manner.9,20 There are many catalysts that promote this reaction and late transition metal catalysts have been shown to be especially efficient.21,22 There are two regioisomeric products alongside E/Z isomers available in this reaction. In the case of propargylic alcohols the two regioisomers are referred to as the α and the β-isomers where the silyl group is located α and β to the alcohol and affords 1,1 or 1,2-substituted vinyl silanes, respectively (eqn (1)).
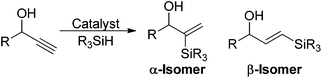 | (1) |
Ruthenium complexes are especially efficient at producing the internal α-isomer with high selectivity. Trost and co-workers have investigated this reaction extensively, firstly using a cyclopentadienyl ruthenium complex to perform the desired hydrosilylation with moderate stereoselectivity.22 Further investigation led to the development of a highly active and selective cationic catalyst, [Cp*Ru(MeCN)3]PF6, which gives the α-vinylsilane in a >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.23 The Lewis acid catalysed hydrosilylation of a range of alkynes with diverse functionality, as developed by Yamamoto,24–27 also affords the α-isomer as a single product, although this method is still in its infancy.
:1 ratio.23 The Lewis acid catalysed hydrosilylation of a range of alkynes with diverse functionality, as developed by Yamamoto,24–27 also affords the α-isomer as a single product, although this method is still in its infancy.
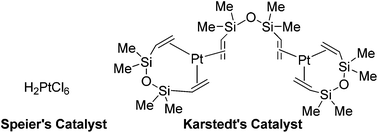
The majority of transition metal catalysed hydrosilylation reactions give the β-isomer as the major product.28,29 Metals such as rhodium,30–35 ruthenium36–38 and titanium39 have proven useful. Platinum complexes are by far the most well developed catalysts for the hydrosilylation of alkynes, affording the β-isomer, in the E-geometry as the major product.21,40 Complexes such as Speier's catalyst [H2PtCl6] and Karstedt's catalyst [Pt2(dvs)3] (dvs = 1,3-divinyl-1,1,3,3-tetramethyldisiloxane) can produce high catalyst turnovers; however, both the regio and E/Z selectivity can be poor.41–43
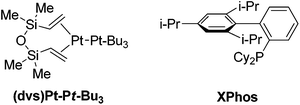
We required a rapid and reliable method for the synthesis of β-silyl alyllic alcohols that was tolerant of significant steric encumbrance.55 Panek previously disclosed the use of the highly reactive (dvs)Pt-Pt-Bu3 catalyst for the regioselective hydrosilylation of propargylic alcohols;56,57 however, no examples of sterically congested or tertiary propargylic alcohols were reported. We speculated that we could modify the reaction reported by Alami to combine the catalytic activity of the bulky Pt(0) catalyst with the selectivity and operational ease of the air stable Pt(II) system. We discovered that the PtCl2/XPhos catalyst system was very efficient for propargylic alcohols vida infra.58 Under these conditions the catalyst loading could be lowered to 1 mol% and 0.5 mol% platinum(II) chloride for the secondary and tertiary propargylic alcohols, respectively.
Herein we report a full investigation into the scope and limits of these hydrosilylation reactions including the use of highly functionalised substrates.
Results and discussion
Optimisation of Pt catalysed hydrosilylation
We began investigating the hydrosilylation of commercially available but-3-yn-2-ol (Table 1). Using 5 mol% PtCl2 and 10 mol% XPhos the reaction afforded the vinyl silane as a single regio- and geometric isomer in excellent yield (entry 1).
When the reaction was carried out in the absence of ligand, the regioselectivity fell to 9:1 (entry 2). Further optimisation of the transformation led to a reduction of the amount of catalyst needed, and at 1 mol% PtCl2 excellent yields were still being obtained (entry 4). If the catalyst loading is dropped further, the reaction becomes sluggish and lower yields are obtained (entry 5). The amount of silane can also be reduced, but once again a noticeable slowing of the reaction is observed (entry 6).
Hydrosilylation of secondary propargylic alcohols
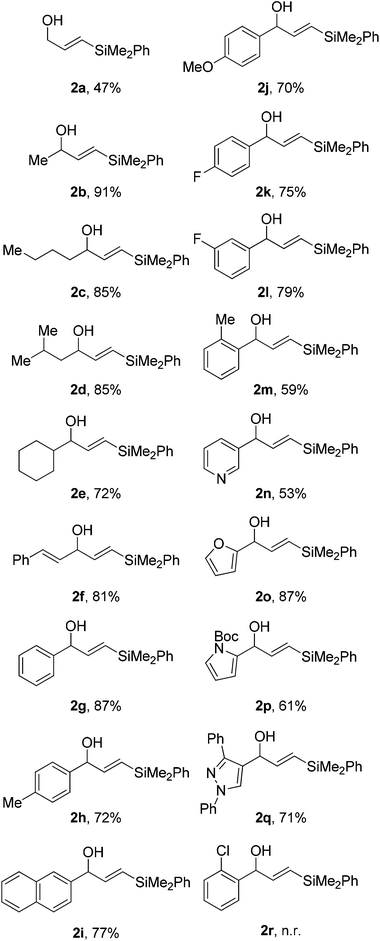
With the optimised reaction conditions in hand, we began by examining the scope of the reaction using secondary propargyl alcohols (Table 2). We soon discovered that propargyl alcohol itself was a very poor substrate for this reaction with only 47% yield of the vinyl silane 2a being produced. Contrary to this, a wide range of alkyl substituents were well tolerated, including linear 2b–c and branched groups 2d–e all providing good yields of the corresponding hydrosilylated product. Our attention then turned to vinyl and aryl substituents. A styrenyl derivative 2f was well tolerated as were many aromatic groups 2g–l even including aryl groups bearing ortho-substituents 2m that are tolerated albeit in a reduced yield. We also examined heterocycles 2n–q, especially those containing basic nitrogen atoms to test whether these would inhibit the catalyst. Gratifyingly, these were all good substrates providing the requisite vinyl silane in moderate to high yields in all cases. In general these heterocyclic substrates afforded slightly lower yields and required prolonged reaction times compared to their less functionalised counterparts. When the hydrosilylation was attempted using the ortho-chloroaryl derivative 1r, no reaction was observed. Both increasing the reaction time and the catalyst loading did not afford any product, with starting material recovered in each case. This class of substrate is the only one we have found so far to be ineffective for this reaction.|
|
|---|
|

Hydrosilylation of tertiary propargylic alcohols
We next turned our attention to tertiary propargylic alcohols as prior to our original communication the hydrosilylation of hindered propargylic alcohols was not widely reported except in the presence of non-commercial specialist ligands.59 We found that the PtCl2/XPhos system was especially efficient for the hydrosilylation of tertiary propargylic alcohols and that 0.5 mol% of PtCl2 could be used to give excellent yields. We have further expanded the scope of the reaction and found that it is tolerant of a wide range of functional groups (Table 3).|
|
|---|

|

We began by examining simple alkyl substituents 4a–c and found that these underwent the hydrosilylation reaction in excellent yields. Similarly, aryl substituents, both with one and two aryl groups 4d–f, were tolerated. Fluoroalkyl groups could be included in 4g as could heterocycles, with thiophene 4h substituents easily incorporated. Our attention then turned to cyclic tertiary propargylic alcohols. A wide range of cyclic alkyl substituents were addressed in this reaction, all of which proceeded in good to excellent yields 4i–n. Substituents on these carbocycles were also tolerated with a difluoromethylene and ketal groups 4o–p, alongside fused aromatics 4q–r being installed with little effect on the yields of the reaction. Finally we examined heterocyclic substrates, with nitrogen containing heterocycles performing well in this reaction, including azetidines 4s, piperidines 4t and even tropanes 4u to afford good yields of vinyl silanes, albeit with a substantial increase in the required reaction time. Chalcogen containing heterocycles 4v–w were also examined, with the reaction proceeding in a similar fashion to their carbocyclic analogues providing the corresponding vinyl silanes in good to excellent yields.
Catalyst loading studies
As the reaction is highly efficient for tertiary propargylic alcohols, using just 0.5 mol% catalyst loading, we examined the effect of further reducing the amount of catalyst and whether this would have a negative effect on the reaction (Table 4). Halving the catalyst loading to 0.25 mol% proved to have very little effect on the reaction and further reductions showed that this system still gave synthetically useful yields when dropped to as low as 0.05 mol%. Further reductions, to 0.025 and 0.01 mol%, were possible; however, the reaction slows significantly. A prolonged reaction time appears to counteract this to some degree, leading to the conclusion that the catalyst is stable for extended periods of time; hence, further reductions in catalyst loading could be possible if required. A TON of 6900 was achieved which shows the catalyst's stability and applicability. It is difficult to directly compare to other catalysts as our studies were performed using PhMe2SiH and most other hydrosilylation catalysts are tested against Et3SiH as a standard. In general, Et3SiH is a more reactive silane with TONs of around 70000 being achieved in certain cases.59 Our catalyst system does, however, benefit from excellent stability and functional group tolerance, making it a very practical method.

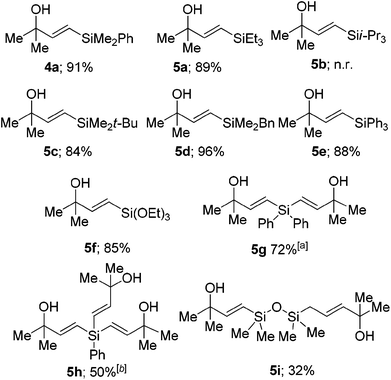
Use of other silanes
As all of the examples shown in Tables 1–4 utilised phenyldimethylsilane, we then began examining the use of other commercially available silanes. We found that the reaction is tolerant of a wide range of silanes, with the triethylsilyl 5a, tert-butyldimethylsilyl 5c, benzyldimethylsilyl 5d triphenylsilyl 5e and triethoxysilyl 5f derivatives formed in good yields. When tri-iso-propylsilane was used no reaction was observed with quantitative recovery of starting material. Presumably the increased steric bulk of the silane prevents the reaction from occurring. Interestingly when diphenyl silane and phenylsilane were used, two or three sequential hydrosilylations could be carried out on the same silicon atom, forming dimers and trimers of the propargylic alcohols (Table 5).60|
|
|---|
| a Using 0.5 equivalent of Ph2SiH2. b Using 0.3 equivalent of PhSiH3. |
|

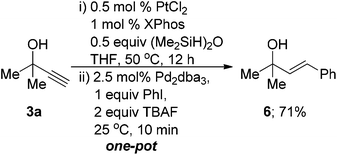
Our attention next turned to the hydrosilylation using bis(dimethylsiloxane) to allow subsequent Hiyama couplings. Unfortunately this afforded poor isolated yields, although there was complete conversion to the vinylsilane from analysis of the crude reaction mixture suggesting that the product 5h was unstable for silica gel chromatography. To circumvent this issue, a one-pot Denmark–Hiyama coupling was attempted.61 The hydrosilylation occurred in good conversion using the standard conditions and then simply cooling the reaction, adding TBAF followed by Pd2dba3 and iodobenzene afforded a very facile cross-coupling which gave 72% yield after just 10 minutes (eqn (2)).
 | (2) |
Internal alkynes
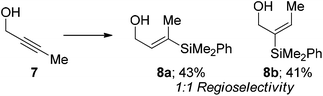
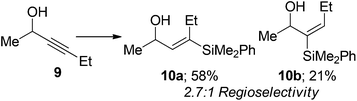
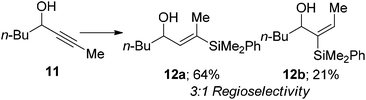
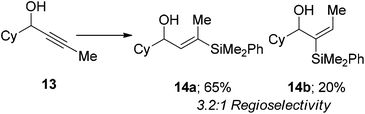
We also examined internal alkynes to investigate whether the Lewis basic oxygen atom was co-ordinating to the metal and directing the addition62 or whether the regioselectivity was based purely on steric factors. The first substrate attempted was but-2-yn-1-ol 7, which gave a 1:1 mixture of regioisomers (eqn (3)). This clearly suggests that the oxygen is not directing the addition and that pure sterics govern the regioselectivity. This also demonstrates that primary propargylic alcohols can participate well in this reaction when extra substitution is present at the β-position as an overall yield of 84% was obtained. When steric bulk is increased in the propargylic position the ratio increases to give moderate β selectivity. Alkyl groups give around 3:1 selectivity with methyl 9 affording 2.7:1 β:α, butyl 11 3.2:1 and when a cyclohexyl group 13 is introduced a β-selectivity of 3.2:1 was observed (eqn (4)–(6)). This is in line with what one would expect based on the steric environment of the alkyne.
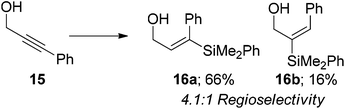
When the terminal alkyne position of propargyl alcohol was substituted with a phenyl group 15, modest selectivity was observed with the β-isomer 16a prevailing in a 4.1:1 ratio (eqn (7)). This is contrary to the steric environment around the alkyne and is clearly showing preference for hydride attack at the more electrophilic terminus of the alkyne.23,47,63,64 Indeed, Alami observed this effect when applied to the palladium catalyzed hydrostannylation of 13, with 4:1 regioselectivity for β-substitution being observed.65 They also showed that the regioselectivity in these systems directly correlated to the Hammett σ-value for substituted benzene rings. When strong electron withdrawing groups were present in the para-position of the benzene ring a single β-regioisomer was obtained whereas electron releasing groups produce selectivities of the order of a 3:1 β:α ratio.
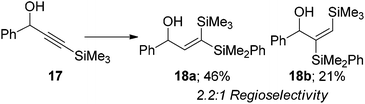
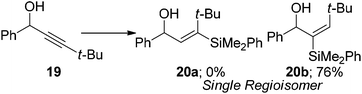
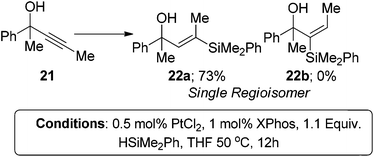
Similarly when a trimethylsilyl group is installed at the β-position 17, the product ratio is contrary to the steric environment (eqn (8)). The mild polarisation of the alkyne group caused by the silane is enough to override the significant steric bias to afford the β-regioisomer 18a as the major product. If the steric environment at the β-position is increased greatly by installing a tert-butyl group 19, the regioselectivity of the reaction can be switched to give solely the α-isomer. No traces of the β-isomer 20a were observed with only the α-isomer 20b being formed in good yield (eqn (9)). A single regioisomer could again be obtained when the propargylic position was highly substituted compared to the β-position 21. In this instance only the β-isomer 20a was observed again in good yield (eqn (10)). The results in eqn (9) and (10) demonstrate that the steric environment around the alkyne plays a pivotal role in determining the selectivity of the products. However, when the alkyne is polarised and an electronic bias is present, such as eqn (7) and (8), the regiochemistry will be dictated by the attack of the hydridic hydrogen at the more electron deficient terminus.
 | (3) |
 | (4) |
 | (5) |
 | (6) |
 | (7) |
 | (8) |
 | (9) |
 | (10) |
Similarly, when a trimethylsilyl substituted alkyne was used the hydrosilylation gave a 2.2:1 mixture of β:α isomers. The reaction proceeds to give a differentially substituted 1,1-vinyldisilane. Again the polarising effect of the silane can rationalise the regiochemical outcome as the β position is more electrophilic than the α.
Mechanistic insights
Fig. 1 highlights our model for regioselectivity. In the cases where there is a steric bias, the large group orientates itself away from the bulky XPhos ligand. The result of this is the selective formation of a product with the hydrogen adjacent to the large group and the silyl group next to the smaller group. In the case of the electronically biased substrates, the phenyl or silyl group creates a polarised alkyne and the hydridic hydrogen on the platinum adds at the more electrophilic terminus thus resulting in the hydrogen adjacent to the less electron withdrawing group and the silyl group next to the electron withdrawing substituent. This polarisation appears to be significant enough to override the inherent steric bias for one product over the other.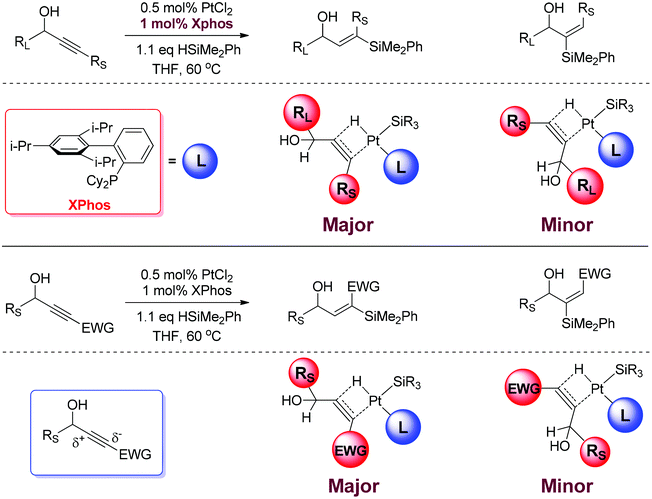 | ||
| Fig. 1 Origin of regioselectivity. | ||
We conducted a competition experiment between propargyl alcohol 1a, but-3-yn-2-ol 1b, and 2-methyl but-3-yn-2-ol 3a where the reaction was conducted with one equivalent of each of the substrates and one equivalent of PhMe2SiH to ascertain the relative rate of reactions. As expected the tertiary alcohol reacted faster than the secondary which in turn was faster than the primary in a krel of 2.7:1.7:1. This proves that the enhanced reactivity observed for tertiary propargylic alcohols is due to an inherently faster reaction rate rather than factors such as product inhibition or catalyst decomposition (Scheme 1).
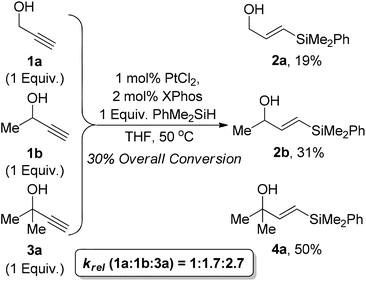 | ||
| Scheme 1 Primary/secondary/tertiary competition experiment. | ||
We next examined whether the incorporation of deuterium at the terminal position of the alkyne led to an observable kinetic isotope effect. Alkyne 23 was readily prepared and was subjected to competition experiment with its non-deuterated analogue. Following conversion to 49%, the product 4k/24 was protioenriched alongside the recovered starting material being deuteroenriched in the same ratio. Both recovered starting material and product conversion gave a kH/kD value of 1.44. This suggests a large secondary kinetic isotope effect consistent with a change in geometry from sp to sp2 during the rate limiting step (Scheme 2).66
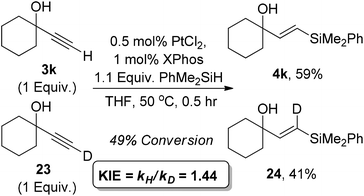 | ||
| Scheme 2 Alkyne kinetic isotope effect. | ||
We observed significant evolution of gas, presumably hydrogen, upon addition of the silane to the reaction mixture. This led us to examine the origin of this gas. There are several possible explanations including: the simple reduction of the Pt(II) salt to Pt(0) producing H2 and a disilane or the OH bond reacting directly with the catalyst producing a Pt(OR) type complex. To probe the effect the O–H bond may have on the reactivity of this reaction a series of experiments were conducted. Firstly we performed a control experiment with benzyl alcohol to determine whether any O-silylation was observed during the reaction (eqn (11)). No reaction or O-silylation was observed with quantitative recovery of the starting material being achieved.
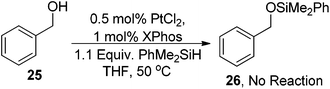 | (11) |
We next investigated the use of the methyl ether 27 and found that the hydrosilylation occurred in a similar overall yield to the hydroxyl analogue 28 (eqn (12)). This suggests that the OH is not required for reactivity and to obtain high yields.
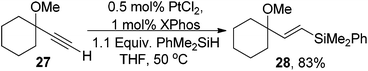 | (12) |
A competition experiment was conducted between the OH and OMe compounds to elucidate the relative rates of the reactions. The OH compound 3k reacted much faster than the OMe analogue 27 with a krel of 2.3 being observed, suggesting that the OH does accelerate the reaction (Scheme 3).
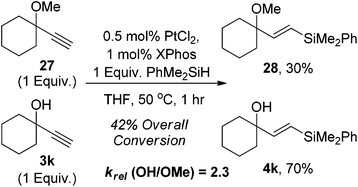 | ||
| Scheme 3 Role of the OH bond. | ||
To further probe this effect, we prepared the O-deuterated analogue and subjected this to the reaction conditions. Again the reaction proceeded with a similar overall yield and with no deuterium incorporated into the resultant alkene (eqn (13)). This suggests that the formation of Pt(OR) complexes is unlikely as deuterium would also be delivered to the platinum catalyst which would in turn be observed in the vinyl silane product.
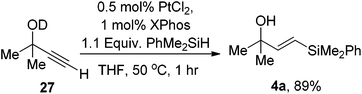 | (13) |
Finally, we examined whether there was a OH/OD kinetic isotope effect which would elucidate the true role of the alcohol moiety. As this is an exchangeable deuterium measuring the product or recovered starting material, deuterium enrichment would be difficult. We therefore took two alkynes, the dimethyl substrate 3a and the cyclohexyl derivative 3k. We first of all performed a control experiment where we measured the relative rate of reactions between the two compounds. The cyclohexyl analogue was found to have a faster rate of reaction with a krel (3k:3a) of 1.24 and this was used as a correction factor in the kinetic isotope effect calculations. When the reaction was performed using equimolar amounts of 3k and d1-3a, which was formed in 86% isotopic purity,67 the order of reactivity was reversed with the dimethyl compound reacting at a faster rate. As the alcohol was only deuterated to 86% we had to adjust this value to 100% isotopic purity assuming a linear relationship and found a krel (3k:d1-3a) of 0.86. This was then used to measure the kinetic isotope effect which equated to 0.69 (Scheme 4).
An inverse kinetic isotope effect could be due to several factors, including a pre-equilibrium; however, the absence of deuterium in the vinyl silane and the rapid evolution of hydrogen gas make this unlikely. A more likely scenario is the OH moiety hydrogen bonding to the hydridic hydrogen bound to the platinum centre and accelerating the hydrometallation step through mild acid catalysis. Indeed, this is consistent with the hydrometallation step being rate limiting. This can be seen in Fig. 2 where the OD/OH group can H-bond to the hydridic hydrogen, thus weakening the Pt–H bond and accelerating the reaction. This interaction does explain the rate acceleration; however, one would predict some regioselectivity based on this, which is not seen in the case of internal alkynes. Either this chelate is not possible in the internal cases or this occurs in an intermolecular fashion.
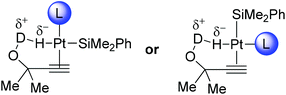 | ||
| Fig. 2 Possible H-bond acceleration of hydrometallation. | ||
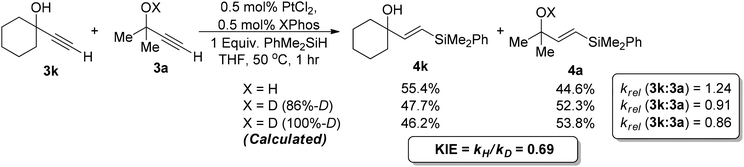 | ||
| Scheme 4 OH kinetic isotope effects. | ||
More sterically hindered substrates react much faster in this reaction and we sought to rationalise this mechanistically. In the standard Chalk–Harrod mechanism28 for hydrosilylation of alkynes, there are two events that can occur to lead to product (Fig. 3): either oxidative addition of the silane followed by alkyne coordination (Pathway 1) or alkyne coordination followed by oxidative addition (Pathway 2). Pathway 1 would favour relatively small alkynes as the coordination event occurs on a sterically congested Pt(II) species C. These types of substrates are excellent with Speier's catalyst and Karstedt's catalyst.
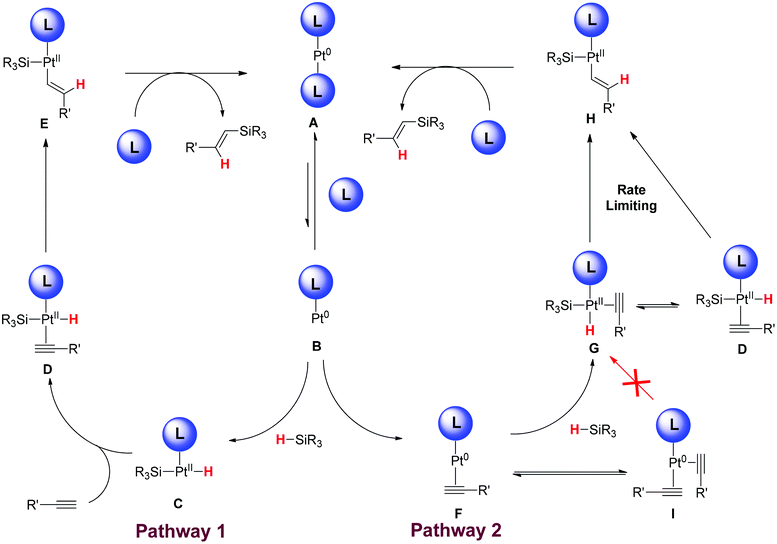 | ||
| Fig. 3 Proposed catalytic cycle. | ||
Our results suggest that Pathway 2 is operating as the major pathway as more hindered substrates are superior. A large alkyne can bind to the coordinatively unsaturated Pt(0) species B thus stabilising this prior to oxidative addition. Alternatively, if a small alkyne is bound to the coordinately unsaturated platinum–ligand complex F, a second alkyne can then bind, thus removing the coordination sites needed for the oxidative addition reaction. The result is that the catalyst is removed from the productive cycle I and the rate of reaction is based on the decomplexation of the second alkyne. When a bulky alkyne is used the complexation of two substrates is disfavoured, thus leaving the catalytically active species available for subsequent oxidative addition to form G, migratory insertion H and reductive elimination. This is very clearly demonstrated in the case of propargyl alcohol 1a.
Our experiments suggest that the migratory insertion event is rate limiting with a large sp–sp2 secondary kinetic isotope effect being observed. This could occur from G or an isomerisation event to D could occur due to the large trans-effect of both silanes and hydrides. Both complexes would give a similar result in terms of kinetic isotope effects and therefore cannot be distinguished.
It is also possible that the silane can react directly with F in a concerted manner to proceed directly to H with the same factors that would govern reactivity as in the two-step complexation migratory insertion process.
Conclusions
We have shown the effectiveness and robustness of PtCl2/XPhos catalysed hydrosilylation. Both secondary and tertiary propargyl alcohols perform well as do internal alkynes. Furthermore, the catalyst system is tolerant of various silanes which improves the usefulness of this protocol. A one-pot Hiyama–Denmark cross-coupling reaction was also performed using the catalyst system which allows access to a range of functionalities. We have examined the use of internal alkynes and have found that this is governed by both steric and electronic factors.Experimental section
Compounds 2a–2g, 2o, 4a–4d, 4e, 4j–4k and 4v were fully characterised and reported previously.58 For the synthesis of the starting alkynes see the ESI.†General methods
All reactions were carried out under an atmosphere of argon in oven-dried glassware with magnetic stirring. All reactions were monitored by thin layer chromatography (TLC) using Merck TLC silica gel 60 sheets, which were visualised with ultraviolet light and then developed with iodine and basic potassium permanganate solution. Flash chromatography was performed on Sigma-Aldrich silica gel 60 as the stationary phase and the solvents employed were of analytical grade. 1H NMR spectra were recorded on a Bruker AVX400 (400 MHz) spectrometer at ambient temperature. Data are reported as follows: chemical shift in parts per million (δ, ppm) from deuterated chloroform (CDCl3) taken as 7.26 ppm, integration, multiplicity (s = singlet; d = doublet; t = triplet; dd = double doublets; m = multiplet), and coupling constant (Hz). 13C NMR spectra were recorded on a Bruker AVX400 (100 MHz) spectrometer. Chemical shifts are reported in ppm from CDCl3 taken as 77.0 ppm. Infrared spectra were recorded on a Perkin Elmer RX I FT-IR spectrometer as liquid films or as dilute solutions between two KBr discs. Mass spectra were recorded on either a Micromass GCT Premier or a Waters Micromass LCT Premier spectrometer using electron ionisation (EI) at 70 eV or electrospray (ES) techniques, respectively. Unless stated otherwise, all commercially available reagents were used as received. When necessary, commonly used organic solvents were dried prior to use according to standard laboratory practices.General procedure A: hydrosilylation secondary propargyl alcohols
To an oven dried 5 mL round bottom flask equipped with a reflux condenser and magnetic stirrer was added PtCl2 (1 mol%) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (2 mol%) (XPhos). The flask was then flushed quickly with argon and dry THF was added. The mixture was then stirred at 50 °C for 20 minutes until a yellow homogeneous mixture was obtained. The corresponding propargyl alcohol (1 eq.) was added followed by the silane (1.1 equivalents) via syringe (CAUTION: Rapid evolution of hydrogen gas) and the solution was stirred at 50 °C overnight. The solvent was evaporated and the crude mixture was applied to the top of a column and chromatographed to afford the requisite (E)-vinyl silane.:1 hexane–EtOAc) afforded 2h (1.38 g, 72%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.29; νmax (thin film)/cm−1; 3001, 3068, 2956, 1619, 1427, 248, 1114, 842, 822, 730, 699; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.48 (2H, m), 7.37–7.32 (3H, m), 7.25–7.21 (2H, m), 7.18–7.13 (2H, m), 6.28 (1H, m), 6.12 (1H, dt, J = 18.8, 1.28 Hz), 5.17 (1H, t, J = 4.3 Hz), 2.43 (3H, s), 1.96 (1H, m), 0.35 (3H, d, J = 2.0 Hz), 0.34 (3H, d, J = 2.0 Hz); 13C NMR (100 MHz, CDCl3) δ 149.0, 139.5, 138.4, 137.5, 133.7, 129.3, 129.0, 127.8, 127.2, 126.5, 76.5, 21.1, −2.65; HRMS (ES+) calcd for C18H22OSiNa [M + Na]+ 305.1330. Found 305.1338.
:1 hexane–EtOAc) afforded 2i (660.6 mg, 77%) as a colourless solid.
R
f (9:1 hexane–EtOAc) = 0.41; νmax (thin film)/cm−1; 3434, 1634, 1247, 1114, 730.8, 699.4; 1H NMR: (400 MHz, CDCl3) δ 7.88–7.83 (4H, m), 7.58–7.47 (5H, m), 7.40–7.36 (3H, m), 6.39 (1H, dd, J = 18.6, 5.0 Hz), 6.22 (1H, dd, J = 18.8, 1.5 Hz), 5.41 (1H, t, J = 4.0 Hz), 2.18 (1H, s), 0.39 (3H, s), 0.39 (3H, s); 13C NMR (100 MHz, CDCl3) δ 148.9, 139.8, 138.4, 133.9, 133.4, 133.1, 129.1, 128.5, 128.1, 128.0, 127.9, 127.7, 126.2, 126.0, 125.2, 124.5, 76.9, −2.6; HRMS (EI) calcd for C21H23OSi [M + H]+ 319.1518. Found 319.1513.
:1 hexane–EtOAc) afforded 2j (643 mg, 70%) as a clear oil.
R
f (9:1 hexane–EtOAc) = 0.29; νmax (thin film)/cm−1; 3412, 2955, 1612, 1512, 1247, 828; 1H NMR: (400 MHz, CDCl3) δ 7.55–7.50 (2H, m), 7.38–7.35 (3H, m), 7.31–7.27 (2H, m) 6.92–6.89 (2H, m), 6.30 (1H, dd, J = 18.6, 4.8 Hz), 6.14 (1H, dd, J = 18.8, 1.5 Hz), 5.19 (1H, t, J = 3.52 Hz), 3.82, (3H, s), 1.95 (1H, d, J = 4.0 Hz), 0.37 (3H, s) 0.37 (3H, s); 13C NMR (100 MHz, CDCl3) δ 159.2, 149.1, 138.4, 134.7, 133.8, 129.0, 127.9, 127.8, 114.0, 76.3, 55.3, −2.6; HRMS (EI) calcd for C18H21O2Si [M − H]+ 297.1311. Found 297.1321.
:1 hexane–EtOAc) afforded 2k (501 mg, 75%) as a clear oil.
R
f (9:1 hexane–EtOAc) = 0.30 νmax (thin film)/cm−1; 3350, 2956, 1604, 1508, 1223, 838; 1H NMR: (400 MHz, CDCl3) δ 7.55–7.50 (2H, m), 7.40–7.30 (5H, m), 7.09–7.03 (2H, m), 6.28 (1H, dd, J = 18.6, 4.8 Hz), 6.14 (1H, dd, J = 18.6, 1.3 Hz), 5.22 (1H, d, J = 4.5 Hz), 2.07 (1H, s), 0.39 (3H, s), 0.38 (3H, s) 13C NMR (100 MHz, CDCl3) δ 162.3 (d, 1JC–F = 244.3 Hz), 158.7, 138.2, 138.1 (d, 4JC–F = 3.3 Hz), 133.8, 129.1, 128.1 (d, 3JC–F = 8.3 Hz), 127.9, 127.8, 115.4 (d, 2JC–F = 21.1 Hz), 76.1, −2.7; HRMS (EI) calcd for C17H17SiF [M − H2O]+ 268.1084. Found 268.1103.
:1 hexane–EtOAc) afforded 2l (1.31 g, 79%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.33; νmax (thin film)/cm−1; 3391, 3069, 2957, 1613, 1591, 1486, 1449, 1428, 1249, 1115, 844, 699; 1H NMR: (400 MHz, CDCl3) δ 7.55–7.49 (2H, m), 7.43–7.30 (4H, m), 7.16–7.08 (2H, m) 7.02–6.96 (1H, m), 6.26 (1H, dd, J = 18.8, 5.0 Hz), 6.15 (1H, dd, J = 18.8, 1.2 Hz), 5.23 (1H, t, J = 5.08 Hz), 2.09 (1H, d, J = 4.0 Hz), 0.38 (3H, s), 0.38 (3H, s); 13C NMR (100 MHz, CDCl3) δ 163.0 (d, 1JC–F = 245.6 Hz), 148.2, 145.0 (d, 3JC–F = 6.6 Hz), 138.1, 133.8, 133.0, 130.0, (d, 3JC–F = 8.4 Hz), 129.1, 128.5, 127.8, 122.0, 121.9, 114.5 (d, 2JC–F = 21.1 Hz), 113.2 (d, 2JC–F = 21.9 Hz), 76.2 (d, 4JC–F = 1.8 Hz), −2.7; HRMS (ES+) calcd for C17H19OFNaSi [M + Na]+ 309.1087. Found 309.1085.
:1 hexane–EtOAc) afforded 2m (109 mg, 56%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.32; νmax (thin film)/cm−1; 3069, 3001, 2954, 1628, 1427, 1114, 842, 830, 738, 699; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.46 (2H, m), 7.41–7.32 (3H, m), 7.23–7.18 (2H, m), 7.17–7.13 (2H, m), 6.27 (1H, dd, J = 18.6, 4.8 Hz), 6.11 (1H, dd, J = 18.6, 4.8 Hz), 5.42 (1H, dt, J = 18.8, 1.28 Hz), 2.35 (3H, s), 0.35 (3H, s), 0.34 (3H, s); 13C NMR (100 MHz, CDCl3) δ 148.4, 134.7, 133.8, 133.0, 130.6, 130.3, 129.7, 128.9, 127.8, 126.6, 73.6, 72.9, 32.1, 19.2, 11.7, −2.6; HRMS (ES+) calcd for C18H22OSi [M]+ 282.1493. Found 282.1440.
:2 hexane–EtOAc) afforded 2n (271 mg, 53%) as a yellow oil.
R
f (9:1 hexane–EtOAc) = 0.35; νmax (thin film)/cm−1; 3290, 2853, 2114, 1581, 1427, 1248, 1041, 1028, 947, 823, 710, 634; 1H NMR: (400 MHz, CDCl3) δ 7.94–7.90 (2H, m), 7.72–7.69 (1H, m), 7.51–7.47 (2H, m), 7.37–7.30 (4H, m), 6.25 (1H, dd, J = 18.6, 4.8 Hz), 6.17 (1H, dd, J = 18.6, 1.1 Hz), 5.27 (1H, d, J = 4.8 Hz), 1.43 (1H, s), 0.35 (3H, s), 0.35 (3H, s) 13C NMR (100 MHz, CDCl3) δ 149.4, 149.2, 148.1, 134.7, 133.8, 129.2, 127.9, 123.6, 75.3, 75.4, 62.1, 61.9, −2.7, −2.7; HRMS (ES+) calcd for C16H19NOSi 269.1236. Found 269.1293.
:1 hexane–EtOAc) afforded 2r (297 mg, 61%) as a yellow oil.
R
f (9:1 hexane–EtOAc) = 0.49; νmax (thin film)/cm−1; 3466, 3068, 2979, 2900, 1724, 1617, 1478, 1427, 1408, 1371, 1342, 1248, 1229, 1162, 1124, 1064, 998, 884, 821, 728, 700; 1H NMR: (400 MHz, CDCl3) δ 7.53 (2H, m), 7.47–7.44 (2H, m), 7.35–7.30 (2H, m), 7.17 (1H, m), 6.40–6.34 (1H, dd, J = 18.6, 4.3 Hz), 6.13 (1H, d, J = 7.8 Hz), 6.10 (1H, d, J = 3 Hz), 6.03–5.99 (1H, m), 1.57 (9H, d, J = 11.5 Hz), 0.36 (6H, s); 13C NMR (100 MHz, CDCl3) δ 146.9, 136.2, 134.1, 128.9, 127.9, 127.6, 122.3, 114.7, 113.4, 110.3, 110.1, 84.8, 69.8, 69.0, 27.9, −2.3; HRMS (EI+) calcd for C16H16Si 236.1021. Found 236.1025.
:1 hexane–EtOAc) afforded 2q (290 mg, 71%) as a colourless oil.
R
f (9:1 hexane–ethyl acetate) = 0.26; IR: νmax (thin film)/cm−1; 3402, 2953, 1600, 1503, 1426, 1355, 1248, 1113, 1060, 820, 773, 756, 698; 1H NMR: (400 MHz, CDCl3) δ 7.87 (2H, d, J = 5.0 Hz), 7.83–7.80 (1H, m), 7.77–7.69 (2H, m), 7.61–7.57 (1H, m), 7.83–7.80 (1H, m), 7.50–7.27 (7H, m), 7.20 (2H, m), 6.37 (1H, dd, J = 18.5, 4.8 Hz), 6.18 (1H, dd, J = 18.5, 1.25 Hz), 5.42 (1H, s), 0.35 (6H, s); 13C NMR (100 MHz, CDCl3) δ 151.6, 148.1, 140.0, 139.9, 138.8, 133.8, 133.2, 133.0, 129.4, 129.2, 128.9, 128.6, 128.3, 127.8, 126.4, 125.7, 124.6, 119.0, 68.6, −2.5, −2.7; HRMS (ES) calcd for C26H27N2OSi [M + H]+ 256.1647. Found 411.1873.
General procedure B: hydrosilylation of disubstituted propargyl alcohols
To an oven dried 5 mL round bottom flask equipped with a reflux condenser and a magnetic stirrer was added PtCl2 (0.5 mol%) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (1 mol%) (XPhos). The flask was then flushed quickly with argon and dry THF was added. The mixture was then stirred at 50 °C for 20 minutes until a yellow homogeneous mixture was obtained. The corresponding propargyl alcohol (1 eq.) was added followed by the silane (1.1 eq.) via syringe (CAUTION: Rapid evolution of hydrogen gas) and the solution was stirred at 50 °C overnight. The solvent was evaporated and the crude mixture was applied to the top of a column and chromatographed to afford the requisite (E)-vinyl silane.:1 hexane–EtOAc) afforded 4e (154 mg, 83%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.39; νmax (thin film)/cm−1; 3479, 3068, 3023, 2960, 1685, 1446, 1427, 1254, 1118, 1048, 998, 832, 795, 729, 699, 469; 1H NMR: (400 MHz, CDCl3) δ 7.98 (1H, d, J = 7.0), 7.55–7.51 (2H, m), 7.50–7.46 (1H, m), 7.44 (2H, d, J = 7.8), 7.39–7.29 (4H, m), 6.53 (1H, dd, J = 18.6 Hz), 6.10 (1H, d, J = 18.8 Hz), 2.24–2.17 (1H, dq, J = 13, 6.8 Hz), 0.90 (6H, d, J = 6.8 Hz), 0.35 (3H, s) 13C NMR (100 MHz, CDCl3) δ 151.5, 144.7, 133.0, 132.7, 132.2, 128.4, 128.2, 127.2, 126.7, 125.2, 124.6, 79.2, 78.9, 16.3, 15.9, −3.2; HRMS (ES+) calcd for C20H26ONaSi [M + Na]+ 333.1651. Found 333.1573.
:1 hexane–EtOAc) afforded 4g (369 mg, 80%) as a yellow oil.
R
f (9:1 hexane–EtOAc) = 0.41; νmax (thin film)/cm−1; 3546, 3070, 2958, 1958, 18883, 1602, 1497, 1450, 1428, 1214, 11163, 1115, 997, 938, 844, 825, 761, 734, 699, 647; 1H NMR: (400 MHz, CDCl3) δ 7.56 (2H, d, J = 8.0 Hz), 7.50–7.46 (2H, m), 7.42–7.33 (6H, m), 6.63 (1H, d, J = 18.8 Hz), 6.42 (1H, d, J = 18.8 Hz), 0.38 (6H, s) 13C NMR (100 MHz, CDCl3) δ 143.3, 137.1, 133.8, 133.5, 131.6, 129.4, 128.7, 128.3, 128.0, 127.9, 126.7 (t, 2JC–F = 223 Hz), 7.4, −2.8, −2.8; HRMS (ES+) calcd for C18H18OF3Si [M − H+]+ 335.1079. Found 335.1081.
:1 hexane–EtOAc) afforded 4h (202 mg, 92%) as a yellow oil.
R
f (9:1 hexane–EtOAc) = 0.39; νmax (thin film)/cm−1; 3399, 3068, 2956, 1617, 1427, 1248, 1114, 991, 844, 827, 730. 698, 644; 1H NMR: (400 MHz, CDCl3) δ 7.52 (2H, dd, J = 5.5, 2.0 Hz), 7.40–7.32 (4H, m), 6.96–6.93 (2H, m), 6.42 (1H, d, J = 18.2 Hz), 6.13 (1H, d, J = 18.2 Hz), 1.72 (3H, s), 0.35 (6H, s) 13C NMR (100 MHz, CDCl3) δ 152.4, 138.3, 133.8, 133.6, 128.9, 127.8, 126.7, 124.7, 124.4, 123.3, 74.2, 30.1, 9.7, −2.6; HRMS (ES+) calcd for C16H21OSSi [M + H+]+ 289.1082. Found 289.1112.
:1 hexane–EtOAc) afforded 4i (287 mg, 88%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.40; νmax (thin film)/cm−1; 3329, 3068, 3049, 2984, 2956, 1616, 1427, 1248, 1169, 1114, 991, 844, 826, 729, 698; 1H NMR: (400 MHz, CDCl3) δ 7.54–7.47 (2H, m), 7.40–7.31 (3H, m), 6.38 (1H, d, J = 18.2 Hz), 5.98 (1H, d, J = 18.2 Hz), 1.72 (2H, s), 2.24–2.10 (1H, m), 2.00–1.92 (1H, m), 1.83–1.76 (1H, m), 1.65–1.52 (2H, m), 0.37 (6H, s) 13C NMR (100 MHz, CDCl3) δ 151.6, 138.7, 133.8, 133.0, 129.0, 127.9, 122.7, 36.1, −2.4, −3.2; HRMS (ES+) calcd for C14H20NaOSi [M + Na+]+ 255.3833. Found 255.3819.
:1 hexane–EtOAc) afforded 4l (562 mg, 79%) as a colourless oil.
R
f (9:1 hexane–ethyl acetate) = 0.48; IR: νmax (thin film)/cm−1; 3372, 3068, 2925, 1616, 1459, 1427, 1246, 1113, 1027, 993, 822, 731, 698; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.50 (2H, m), 7.38–7.34 (3H, m), 6.28 (1H, d, J = 20 Hz), 5.96 (1H, d, J = 20 Hz), 1.79–1.40 (12H, m), 0.36 (6H, s); 13C NMR (100 MHz, CDCl3) δ 158.3, 136.3, 131.4, 130.2, 123.9, 43.5, 33.4, 32.3, 31.9, 24.9, 24.7, −2.5; HRMS (ES) calcd for C17H26OSiNa [M + Na]+ 297.1651. Found 297.1620.
:1 hexane–EtOAc) afforded 4m (303 mg, 80%) as a colourless oil.
R
f (9:1 hexane–ethyl acetate) = 0.41; IR: νmax (thin film)/cm−1; 3362, 3068, 2922, 2854, 1613, 1470, 1446, 1427, 1264, 1246, 1113, 1064, 994, 843, 828, 733, 698, 642; 1H NMR: (400 MHz, CDCl3) δ 7.52–7.49 (2H, m), 7.36–7.33 (3H, m), 6.27 (1H, d, J = 18.8 Hz), 5.97 (1H, d, J = 18.8 Hz), 1.80–1.60 (9H, m), 1.57–1.41 (5H, m), 0.34 (6H, s); 13C NMR (100 MHz, CDCl3) δ 157.6, 141.4, 136.2, 135.4, 131.3, 130.2, 125.4, 78.5, 38.7, 30.7, 27.1, 24.5, 2.4, −0.27, −0.71; HRMS (ES) calcd for C8H28OSiNa [M + Na]+ 311.1807. Found 311.1787.
:1 hexane–EtOAc) afforded 4n (285 mg, 91%) as a yellow oil.
R
f (9:1 hexane–ethyl acetate) = 0.46; IR: νmax (thin film)/cm−1; 3364, 3070, 2940, 2848, 1615, 1469, 1427, 1246, 1114, 994, 844, 823, 730, 698; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.49 (2H, m), 7.36–7.34 (3H, m), 6.24 (1H, d, J = 18.8 Hz), 5.97 (1H, d, J = 18.8 Hz), 1.66–1.58 (2H, m), 1.53–1.42 (5H, m), 1.40–1.32 (18H, m), 0.34 (6H, s); 13C NMR (100 MHz, CDCl3) δ 157.0, 141.3, 136.3, 131.3, 130.1, 125.4, 78.7, 37.1, 28.9, 28.4, 25.0, 24.6, 22.1, 2.5, 0.3, −0.3; HRMS (ES) calcd for C22H35OSi [M − H+]+ 343.2457. Found 343.2454.
:1 hexane–EtOAc) afforded 4o (136 mg, 76%) as a colourless oil.
R
f (9:1 hexane–ethyl acetate) = 0.43; IR: νmax (thin film)/cm−1; 3419, 3069, 2953, 1700, 1618, 1443, 1427, 1377, 1358, 1250, 1117, 1113, 991, 844, 825, 721, 700, 667; 1H NMR: (400 MHz, CDCl3) δ 7.51–7.48 (2H, m), 7.40–7.33 (3H, m), 6.28 (1H, d, J = 18.8 Hz), 5.96 (1H, d, J = 18.8 Hz), 2.16–2.07 (4H, m), 1.86–1.76 (2H, m), 1.70–1.61 (2H, m) 0.35 (6H, s); 13C NMR (100 MHz, CDCl3) δ 153.4, 133.9, 133.5, 129.1, 129.0, 127.9 (t, 1JC–F = 241.6 Hz), 127.8, 33.8, 33.7, −2.4, −3.6; HRMS (ES) calcd for C16H21F2OSi [M − H+]+ 295.1337. Found 295.1330.
:1 hexane–EtOAc) afforded 4p (240 mg, 64%) as a yellow oil.
R
f (4:1 hexane–ethyl acetate) = 0.41; IR: νmax (thin film)/cm−1; 3444, 2922, 2854, 1640, 1397, 1268, 1248, 1102, 845, 749; 1H NMR: (400 MHz, CDCl3) δ 7.51–7.48 (2H, m), 7.36–7.32 (3H, m), 6.21 (1H, dd, J = 18.8, 5.8 Hz), 6.03 (1H, d, J = 18.8 Hz), 4.03, (1H, s), 3.95 (4H, ddd, J = 5.5, 3.5, 1.7 Hz), 2.03–1.79 (4H, m), 1.65–1.57 (4H, m), 0.34 (6H, s); 13C NMR (100 MHz, CDCl3) δ 154.5, 138.5, 133.8, 129.1, 127.8, 123.7, 108.5, 71.9, 64.3, 64.2, 34.9, 30.4, −2.5; HRMS (ES) calcd for C18H30NO3Si [M + NH4]+ 336.1995. Found 336.1996.
:1 hexane–EtOAc) afforded 4q (47 mg, 77%) as a yellow oil.
R
f (9:1 hexane–ethyl acetate) = 0.26; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.47 (2H, m), 7.40–7.35 (3H, m), 7.17 (3H, m), 7.10 (1H, m), 6.31 (1H, dd, J = 18.8 Hz), 6.12 (1H, d, J = 18.8 Hz), 2.84–2.78 (2H, m), 2.21 (2H, m), 2.07–1.88 (2H, ddddd, J = 18.1, 12.3, 7.8, 6.0, 5.8 Hz), 0.35 (6H, s); 13C NMR (100 MHz, CDCl3) δ 156.2, 136.3, 131.5, 131.4, 130.6, 130.2, 130.0, 128.8, 127.1, 76.7, 40.1, 32.4, 32.2, 21.7, 12.2, 0.1, −0.6, −0.9; HRMS (ES) calcd for C20H25OSi [M + H]+ 309.1675. Found 309.1678.
:1 hexane–EtOAc) afforded 4r (55 mg, 76%) as a colourless solid.
R
f (4:1 hexane–ethyl acetate) = 0.35; IR: νmax (thin film)/cm−1; 3402, 3284, 2340, 2275, 1704, 1667, 1604, 1468, 1329, 1052, 962, 748, 667; 1H NMR: (400 MHz, CDCl3) δ 7.76 (1H, s), 7.55–7.49 (2H, d, J = 6.0, 3.6 Hz), 7.44–7.33 (3H, m), 7.25 (1H, m), 7.15–7.14 (2Hm), 6.37 (1H, d, J = 18.7 Hz), 6.17 (1H, d, J = 18.7 Hz), 2.95 (2H, d, J = 15.6 Hz), 2.88–2.70 (2H, dd, J = 14.4, 3.9 Hz), 2.04–1.87 (2H, m), 0.34 (6H, s); 13C NMR (100 MHz, CDCl3) δ153.0, 138.3, 136.0, 133.4, 132.2, 128.6, 127.5, 127.5, 124.3, 121.1, 118.9, 117.4, 110.0, 106.9, 76.9, 72.2, 34.1, 33.7, 19.7, −3.0; HRMS (ES) calcd for C22H26NOSi [M + H]+ 348.1784. Found 348.1778.
:1 hexane–EtOAc) afforded 4s (163 mg, 72%) as a yellow oil.
R
f (4:1 hexane–ethyl acetate) = 0.21; IR: νmax (thin film)/cm−1; 2977, 2881, 1679, 1427, 1367, 1248, 1158, 1114, 825, 764, 699, 667; 1H NMR: (400 MHz, CDCl3) δ 7.53–7.47 (2H, m), 7.40–7.35 (3H, m), 6.36 (1H, dd, J = 18.8 Hz), 6.12 (1H, d, J = 18.8 Hz), 3.98 (1H, d, J = 9.3 Hz), 3.92 (1H, d, J = 9.3 Hz), 1.44 (9H, s), 0.38 (6H, s); 13C NMR (100 MHz, CDCl3) δ 156.5, 148.5, 137.7, 128.9, 127.9, 127.8, 126.5, 79.8, 70.6, 62.3, 28.4, −2.8; HRMS (ES) calcd for C18H27NO3SiNa [M + NH4]+ 356.1658. Found 356.1661
R
f (9:1 hexane–ethyl acetate) = 0.35; IR: νmax (thin film)/cm−1; 3435, 3069, 2957, 1656, 1639, 1427, 1253, 1119, 1054, 1027, 998, 830, 789, 725, 698, 649; 1H NMR: (400 MHz, CDCl3) δ 7.54–7.44 (2H, m), 7.38–7.31 (3H, m), 6.06 (1H, d, J = 18.8 Hz), 5.90 (1H, d, J = 18.8 Hz), 2.51 (1H, s), 2.30–2.27 (3H, d, J = 7.4 Hz), 2.20–2.11 (3H, m), 2.07–1.90 (6 H, m), 0.33 (6H, s); 13C NMR (100 MHz, CDCl3) δ 157.0, 141.2, 136.3, 132.1, 130.1, 125.3, 37.2, 28.9, 25.0, 24.4, 22.0, −0.69; HRMS (ES) calcd for C16H24NOSi [M − CH3]+ 286.1627. Found 286.1661.
:1 hexane–EtOAc) afforded 4w (173 mg, 82%) as a yellow oil.
R
f (9:1 hexane–ethyl acetate) = 0.43; IR: νmax (thin film)/cm−1; 3426, 2922, 2854, 1640, 1397, 1268, 1247, 1113, 849, 825, 748, 699; 1H NMR: (400 MHz, CDCl3) δ 7.49 (2H, m), 7.41–7.33 (3H, m), 6.15 (1H, d, J = 18.8 Hz), 6.00 (1H, d, J = 18.8 Hz), 3.06–2.97 (4H, ddd, J = 10.3, 4.0, 3.8 Hz), 1.86–1.80 (4H, ddd, J = 13.5, 3.5, 1.8 Hz), 0.39 (6H, s); 13C NMR (100 MHz, CDCl3) δ 154.4, 138.4, 133.8, 132.9. 129.7, 129.1, 127.8, 115.3, 71.3, 38.0, 24.0, −2.6; HRMS (ES) calcd for C15H2721OSSi [M − H]+ 277.1082. Found 277.1062.
:1 hexane–EtOAc) afforded 5c (173 mg, 84%) as a yellow oil.
R
f (9:1 hexane–ethyl acetate) = 0.76; IR: νmax (thin film)/cm−1; 3362, 2953, 2928, 2856, 1616, 1470, 1361, 1390, 1247, 1147, 963, 908, 830; 1H NMR: (400 MHz, CDCl3) δ 6.18 (1H, d, J = 18.8 Hz), 5.84 (1H, d, J = 18.8 Hz), 1.31 (6H, s), 0.88 (9H, s), 0.04 (6H, s); 13C NMR (100 MHz, CDCl3) δ 154.6, 121.8, 72.1, 29.5, 26.4, 16.6, −6.14; HRMS (ES) calcd for C11H24NaOSi [M + Na]+ 223.1494. Found 223.1397.
:1 hexane–EtOAc) afforded 5f (808 mg, 97%) as a colourless oil.
R
f (9:1 hexane–EtOAc) = 0.45; IR: νmax (thin film)/cm−1; 3496, 2900, 2655, 1644, 1066, 978; 1H NMR: (400 MHz, CDCl3) δ 7.24–7.17 (2H, m), 7.10–7.04 (1H, m), 7.01–6.97 (2H, m), 6.09 (1H, d, J = 19.1 Hz), 5.77 (1H, d, J = 19.1 Hz), 2.15 (2H, s), 1.42 (1H, s), 1.29 (6H, s), 0.06 (6H, s); 13C NMR (100 MHz, CDCl3) δ154.5, 139.9, 128.3, 128.0, 124.0, 122.6, 74.0, 30.9, 29.3, 26.1, −3.4; HRMS (ES) calcd for C14H23OSi [M + H]+ 235.1518. Found 235.1528.
Acknowledgements
We thank Queen's University Belfast and EPSRC (Grant Number: EP/I006605/1) for support. We are also grateful to Eli Lilly for a CASE award (CAM) and the Department of Employment and Learning, Northern Ireland for a studentship (MGM). GlaxoSmithKline and AstraZeneca are also thanked for their generous donation of laboratory equipment. We also gratefully acknowledge the reviewers for insightful comments.Notes and references
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS.
- A. Hosomi, M. Endo and H. Sakurai, Chem. Lett., 1976, 941 CrossRef CAS.
- R. R. Teizeira, L. C. A. Barbosa and D. Pilo-Veloso, Quim. Nova, 2007, 30, 1704–1720 CrossRef CAS.
- S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS.
- J. P. Pezacki, P. G. Loncke, J. P. Ross, J. Warkentin and T. A. Gadosy, Org. Lett., 2000, 2, 2733–2736 CrossRef CAS.
- S. S. Magar and P. L. Fuchs, Tetrahedron Lett., 1991, 32, 7513–7516 CrossRef CAS.
- K. Suzuki, K. Inomata and Y. Endo, Org. Lett., 2004, 6, 409–411 CrossRef CAS.
- G. B. Dudley, D. A. Engel, I. Ghiviriga, H. Lam, K. W. C. Poon and J. A. Singletary, Org. Lett., 2007, 9, 2839–2842 CrossRef CAS.
- D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 983–1010 CAS.
- I. Fleming, T. W. Newton and F. Roessler, J. Chem. Soc., Perkin Trans. 1, 1981, 2527 RSC.
- I. Fleming and N. J. Lawrence, J. Chem. Soc., Perkin Trans. 1, 1998, 2679 RSC.
- B. H. Lipshutz, J. A. Sclafini and T. Takanami, J. Am. Chem. Soc., 1998, 120, 4021–4022 CrossRef CAS.
- G. Reginato, A. Mordini, M. Valacchi and E. Grandini, J. Org. Chem., 1999, 64, 9211–9216 CrossRef CAS.
- G. Auer and M. Oestreich, Chem. Commun., 2006, 311–313 RSC.
- E. S. Schmidtmann and M. Oestreich, Chem. Commun., 2006, 3643–3645 RSC.
- K. Takai, Y. Kataoka, T. Okazoe and K. Utimoto, Tetrahedron Lett., 1987, 28, 1443–1446 CrossRef CAS.
- D. S. W. Lim and E. A. Anderson, Org. Lett., 2011, 13, 4806 CrossRef CAS.
- Y. Kita, M. Tobisu and N. Chatani, Org. Lett., 2010, 12, 1864 CrossRef CAS.
- K. Itami, T. Nokami and J.-i. Yoshida, Org. Lett., 2000, 2, 1299 CrossRef CAS.
- A. L. Moure, R. Gómez Arrayás, D. J. Cárdenas, I. Alonso and J. C. Carretero, J. Am. Chem. Soc., 2012, 134, 7219–7222 CrossRef CAS.
- B. M. Trost and Z. T. Ball, Synthesis, 2005, 887 Search PubMed.
- B. Marciniec, in Advances in Silicon Science, Springer Science, 2009 Search PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS.
- N. Asao, T. Sudo and Y. Yamamoto, J. Org. Chem., 1996, 61, 7654–7655 CrossRef CAS.
- E. Yoshikawa, V. Gevorgyan, N. Asao and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 6781–6786 CrossRef CAS.
- V. Gevorgyan, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 1997, 62, 2963–2967 CrossRef CAS.
- T. Sudo, N. Asao, V. Gevorgyan and Y. Yamamoto, J. Org. Chem., 1999, 64, 2494–2499 CrossRef CAS.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16–21 CrossRef CAS.
- S. B. Duckett and R. N. Perutz, Organometallics, 1992, 11, 90–98 CrossRef CAS.
- A. K. Roy, in Adv. Organomet. Chem., ed. A. F. H. Robert West and J. F. Mark, Academic Press, 2007, vol. 55, pp. 1–59 Search PubMed.
- S. Clarson, Silicon, 2009, 1, 57–58 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2007, 27, 224–234 CrossRef.
- R. Takeuchi, S. Nitta and D. Watanabe, J. Org. Chem., 1995, 60, 3045–3051 CrossRef CAS.
- R. Takeuchi and N. Tanouchi, J. Chem. Soc., Chem. Commun., 1993, 1319–1320 RSC.
- R. Takeuchi and N. Tanouchi, J. Chem. Soc., Perkin Trans. 1, 1994, 2909–2913 RSC.
- M. A. Esteruelas, J. Herrero and L. A. Oro, Organometallics, 1993, 12, 2377–2379 CrossRef CAS.
- M. Rivera-Claudio, J. Rozell, E. Ramirez-Oliva, J. Cervantes and K. H. Pannell, J. Organomet. Chem., 1996, 521, 267–270 CrossRef CAS.
- Y. Na and S. Chang, Org. Lett., 2000, 2, 1887–1889 CrossRef CAS.
- T. Takahashi, F. Bao, G. Gao and M. Ogasawara, Org. Lett., 2003, 5, 3479–3481 CrossRef CAS.
- R. Cano, M. Yus and D. J. Ramón, ACS Catal., 2012, 2, 1070–1078 CrossRef CAS.
- L. N. Lewis, K. G. Sy, G. L. Bryant and P. E. Donahue, Organometallics, 1991, 10, 3750–3759 CrossRef CAS.
- M. G. Voronkov, V. B. Pukhnarevich, I. I. Tsykhanskaya, N. I. Ushakova, Y. L. Gaft and I. A. Zakharova, Inorg. Chim. Acta, 1983, 68, 103–105 CrossRef CAS.
- J. Stein, L. N. Lewis, Y. Gao and R. A. Scott, J. Am. Chem. Soc., 1999, 121, 3693–3703 CrossRef CAS.
- G. Chandra, P. Y. Lo, P. B. Hitchcock and M. F. Lappert, Organometallics, 1987, 6, 191–192 CrossRef CAS.
- P. J. Murphy, J. L. Spencer and G. Procter, Tetrahedron Lett., 1990, 31, 1051–1054 CrossRef CAS.
- D. A. Rooke and E. M. Ferreira, Angew. Chem., Int. Ed., 2012, 51, 3225–3230 CrossRef CAS.
- D. A. Rooke and E. M. Ferreira, J. Am. Chem. Soc., 2010, 132, 11926–11928 CrossRef CAS.
- G. Berthon-Gelloz and I. E. Markò, N-Heterocyclic Carbenes in Synthesis, Wiley-VCH Verlag GmbH & Co. KgaA, 2006 Search PubMed.
- S. Dìez-Gonzàlez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef.
- A. S. K. Hashmi, C. Lothschütz, C. Böhling, T. Hengst, F. Hubbert and F. Rominger, Adv. Synth. Catal., 2010, 352, 3001 CrossRef CAS.
- P. D. Newman, K. J. Cavell and B. M. Kariuki, Dalton Trans., 2012, 41, 12395–12407 RSC.
- A. S. K. Hashmi, C. Lothschuetz, K. Graf, T. Haeffner, A. Schuster and F. Rominger, Adv. Synth. Catal., 2011, 353, 1407–1412 CrossRef CAS.
- X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. LBuchwald, J. Am. Chem. Soc., 2003, 125, 6653–6655 CrossRef CAS.
- A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789–2797 CrossRef CAS.
- M. G. McLaughlin and M. J. Cook, J. Org. Chem., 2012, 77, 2058–2063 CrossRef CAS.
- C. E. Masse, L. A. Dakin, B. S. Knight and J. S. Panek, J. Org. Chem., 1997, 62, 9335–9338 CrossRef CAS.
- R. T. Beresis, J. S. Solomon, M. G. Yang, N. F. Jain and J. S. Panek, Org. Synth., 2004, Coll. Vol. 10, 531 Search PubMed.
- M. G. McLaughlin and M. J. Cook, Chem. Commun., 2011, 47, 11104–11106 RSC.
- W. Wu and C.-J. Li, Chem. Commun., 2003, 1668–1669 RSC.
- H. Sasabe, N. Kihara, K. Mizuno, A. Ogawa and T. Takata, Tetrahedron Lett., 2005, 46, 3851 CrossRef CAS.
- S. E. Denmark and J. H. C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978–2986 CrossRef CAS.
- G. Berthon-Gelloz, J.-M. Schumers, G. De Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190–4197 CrossRef CAS.
- A. Hamze, O. Provot, M. Alami and J.-D. Brion, Org. Lett., 2005, 7, 5625–5628 CrossRef CAS.
- T. Konno, K.-I. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167–1170 CAS.
- F. Liron, P. Le Garrec and M. Alami, Synlett, 1999, 248 Search PubMed.
- A. Ishii and N. Nakata, Top. Organmet. Chem., 2013, 43, 21–50 CrossRef CAS.
- Y. Inoue, J. Ishikawa, M. Taniguchi and H. Hashimoto, Bull. Chem. Soc. Jpn., 1987, 60, 1204–1206 CrossRef CAS.
- D. A. S. El, Z. M. A. Judeh, H. Basri, Z. Moussa, M. Messali and G. Qi, Synth. Commun., 2011, 41, 533–540 CrossRef.
- C.-F. Xu, M. Xu, L.-Q. Yang and C.-Y. Li, J. Org. Chem., 2012, 77, 3010–3016 CrossRef CAS.
- U. Galli, E. Ercolano, L. Carraro, C. R. B. Roman, G. Sorba, P. L. Canonico, A. A. Genazzani, G. C. Tron and R. A. Billington, ChemMedChem, 2008, 3, 771–779 CrossRef CAS.
- V. Singh and V. Singh, Synth. Commun., 2010, 40, 1280–1291 CrossRef CAS.
- J.-J. Liu, F. Konzelmann and K.-C. Luk, Tetrahedron Lett., 2003, 44, 2545–2548 CrossRef CAS.
- E. A. Shapiro, A. V. Kalinin, B. I. Ugrak and O. M. Nefedov, J. Chem. Soc., Perkin Trans. 2, 1994, 709–713 RSC.
- L. R. M. De, WO, 2010135536A2, 2010 Search PubMed.
- F. Iqbal, H. S. Ateeq, A. Malik, Z. Ali and Z. Ali, Z. Naturforsch., B: Chem. Sci., 1998, 53, 1244–1245 CAS.
- G. Frater and M. Nagel, EP, 1236707A1, 2002 Search PubMed.
- C. Yoshida and K. Nakao, WO, 2011125836A1, 2011 Search PubMed.
- J. A. Moore, J. W. Roche, P. Prince, R. D. Gandour and F. R. Fronczek, Org. Prep. Proced. Int., 1989, 21, 386–388 CrossRef CAS.
- J. A. Burkhard, C. Guerot, H. Knust and E. M. Carreira, Org. Lett., 2012, 14, 66–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of starting material synthesis and spectra of the compounds reported (1H and 13C NMR) are included. See DOI: 10.1039/c3ob40496j |
| This journal is © The Royal Society of Chemistry 2013 |